
Development, Optimization and Validation of a Sustainable and Quantifiable Methodology for the Determination of 2,4,6-Trichloroanisole, 2,3,4,6-Tetrachloroanisole, 2,4,6-Tribromoanisole, Pentachloroanisole, 2-Methylisoborneole and Geosmin in Air



Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents and Solutions
2.2. Sorbent Tubes
2.3. Optimization and Validation of the TD-GCMS Method
2.4. Determination of R-Values
2.4.1. Passive Sampling Theory
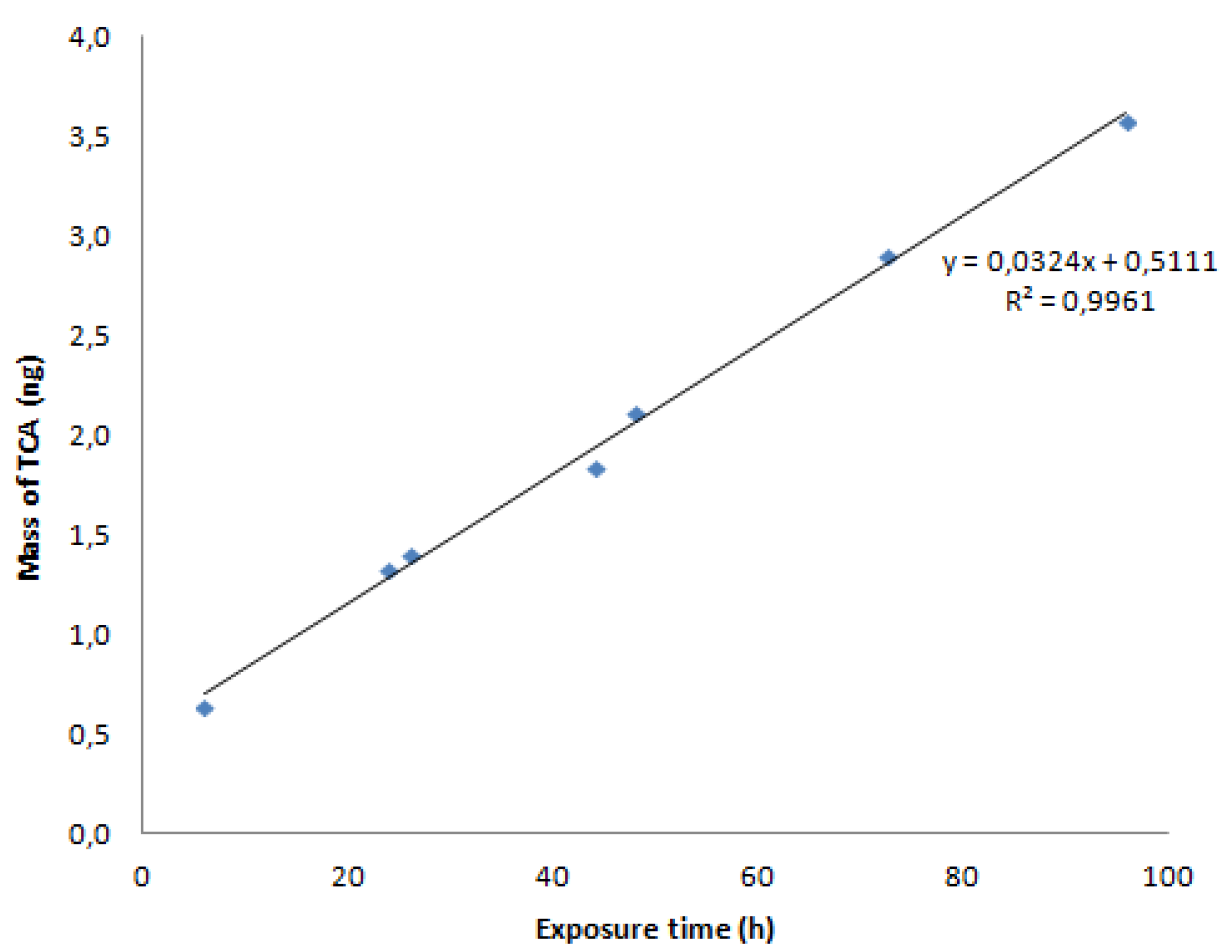
2.4.2. Experimental Determination of R-Values
2.5. Data Analysis
2.5.1. Optimization
2.5.2. Determination of R-Values
3. Results
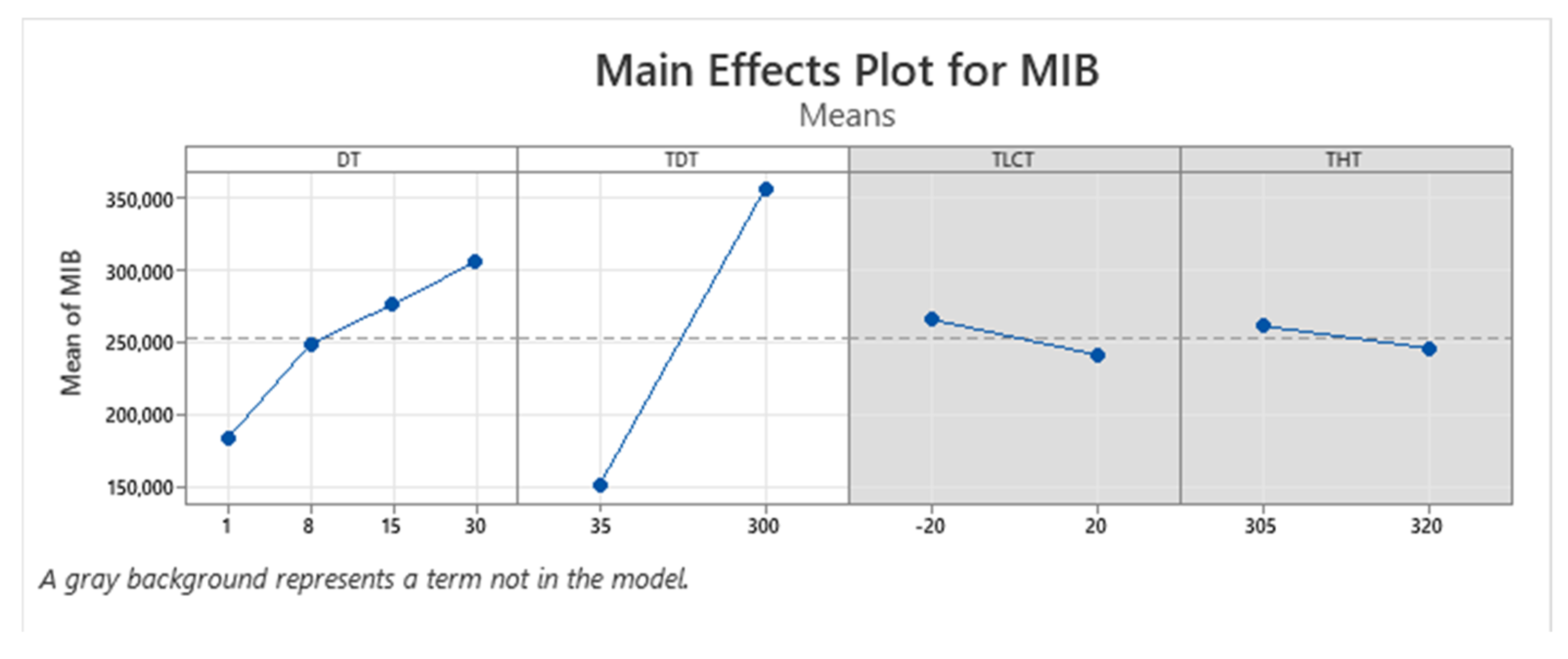
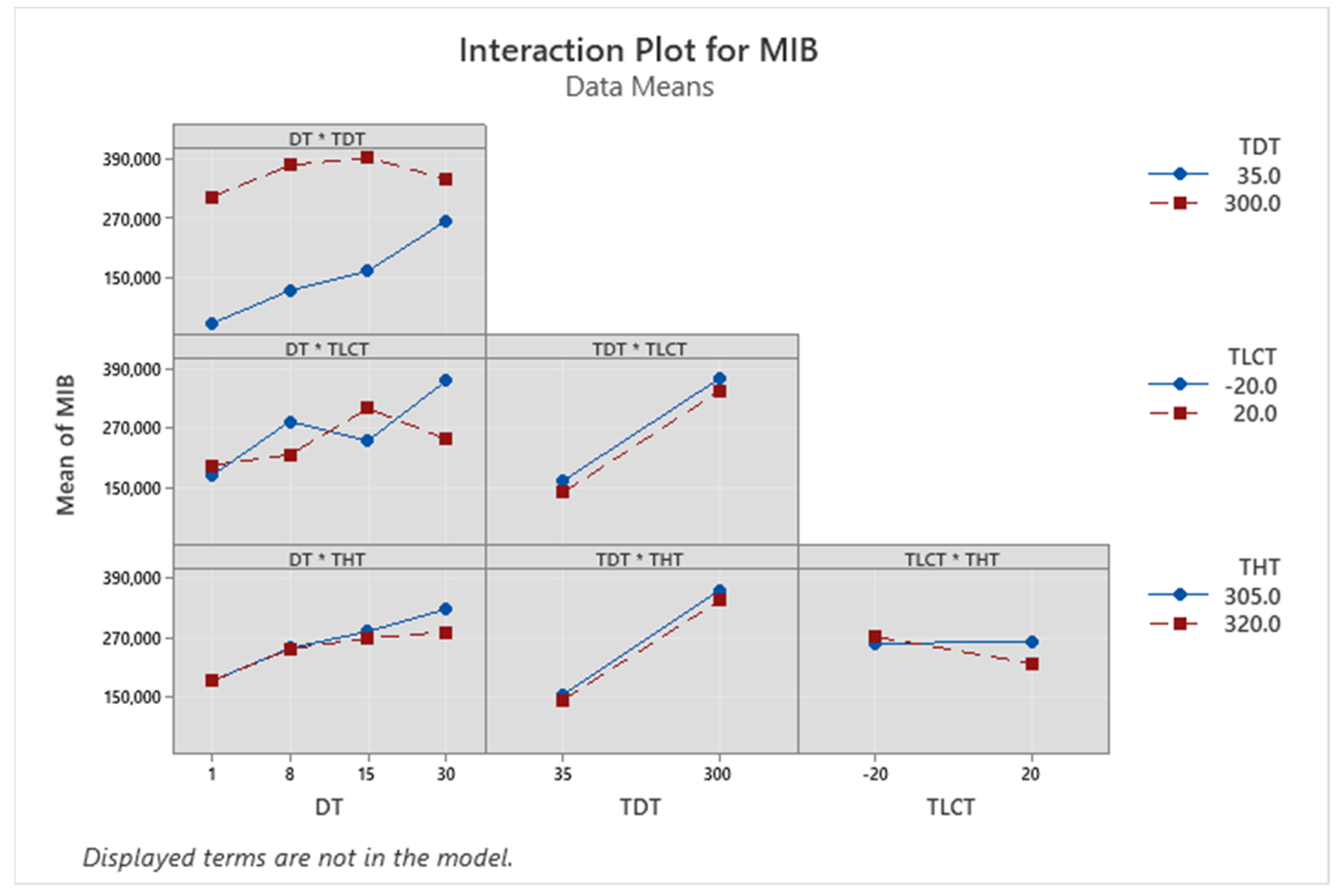
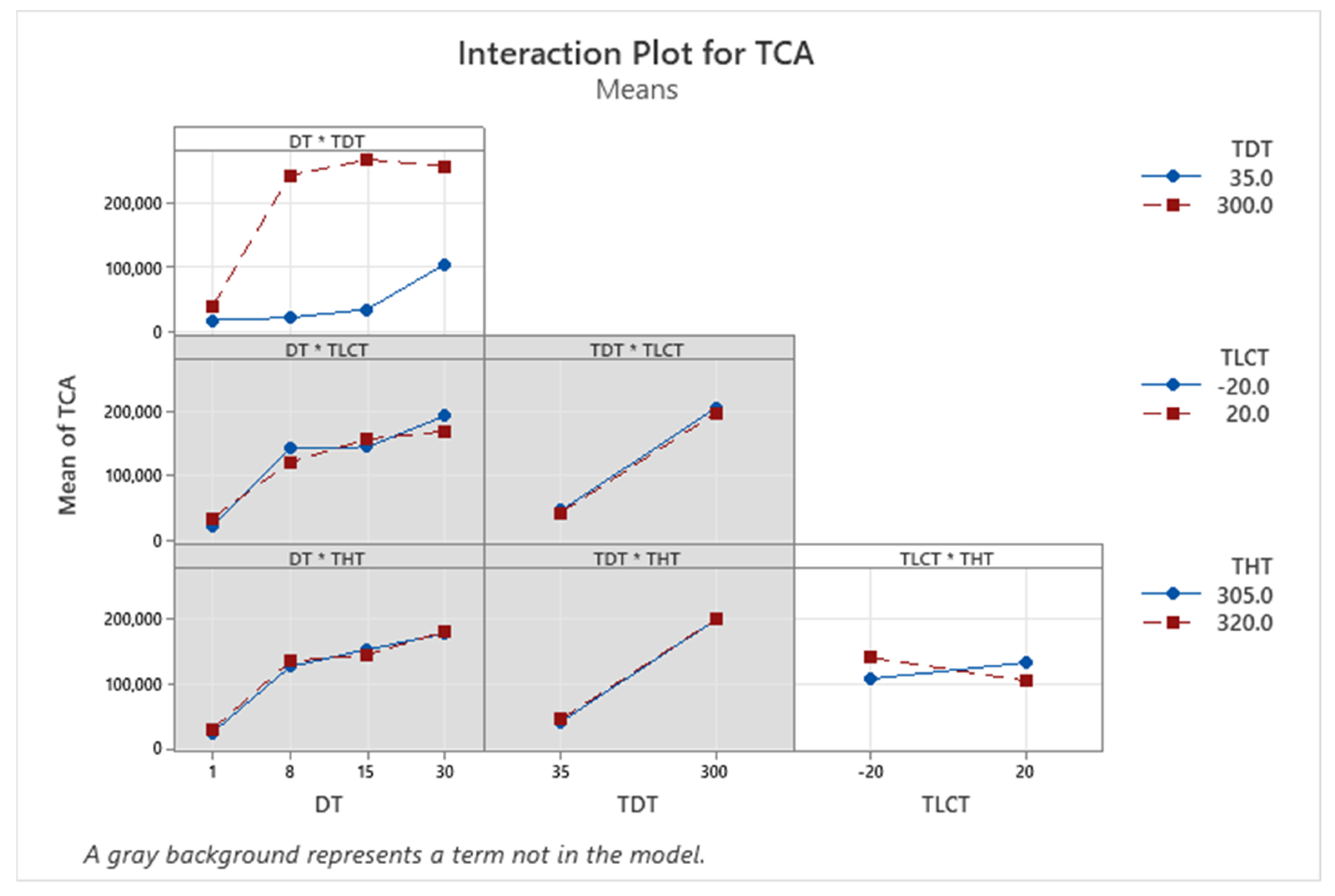
3.1. Optimization of Thermal Desorption Method (TD)
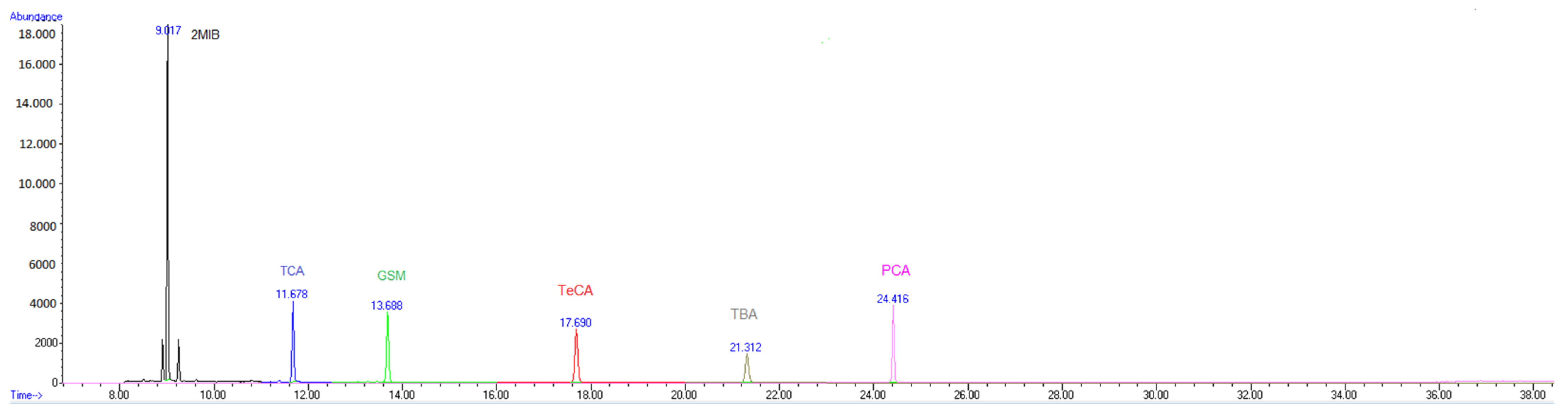
3.2. TD-GC-MS Method Validation
3.3. R-Values
3.4. Application of the Method in Wineries
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Term | Coef | SE Coef | p-Value |
|---|---|---|---|
| Constant | 253,509 | 14,785 | 0.000 |
| DT = 1 | −69,950 | 25,609 | 0.008 |
| DT = 8 | −4656 | 25,609 | 0.856 |
| DT = 15 | 22,802 | 25,609 | 0.377 |
| TDT = 35 | −102,599 | 14,785 | 0.000 |
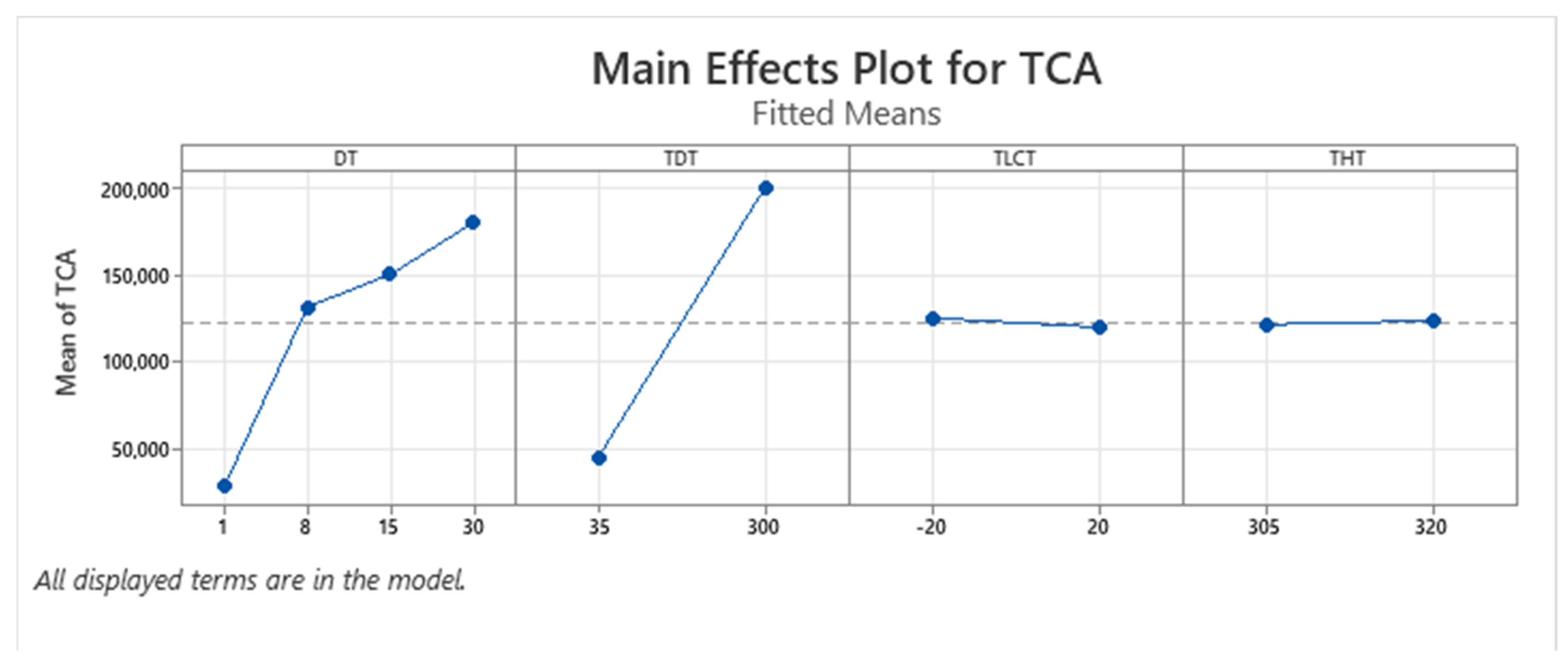
| Term | Coef | SE Coef | p-Value |
|---|---|---|---|
| Constant | 122,107 | 7947 | 0.000 |
| DT = 1 | −94,342 | 13,764 | 0.000 |
| DT = 8 | 9002 | 13,764 | 0.516 |
| DT = 15 | 27,766 | 13,764 | 0.049 |
| TDT = 35 | −77,931 | 7947 | 0.000 |
| TLCT = -20 | 2701 | 7947 | 0.735 |
| THT = 305 | −1109 | 7947 | 0.889 |
| DT = 1 TDT = 35 | 67,602 | 13,764 | 0.000 |
| DT = 8 TDT = 35 | −31,962 | 13,764 | 0.024 |
| DT = 15 TDT = 35 | −37,698 | 13,764 | 0.008 |
| TLCT = -20 THT = 305 | −15,219 | 7947 | 0.061 |
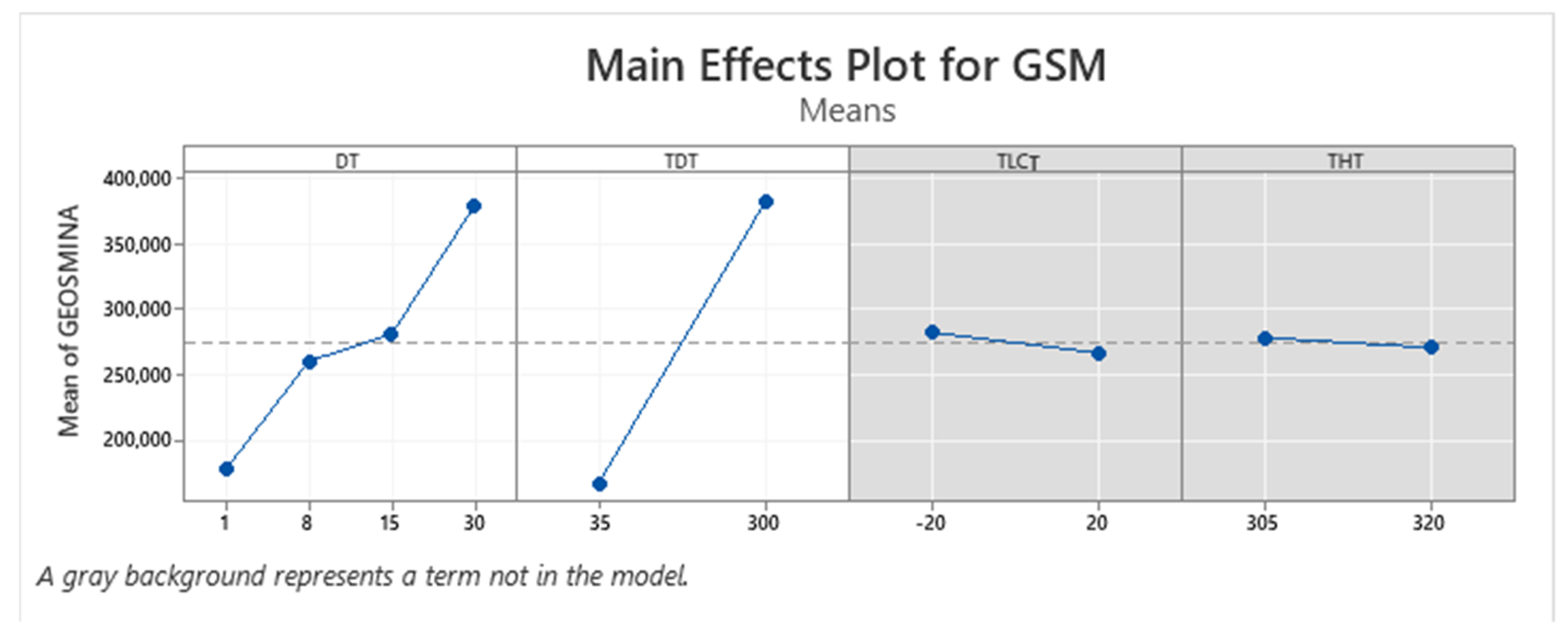
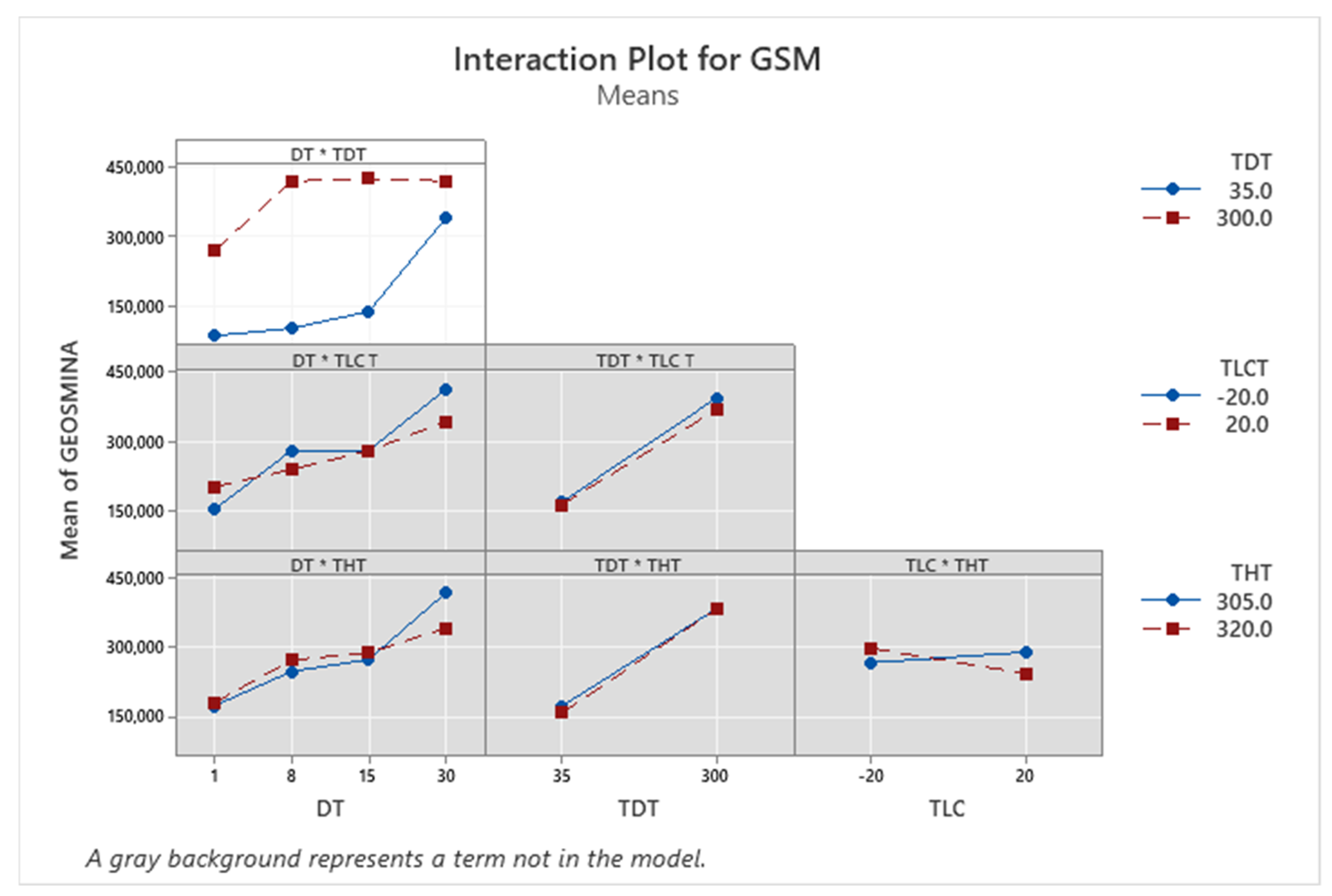
| Term | Coef | SE Coef | p-Value |
|---|---|---|---|
| Constant | 274,084 | 14,858 | 0.000 |
| DT = 1 | −96,736 | 25,735 | 0.000 |
| DT = 8 | −14,084 | 25,735 | 0.586 |
| DT = 15 | 6418 | 25,735 | 0.804 |
| TDT = 35 | −107,638 | 14,858 | 0.000 |
| DT = 1 TDT = 35 | 16,419 | 25,735 | 0.526 |
| DT = 8 TDT = 35 | −49,966 | 25,735 | 0.057 |
| DT = 15 TDT = 35 | −34,701 | 25,735 | 0.183 |
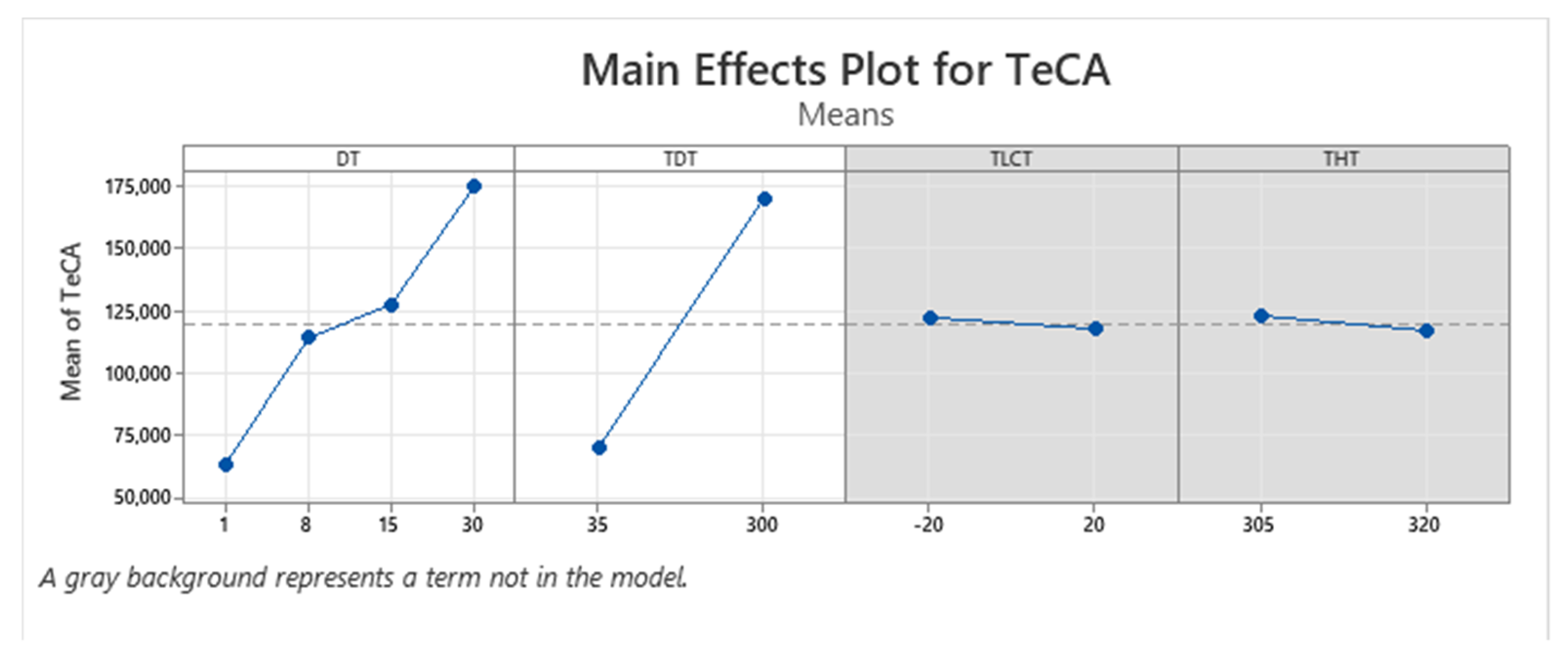
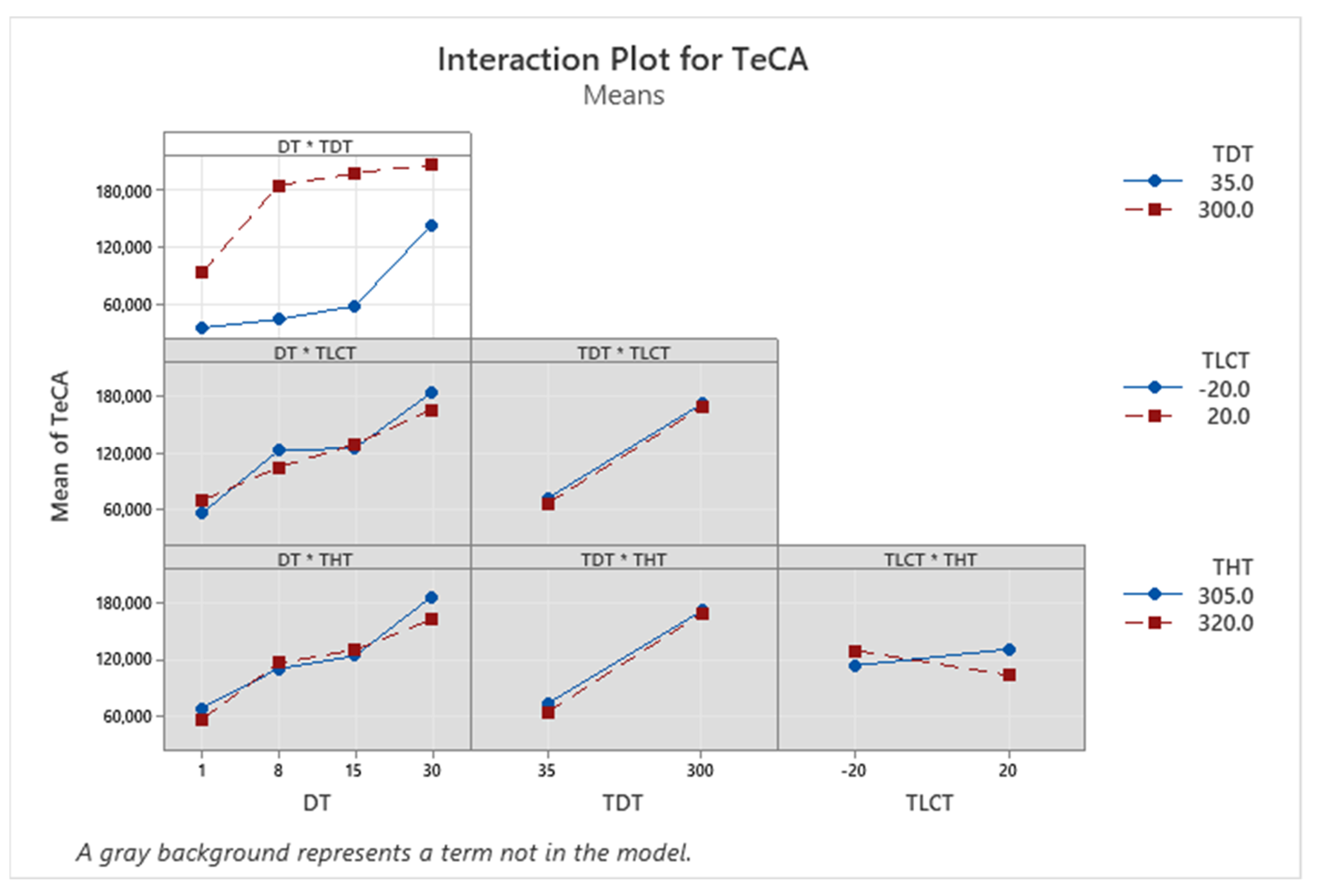
| Term | Coef | SE Coef | p-Value |
|---|---|---|---|
| Constant | 119,559 | 6706 | 0.000 |
| DT = 1 | −56,416 | 11,615 | 0.000 |
| DT = 8 | −5993 | 11,615 | 0.608 |
| DT = 15 | 7603 | 11,615 | 0.515 |
| TDT = 35 | −50,143 | 6706 | 0.000 |
| DT = 1 TDT = 35 | 21,250 | 11,615 | 0.073 |
| DT = 8 TDT = 35 | −20,271 | 11,615 | 0.086 |
