
Diversity and Evolution of Clostridium beijerinckii and Complete Genome of the Type Strain DSM 791T


 , ,
, , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Bacterial Strain
2.2. Cultivation
2.3. DNA Extraction and Sequencing
2.4. Genome Assembly
2.5. Genome Annotation and Analysis
3. Results
3.1. Genome Sequencing and Assembly
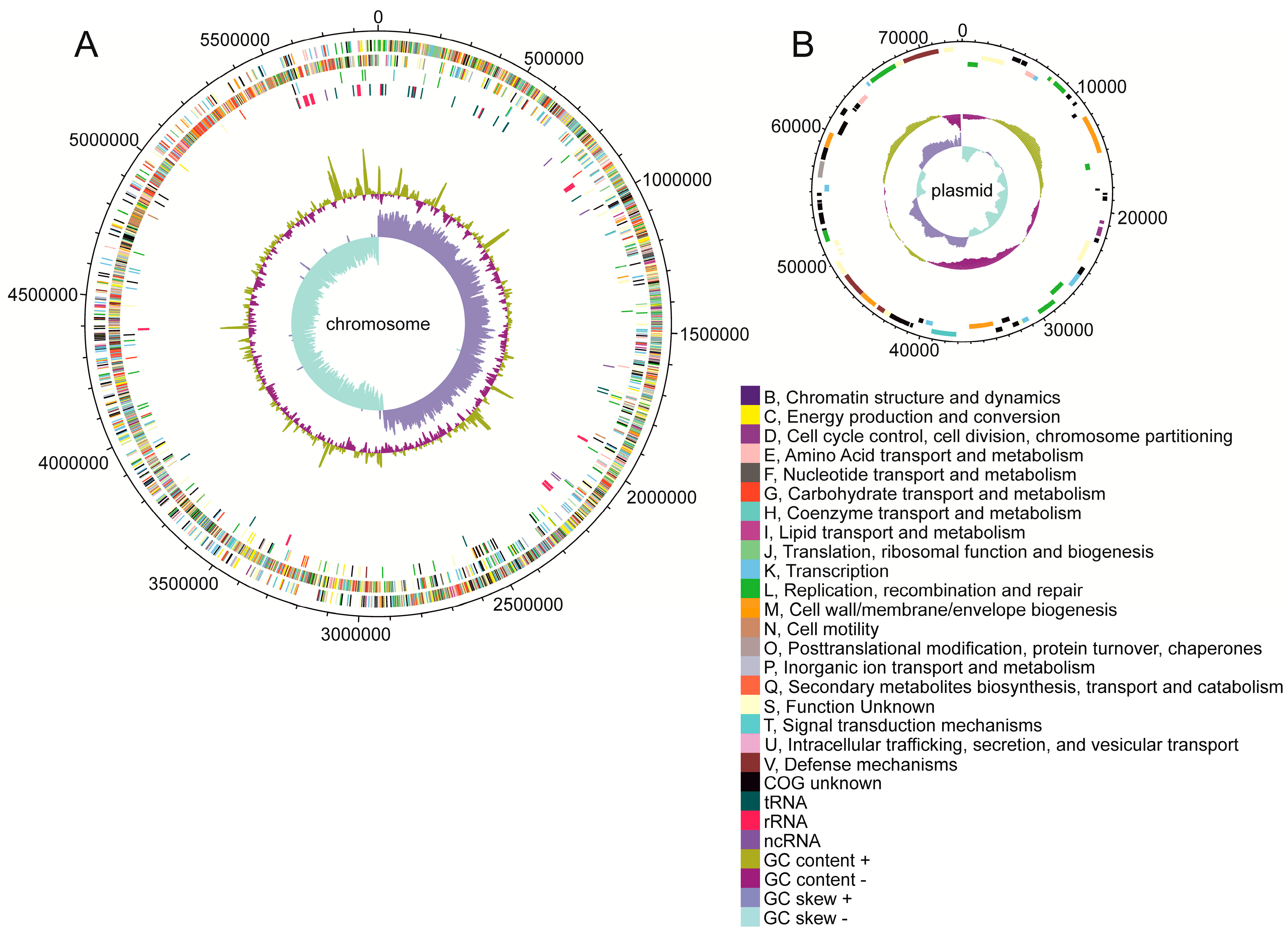