| DT = 15 TDT = 35 | −19,625 | 11615 | 0.097 |
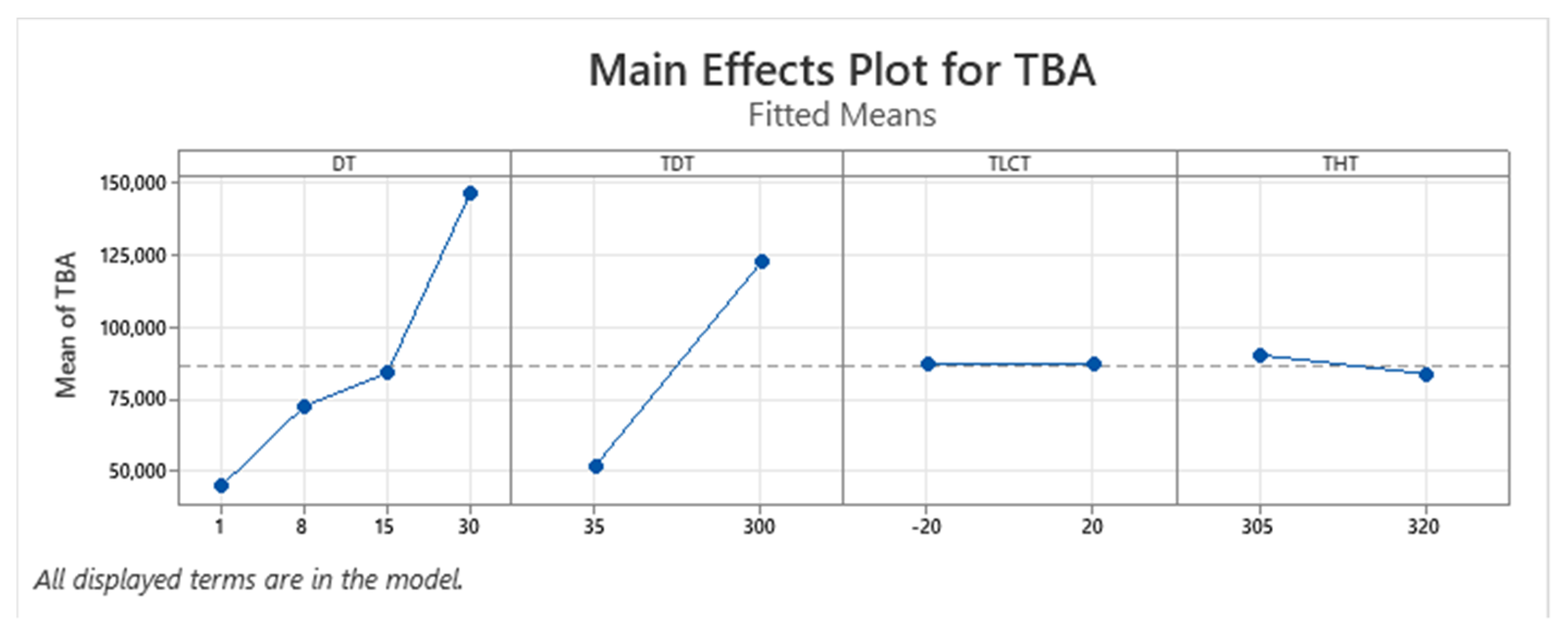
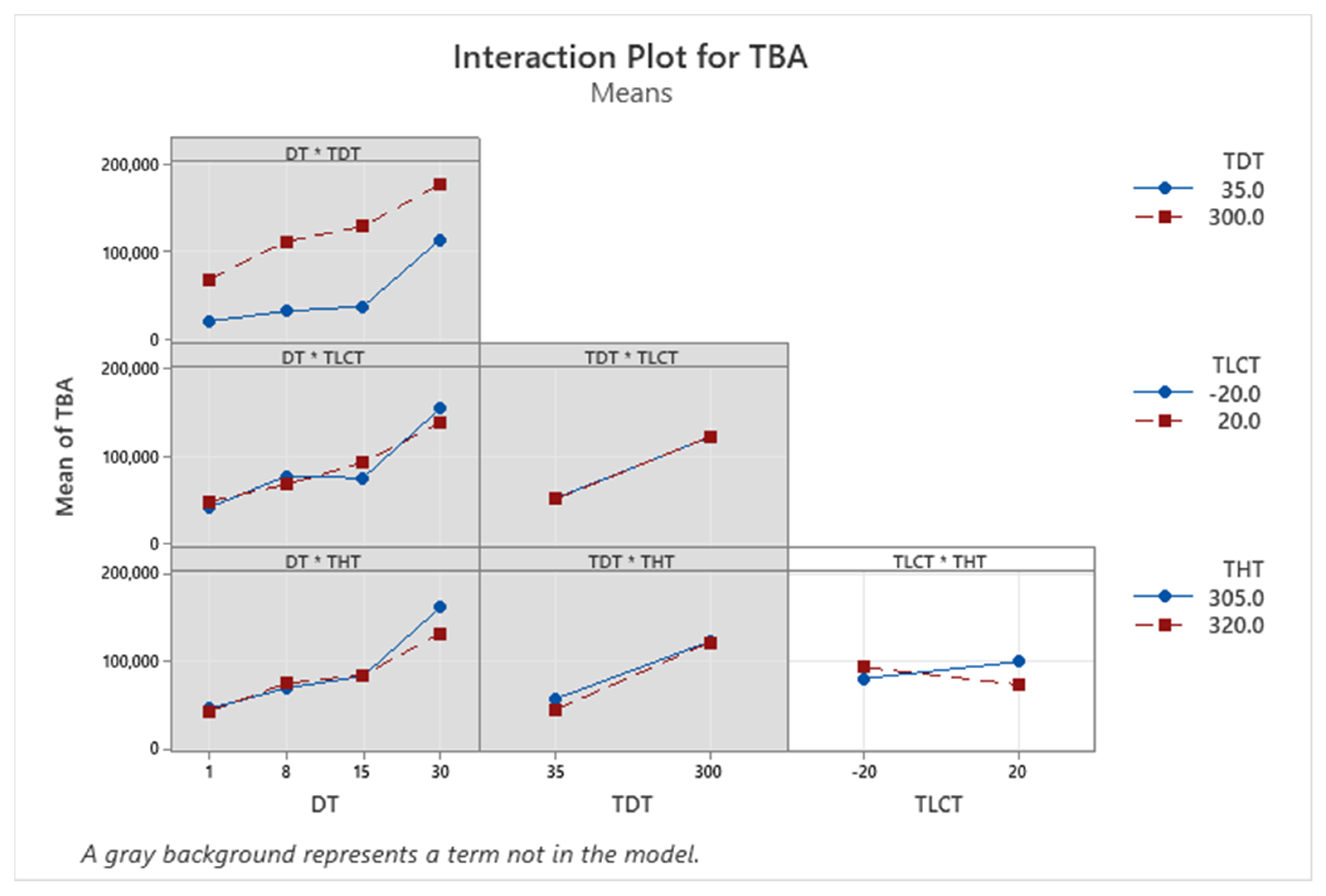
| Term | Coef | SE Coef | p-Value |
|---|---|---|---|
| Constant | 86,714 | 5317 | 0.000 |
| DT = 1 | −42,159 | 9210 | 0.000 |
| DT = 8 | −14,411 | 9210 | 0.123 |
| DT = 15 | −2959 | 9210 | 0.749 |
| TDT = 35 | −35,341 | 5317 | 0.000 |
| TLCT = −20 | 124 | 5317 | 0.981 |
| THT = 305 | 3251 | 5317 | 0.543 |
| TLCT = −20 THT = 305 | −9584 | 5317 | 0.077 |
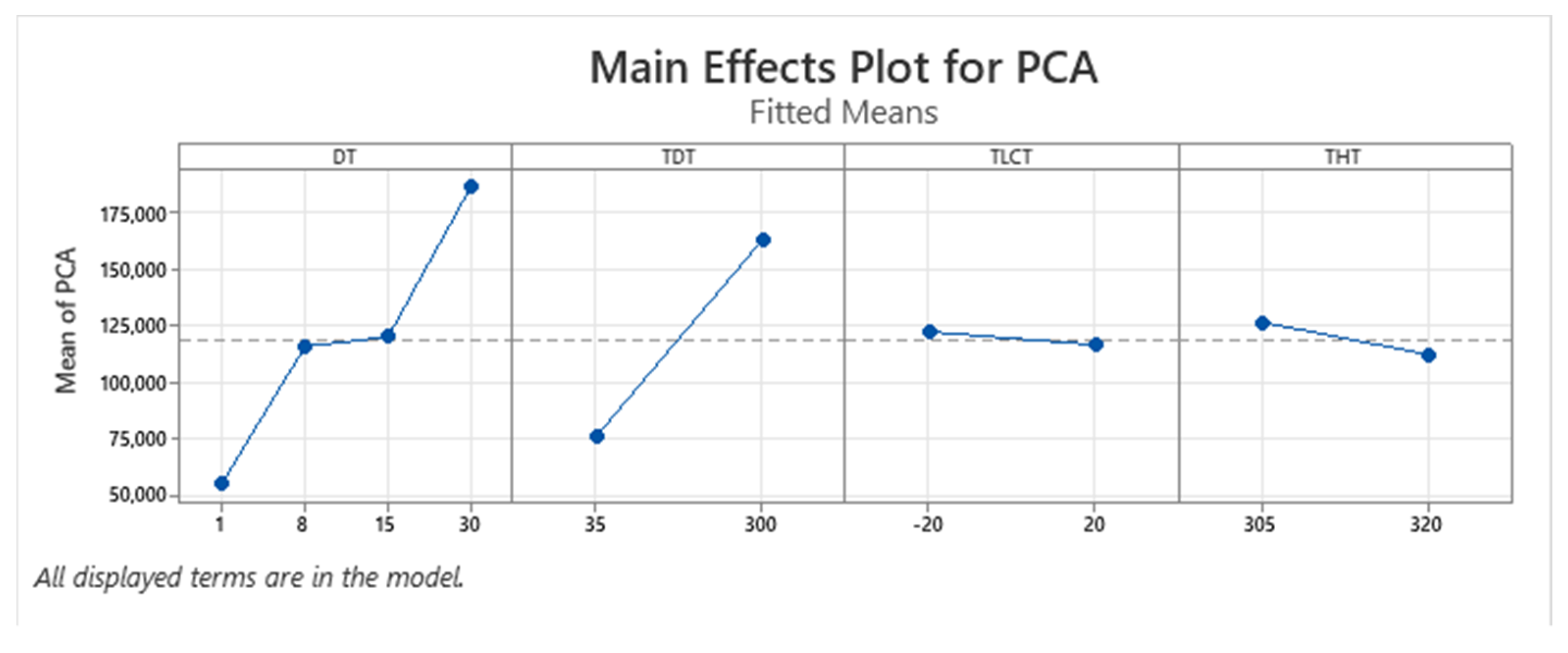
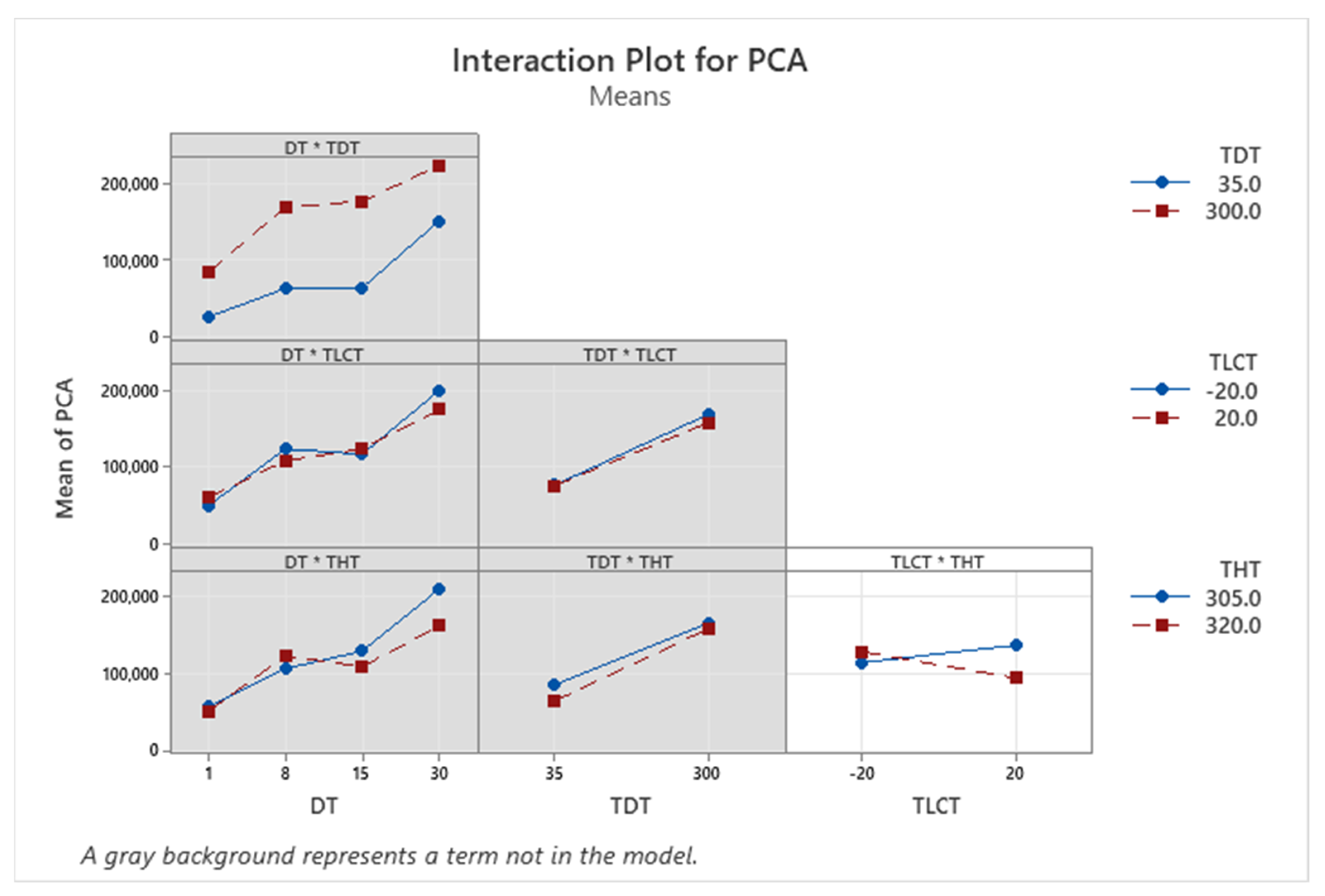
| Term | Coef | SE Coef | p-Value |
|---|---|---|---|
| Constant | 118,958 | 7386 | 0.000 |
| DT = 1 | −64,400 | 12,794 | 0.000 |
| DT = 8 | −3661 | 12,794 | 0.776 |
| DT = 15 | 620 | 12,794 | 0.962 |
| TDT = 35 | −43,603 | 7386 | 0.000 |
| TLCT = −20 | 2866 | 7386 | 0.699 |
| THT = 305 | 7159 | 7386 | 0.337 |
| TLCT = −20 THT = 305 | −14,159 | 7386 | 0.060 |
Appendix B
| Run (Standard Order) | DT | TDT | TLCT | THT | MIB | TCA | GSM | TeCA | TBA | PCA |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 1 | 35 | −20 | 305 | 39,729 | 5980 | 51,859 | 30,126 | 24,167 | 29,759 |
| 2 | 1 | 35 | −20 | 305 | 41,691 | 2051 | 26,508 | 37,057 | 19,137 | 41,719 |
| 3 | 8 | 35 | −20 | 305 | 1062 | 5329 | 2128 | 7549 | 4438 | 8390 |
| 4 | 8 | 35 | −20 | 305 | 113,150 | 27,479 | 123,655 | 67,597 | 45,365 | 79,090 |
| 5 | 15 | 35 | −20 | 305 | 287,128 | 73,984 | 282,752 | 122,206 | 68,384 | 108,877 |
| 6 | 15 | 35 | −20 | 305 | 29,209 | 13,847 | 38,480 | 15,355 | 9794 | 18,525 |
| 7 | 30 | 35 | −20 | 305 | 356,711 | 102,751 | 443,958 | 177,078 | 152,746 | 207,312 |
| 8 | 30 | 35 | −20 | 305 | 528,734 | 130,252 | 480,067 | 167,733 | 140,451 | 205,729 |
| 9 | 1 | 300 | −20 | 305 | 321,284 | 31,773 | 249,483 | 74,043 | 73,626 | 101,608 |
| 10 | 1 | 300 | −20 | 305 | 223,313 | 26,527 | 198,734 | 75,342 | 47,031 | 46,104 |
| 11 | 8 | 300 | −20 | 305 | 506,839 | 291,312 | 519,946 | 225,261 | 139,775 | 208,774 |
| 12 | 8 | 300 | −20 | 305 | 410,427 | 241,360 | 429,038 | 184,862 | 111,428 | 162,671 |
| 13 | 15 | 300 | −20 | 305 | 352,257 | 245,729 | 418,726 | 191,182 | 114,042 | 176,334 |
| 14 | 15 | 300 | −20 | 305 | 457,017 | 253,548 | 470,958 | 190,315 | 115,926 | 165,099 |
| 15 | 30 | 300 | −20 | 305 | 430,410 | 276,330 | 441,079 | 219,637 | 184,751 | 229,804 |
| 16 | 30 | 300 | −20 | 305 | 41,811 | 7421 | 88,853 | 38,062 | 37,029 | 47,382 |
| 17 | 1 | 35 | 20 | 305 | 12,286 | 6455 | 17,981 | 3676 | 800 | 1225 |
| 18 | 1 | 35 | 20 | 305 | 57,049 | 10,238 | 97,838 | 48,081 | 39,478 | 53,325 |
| 19 | 8 | 35 | 20 | 305 | 165,375 | 16,977 | 125,683 | 35,766 | 35,003 | 74,158 |
| 20 | 8 | 35 | 20 | 305 | 95,354 | 14,450 | 81,251 | 55,176 | 41,365 | 63,699 |
| 21 | 15 | 35 | 20 | 305 | 256,854 | 31,085 | 168,048 | 55,463 | 48,429 | 88,828 |
| 22 | 15 | 35 | 20 | 305 | 66,263 | 65,907 | 152,733 | 69,075 | 26,870 | 38,613 |
| 23 | 30 | 35 | 20 | 305 | 200,276 | 83,481 | 314,728 | 144,049 | 113,449 | 146,723 |
| 24 | 30 | 35 | 20 | 305 | 239,546 | 86,190 | 372,967 | 143,877 | 149,889 | 211,230 |
| 25 | 1 | 300 | 20 | 305 | 457,141 | 25,281 | 475,253 | 164,570 | 88,351 | 88,197 |
| 26 | 1 | 300 | 20 | 305 | 316,784 | 93,303 | 275,923 | 119,945 | 77,313 | 99,826 |
| 27 | 8 | 300 | 20 | 305 | 371,922 | 216,136 | 350,845 | 151,718 | 85,765 | 117,775 |
| 28 | 8 | 300 | 20 | 305 | 332,791 | 202,924 | 350,717 | 156,955 | 93,797 | 140,004 |
| 29 | 15 | 300 | 20 | 305 | 467,305 | 306,122 | 238,279 | 154,174 | 154,174 | 250,276 |
| 30 | 15 | 300 | 20 | 305 | 350,350 | 240,497 | 416,198 | 194,518 | 127,276 | 190,512 |
| 31 | 30 | 300 | 20 | 305 | 446,550 | 401,763 | 645,897 | 325,578 | 275,582 | 346,003 |
| 32 | 30 | 300 | 20 | 305 | 380,842 | 335,447 | 542,584 | 274,466 | 233,260 | 288,172 |
| 33 | 1 | 35 | −20 | 320 | 34,947 | 6040 | 51,342 | 28,349 | 19,429 | 20,902 |
| 34 | 1 | 35 | −20 | 320 | 74,483 | 30,764 | 147,709 | 52,043 | 28,349 | 23,773 |
| 35 | 8 | 35 | −20 | 320 | 263,973 | 38,899 | 187,344 | 82,755 | 51,849 | 87,776 |
| 36 | 8 | 35 | −20 | 320 | 135,191 | 13,940 | 75,521 | 21,590 | 27,166 | 69,227 |
| 37 | 15 | 35 | −20 | 320 | 6711 | 2025 | 2963 | 1651 | 1240 | 2013 |
| 38 | 15 | 35 | −20 | 320 | 177,200 | 33,002 | 156,131 | 68,055 | 41,884 | 71,770 |
| 39 | 30 | 35 | −20 | 320 | 272,980 | 97,014 | 339,391 | 136,651 | 106,183 | 140,807 |
| 40 | 30 | 35 | −20 | 320 | 240,804 | 147,960 | 297,407 | 138,987 | 88,768 | 96,628 |
| 41 | 1 | 300 | −20 | 320 | 209,496 | 29,537 | 199,364 | 58,513 | 43,207 | 51,021 |
| 42 | 1 | 300 | −20 | 320 | 438,840 | 38,076 | 294,124 | 95,677 | 72,674 | 80,455 |
| 43 | 8 | 300 | −20 | 320 | 522,384 | 315,696 | 558,821 | 237,272 | 146,785 | 229,281 |
| 44 | 8 | 300 | −20 | 320 | 307,007 | 206,712 | 347,495 | 151,281 | 89,498 | 138,143 |
| 45 | 15 | 300 | −20 | 320 | 211,628 | 255,560 | 392,838 | 198,331 | 117,156 | 181,967 |
| 46 | 15 | 300 | −20 | 320 | 427,898 | 272,181 | 484,278 | 214,030 | 131,009 | 204,106 |
| 47 | 30 | 300 | −20 | 320 | 585,382 | 411,957 | 646,920 | 335,835 | 281,271 | 357,102 |
| 48 | 30 | 300 | −20 | 320 | 453,946 | 358,835 | 570,533 | 253,783 | 244,263 | 306,228 |
| 49 | 1 | 35 | 20 | 320 | 59,301 | 40,814 | 152,110 | 47,271 | 22,924 | 22,668 |
| 50 | 1 | 35 | 20 | 320 | 128,583 | 37,143 | 143,682 | 27,395 | 11,213 | 9617 |
| 51 | 8 | 35 | 20 | 320 | 134,804 | 13,087 | 97,797 | 25,040 | 28,257 | 74,345 |
| 52 | 8 | 35 | 20 | 320 | 72,361 | 39,568 | 125,790 | 49,748 | 26,338 | 42,522 |
| 53 | 15 | 35 | 20 | 320 | 202,770 | 26,348 | 132,699 | 59,221 | 38,731 | 59,152 |
| 54 | 15 | 35 | 20 | 320 | 268,232 | 27,753 | 171,495 | 68,128 | 67,211 | 118,823 |
| 55 | 30 | 35 | 20 | 320 | 36,275 | 5026 | 47,982 | 28,455 | 29,874 | 40,194 |
| 56 | 30 | 35 | 20 | 320 | 230,066 | 177,791 | 416,276 | 206,119 | 134,749 | 153,933 |
| 57 | 1 | 300 | 20 | 320 | 203,962 | 22,891 | 182,810 | 61,889 | 64,813 | 89,814 |
| 58 | 1 | 300 | 20 | 320 | 318,045 | 37,369 | 272,839 | 86,310 | 80,373 | 112,924 |
| 59 | 8 | 300 | 20 | 320 | 186,840 | 223,351 | 377,598 | 180,314 | 112,725 | 174,757 |
| 60 | 8 | 300 | 20 | 320 | 362,158 | 230,536 | 406,363 | 184,179 | 117,303 | 174,143 |
| 61 | 15 | 300 | 20 | 320 | 356,928 | 249,009 | 434,678 | 197,170 | 121,469 | 189,884 |
| 62 | 15 | 300 | 20 | 320 | 503,220 | 301,370 | 526,770 | 235,723 | 156,489 | 48,466 |
| 63 | 30 | 300 | 20 | 320 | 62,800 | 13,215 | 17,413 | 5716 | 4469 | 4931 |
| 64 | 30 | 300 | 20 | 320 | 377,885 | 239,466 | 389,722 | 193,828 | 163,154 | 200,208 |
References
- Jensen, S.E.; Anders, C.L.; Goatcher, L.J.; Perley, T.; Kenefick, S.; Hrudey, S.E. Actinomycetes as a factor in odour problems affecting drinking water from the North Saskatchewan River. Water Res. 1994, 8, 1393–1401. [Google Scholar] [CrossRef]
- Boutou, S.; Chatonnet, P. Rapid headspace solid-phase microextraction/gas chromatographic/mass spectrometric assay for the quantitative determination of some of the main odorants causing off-flavours in wine. J. Chromatogr. A 2007, 1141, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Callejón, R.M.; Ubeda, C.; Ríos-Reina, R.; Morales, M.L.; Troncoso, A.M. Recent developments in the analysis of musty odour compounds in water and wine: A review. J. Chromatogr. A 2016, 1428, 72–85. [Google Scholar] [CrossRef] [PubMed]
- Azzi-Achkouty, S.; Estephan, N.; Ouaini, N.; Rutledge, D.N. Headspace solid-phase microextraction for wine volatile analysis. Crit. Rev. Food Sci. Nutr. 2017, 57, 2009–2020. [Google Scholar] [CrossRef] [PubMed]
- Jové, P.; Pareras, A.; De Nadal, R.; Verdum, M. Development and optimization of a quantitative analysis of main odorants causing off flavours in cork stoppers using headspace solid-phase microextraction gas chromatography tandem mass spectrometry. J. Mass Spectrom. 2021, 56, e4728. [Google Scholar] [CrossRef] [PubMed]
- Camino-Sánchez, F.J.; Ruiz-García, J.; Zafra-Gómez, A. Development of a thermal desorption gas chromatography–mass spectrometry method for quantitative determination of haloanisoles and halophenols in wineries ambient air. J. Chromatogr. A 2013, 1305, 259–266. [Google Scholar] [CrossRef]
- Sung, Y.H.; Li, T.Y.; Huang, S.D. Analysis of earthy and musty odors in water samples by solid-phase microextraction coupled with gas chromatography/ion trap mass spectrometry. Talanta 2005, 65, 518–524. [Google Scholar] [CrossRef] [PubMed]
- Fontana, A.R. Analytical methods for determination of cork-taint compounds in wine. TrAC Trends Anal. Chem. 2012, 37, 135–147. [Google Scholar] [CrossRef]
- Cravero, M.C. Musty and Moldy Taint in Wines: A Review. Beverages 2020, 6, 41. [Google Scholar] [CrossRef]
- Tarasov, A.; Rauhut, D.; Jung, R. “Cork taint”, responsible compounds. Determination of haloanisoles and halophenols in cork matrix: A review. Talanta 2017, 175, 82–92. [Google Scholar] [CrossRef]
- Žnideršič, L.; Mlakar, A.; Prosen, H. Development of a SPME-GC-MS/MS method for the determination of some contaminants from food contact material in beverages. Food Chem. Toxicol. 2019, 134, 110829. [Google Scholar] [CrossRef]
- Gallego, E.; Roca, F.J.; Perales, J.F.; Sánchez, G.; Esplugas, P. Characterization and determination of the odorous charge in the indoor air of a waste treatment facility through the evaluation of volatile organic compounds (VOCs) using TD–GC/MS. Waste Manag. 2012, 32, 2469–2481. [Google Scholar] [CrossRef]
- Camino-Sánchez, F.J.; Bermúdez-Peinado, R.; Zafra-Gómez, A.; Ruíz-García, J.; Vílchez-Quero, J.L. Determination of trichloroanisole and trichlorophenol in wineries’ ambient air by passive sampling and thermal desorption–gas chromatography coupled to tandem mass spectrometry. J. Chromatogr. A 2015, 1380, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Hellén, H.; Schallhart, S.; Praplan, A.P.; Petäjä, T.; Hakola, H. Using in situ GC-MS for analysis of C-2-C-7 volatile organic acids in ambient air of a boreal forest site. Atmos. Meas. Tech. 2017, 10, 281–289. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Roux, S.; Descharles, N.; Bonazzi, C.; Rega, B. Quantitative determination of volatile compounds using TD-GC-MS and isotope standard addition for application to the heat treatment of food. Food Control 2021, 121, 107635. [Google Scholar] [CrossRef]
- Rosli, A.S.; Abdullah, F. Determination of Volatile Organic Compounds in Ambient Air of Pasir Gudang by using Gas Chromatogrpahy-Mass Spectrometry. Eproc. Chem. 2020, 5, 41–48. [Google Scholar]
- Jia, C.; Fu, X. Diffusive uptake rates of volatile organic compounds on standard ATD tubes for environmental and workplace applications. Environments 2017, 4, 87. [Google Scholar] [CrossRef] [Green Version]
- Ramos, T.D.; de la Guardia, M.; Pastor, A.; Esteve-Turrillas, F.A. Assessment of air passive sampling uptakes for volatile organic compounds using VERAM devices. Sci. Total Environ. 2018, 619, 1014–1021. [Google Scholar] [CrossRef]
- Batterman, S.; Metts, T.; Kalliokoski, P. Diffusive uptake in passive and active adsorbent sampling using thermal desorption tubes. J. Environ. Monit. 2002, 4, 870–878. [Google Scholar] [CrossRef] [PubMed]
- Hellén, H.; Hakola, H.; Laurila, T.; Hiltunen, V.; Koskentalo, T. Aromatic hydrocarbon and methyl tert-butyl ether measurements in ambient air of Helsinki (Finland) using diffusive samplers. Sci. Total Environ. 2002, 298, 55–64. [Google Scholar] [CrossRef]
- Martin, N.A.; Marlow, D.J.; Henderson, M.H.; Goody, B.A.; Quincey, P.G. Studies using the sorbent Carbopack X for measuring environmental benzene with Perkin–Elmer-type pumped and diffusive samplers. Atmos. Environ. 2003, 37, 871–879. [Google Scholar] [CrossRef]
- McClenny, W.A.; Oliver, K.D.; Jacumin, H.H., Jr.; Daughtrey, E.H., Jr.; Whitaker, D.A. 24 h diffusive sampling of toxic VOCs in air onto Carbopack X solid adsorbent followed by thermal desorption/GC/MS analysis—laboratory studies. J. Environ. Monit. 2005, 7, 248–256. [Google Scholar] [CrossRef]
- Simpson, A.T.; Wright, M.D. Diffusive sampling of c7–c16 hydrocarbons in workplace air: Uptake rates, wall effects and use in oil mist measurements. Ann. Occup. Hyg. 2008, 52, 249–257. [Google Scholar] [PubMed] [Green Version]
- Batterman, S.; Metts, T.; Kalliokoski, P.; Barnett, E. Low-flow active and passive sampling of VOCs using thermal desorption tubes: Theory and application at an offset printing facility. J. Environ. Monit. 2002, 4, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Hoed, N.V.D.; Halmans, M.T.H. Sampling and thermal desorption efficiency of tube-type diffusive samplers: Selection and performance of adsorbents. Am. Ind. Hyg. Assoc. J. 1987, 48, 364–373. [Google Scholar] [CrossRef]
- Lewis, R.G.; Mulik, J.D.; Coutant, R.W.; Wooten, G.W.; McMillin, C.R. Thermally desorbable passive sampling device for volatile organic chemicals in ambient air. Anal. Chem. 1985, 57, 214–219. [Google Scholar] [CrossRef]
- Harper, M.; Purnell, C.J. Diffusive sampling—A review. Am. Ind. Hyg. Assoc. J. 1987, 48, 214–218. [Google Scholar] [CrossRef]
- Ochiai, N.; Sasamoto, K.; Takino, M.; Yamashita, S.; Daishima, S.; Heiden, A.; Hoffman, A. Determination of trace amounts of off-flavor compounds in drinking water by stir bar sorptive extraction and thermal desorption GC-MS. Analyst 2001, 126, 1652–1657. [Google Scholar] [CrossRef]
- Benanou, D.; Acobas, F.; De Roubin, M.R. Optimization of stir bar sorptive extraction applied to the determination of odorous compounds in drinking water. Water Sci. Technol. 2004, 49, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.H. Environmental use of diffusive samplers: Evaluation of reliable diffusive uptake rates for benzene, toluene and xylene. J. Environ. Monit. 1999, 1, 115–116. [Google Scholar] [CrossRef]
- Persoon, C.; Hornbuckle, K.C. Calculation of passive sampling rates from both native PCBs and depuration compounds in indoor and outdoor environments. Chemosphere 2009, 74, 917–923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
| Parameter | Value | Comments |
|---|---|---|
| Desorption time (min) (DT) | 1 to 30 | To be optimized |
| Desorption temperature (°C) (TDT) | 35 to 300 | To be optimized |
| Desorption mode | splitless | Necessary for maximum sensibility |
| Trap purge time (min) | 1.0 | Recommended for Tenax® TA tubes |
| Trap low cryo-temperature (°C) (TLCT) | −20 to 20 | To be optimized |
| Trap heating rate (°C s−1) | 50 (MAX) | Previously reported (Camino et al., 2013) |
| Trap high temperature (°C) (THT) | 305 to 320 | To be optimized |
| Main Effects | Interactions | |||||
|---|---|---|---|---|---|---|
| Response | DT | TDT | TLCT | THT | DT*TDT | TLCT*THT |
| MIB | + | + | NA | NA | NA | NA |
| TCA | + | + | NA | NA | Yes | Yes |
| GSM | + | + | NA | NA | Yes | NA |
| TeCA | + | + | NA | NA | Yes | NA |
| TBA | + | + | + | − | NA | Yes |
| PCA | + | + | + | − | NA | Yes |
| 2MIB | TCA | GSM | TeCA | TBA | |
|---|---|---|---|---|---|
| TCA | 0.776 | ||||
| GSM | 0.899 | 0.875 | |||
| TeCA | 0.848 | 0.937 | 0.972 | ||
| TBA | 0.816 | 0.889 | 0.916 | 0.952 | |
| PCA | 0.790 | 0.865 | 0.868 | 0.906 | 0.953 |
| Predicted Response (95% CI) | ||
|---|---|---|
| DT = 30 | DT = 8 | |
| PCA | 248,455 (122,899; 374,011) | 177,353 (135,501; 219,205) |
| TBA | 194,295 (103,912; 284,678) | 120,356 (90,228; 150,484) |
| TeCa | 205,863 (91,872; 319,854) | 183,980 (145,983; 221,977) |
| GSM | 417,875 (165,319; 670,431) | 417,603 (333,418; 501,788) |
| TCA | 266,963 (128,930; 404,996) | 252,412 (199,550; 305,275) |
| 2MIB | 407,913 (162,156; 653,670) | 351,452 (285,296; 417,607) |
| Target Compounds | Quantification Ions | R2 | LOD (ng tube−1) | LOQ (ng tube−1) | Repeatability (%RSD at 1 ng tube−1) a | Reproducibility (%RSD at 1 ng tube−1) a | |
|---|---|---|---|---|---|---|---|
| m/z 1 | m/z 2 | ||||||
| TCA | 195 | 210 | 0.995 | 0.01 | 0.05 | 9.3 | 9.9 |
| TeCA | 231 | 246 | 0.994 | 0.06 | 0.1 | 6.0 | 9.6 |
| TBA | 346 | 331 | 0.995 | 0.05 | 0.1 | 6.8 | 5.7 |
| PCA | 280 | 265 | 0.994 | 0.03 | 0.05 | 3.5 | 3.9 |
| GSM | 112 | 125 | 0.995 | 0.03 | 0.05 | 8.9 | 9.0 |
| 2MIB | 95 | - b | 0.994 | 0.03 | 0.06 | 2.5 | 3.9 |
| Exposure Time (h) | Mad, ng (Mean ± Standard Deviation) | |||||
|---|---|---|---|---|---|---|
| TCA | TeCA | TBA | PCA | GSM | 2MIB | |
| 2 | 0.3 ± 0.1 | 1.2 ± 0.20 | 1.8 ± 0.0 | 1.3 ± 1.1 | 0.8 ± 0.3 | 0.1 ± 0.0 |
| 6 | 1.2 ± 0.8 | 3.0 ± 0.42 | 3.9 ± 2.5 | 2.7 ± 0.0 | 0.9 ± 0.2 | 0.7 ± 0.0 |
| 24 | 1.7 ± 0.6 | 6.32 ± 0.01 | 17.6 ± 0.0 | 13.2 ± 0.8 | 3.4 ± 1.2 | 1.3 ± 0.1 |
| 48 | 2.6 ± 1.1 | 10.12 ± 0.01 | 20.1 ± 0.0 | 23.4 ± 0.5 | 5.5 ± 2.0 | 1.8 ± 0.0 |
| 54 | 2.7 ± 0.8 | 11.86 ± 0.0 | 17.4 ± 0.0 | 20.9 ± 0.2 | 6.9 ±0.1 | 2.1 ± 0.1 |
| 72 | 2.8 ± 0.1 | 15.48 ± 0.13 | 16.6 ± 0.6 | 20.4 ± 0.7 | 9.3 ± 2.2 | 2.2 ± 0.1 |
| 96 | 3.2 ± 0.09 | 14.00 ± 0.0 | 13.4 ± 0.0 | 18.1 ± 0.5 | 12.8 ± 3.2 | 0.9 ± 0.1 |
| 120 | 4.1 ± 0.6 | 12.2 ± 0.0 | 8.0 ± 0.0 | 16.2 ± 0.1 | 7.2 ± 1.2 | 0.8 ± 0.2 |
| R-values (m3 h−1) | 0.035 ± 17.1 | 0.028 ± 0.8 | 0.013 ± 26.2 | 0.071.3 ± 3.9 | 0.032.3 ± 13.4 | 0.025 ± 28.8 |
| TCA | TeCA | TBA | PCA | GSM | 2MIB | |
|---|---|---|---|---|---|---|
| w1 | 0.008 ± 0.006 | <LD | <LD | 0.003 ± 0.003 | <LD | <LD |
| w2 | <LD | <LD | <LD | <LD | <LD | <LD |
| w3 | <LD | <LD | <LD | <LD | <LD | <LD |
| w4 | <LD | <LD | <LD | <LD | <LD | <LD |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jové, P.; Vives-Mestres, M.; Nadal, R.D.; Verdum, M. Development, Optimization and Validation of a Sustainable and Quantifiable Methodology for the Determination of 2,4,6-Trichloroanisole, 2,3,4,6-Tetrachloroanisole, 2,4,6-Tribromoanisole, Pentachloroanisole, 2-Methylisoborneole and Geosmin in Air. Processes 2021, 9, 1571. https://doi.org/10.3390/pr9091571
Jové P, Vives-Mestres M, Nadal RD, Verdum M. Development, Optimization and Validation of a Sustainable and Quantifiable Methodology for the Determination of 2,4,6-Trichloroanisole, 2,3,4,6-Tetrachloroanisole, 2,4,6-Tribromoanisole, Pentachloroanisole, 2-Methylisoborneole and Geosmin in Air. Processes. 2021; 9(9):1571. https://doi.org/10.3390/pr9091571
Chicago/Turabian StyleJové, Patricia, Marina Vives-Mestres, Raquel De Nadal, and Maria Verdum. 2021. "Development, Optimization and Validation of a Sustainable and Quantifiable Methodology for the Determination of 2,4,6-Trichloroanisole, 2,3,4,6-Tetrachloroanisole, 2,4,6-Tribromoanisole, Pentachloroanisole, 2-Methylisoborneole and Geosmin in Air" Processes 9, no. 9: 1571. https://doi.org/10.3390/pr9091571
APA StyleJové, P., Vives-Mestres, M., Nadal, R. D., & Verdum, M. (2021). Development, Optimization and Validation of a Sustainable and Quantifiable Methodology for the Determination of 2,4,6-Trichloroanisole, 2,3,4,6-Tetrachloroanisole, 2,4,6-Tribromoanisole, Pentachloroanisole, 2-Methylisoborneole and Geosmin in Air. Processes, 9(9), 1571. https://doi.org/10.3390/pr9091571