3.2. The Characteristics of the C. beijerinckii DSM 791 Genome
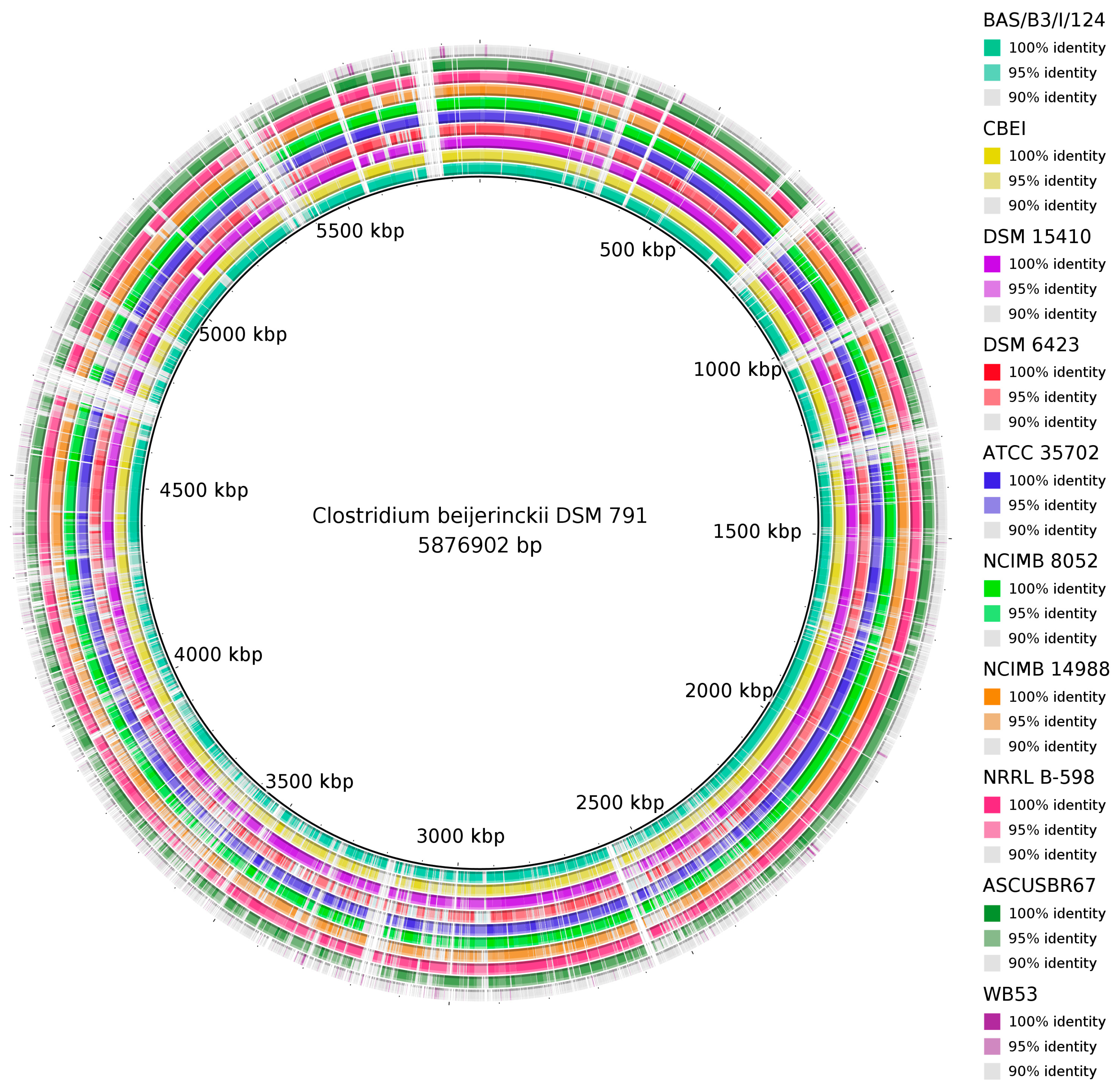
3.3. Diversity of C. beijerinckii Strains
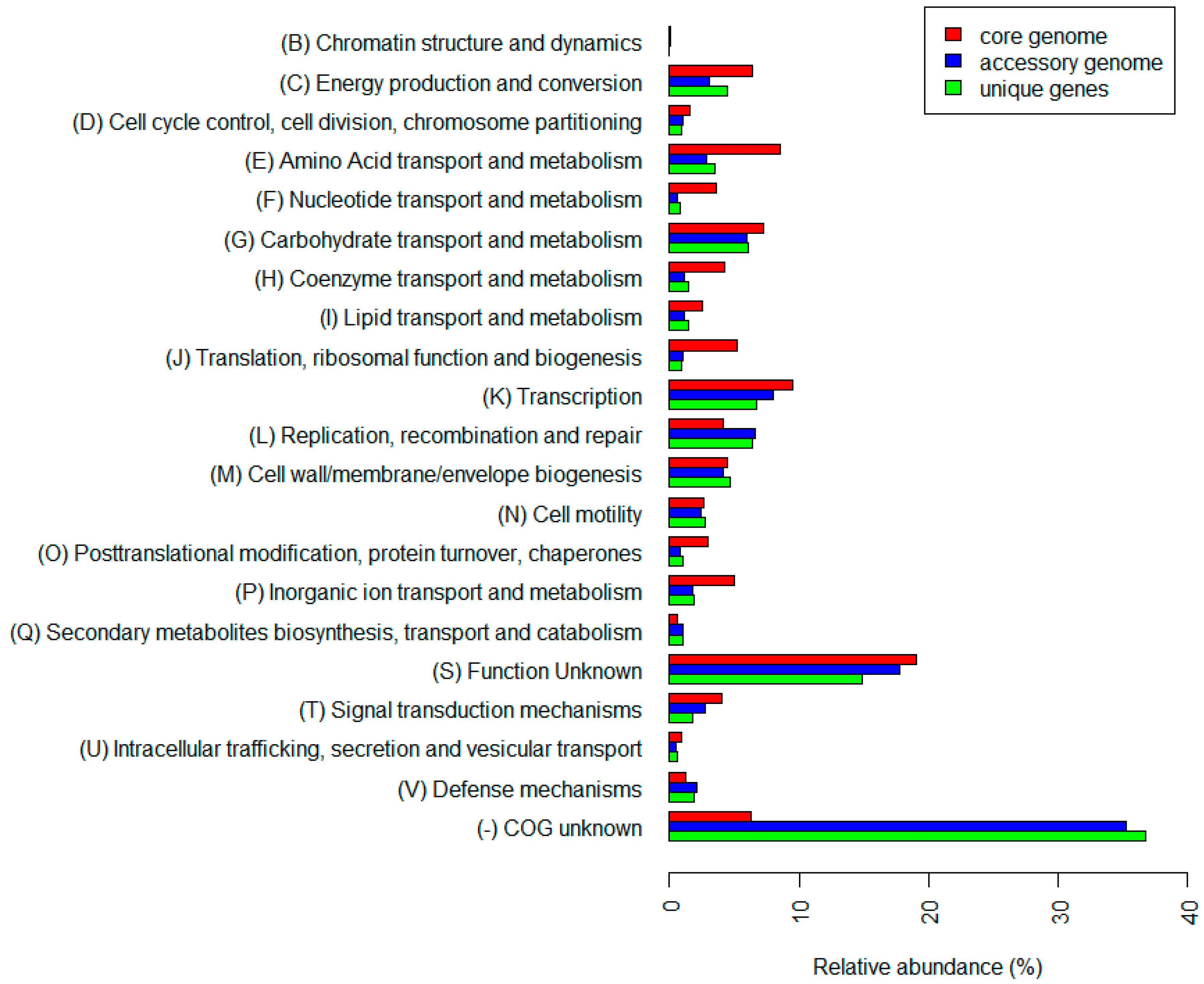
3.4. Pangenome of C. beijerinckii
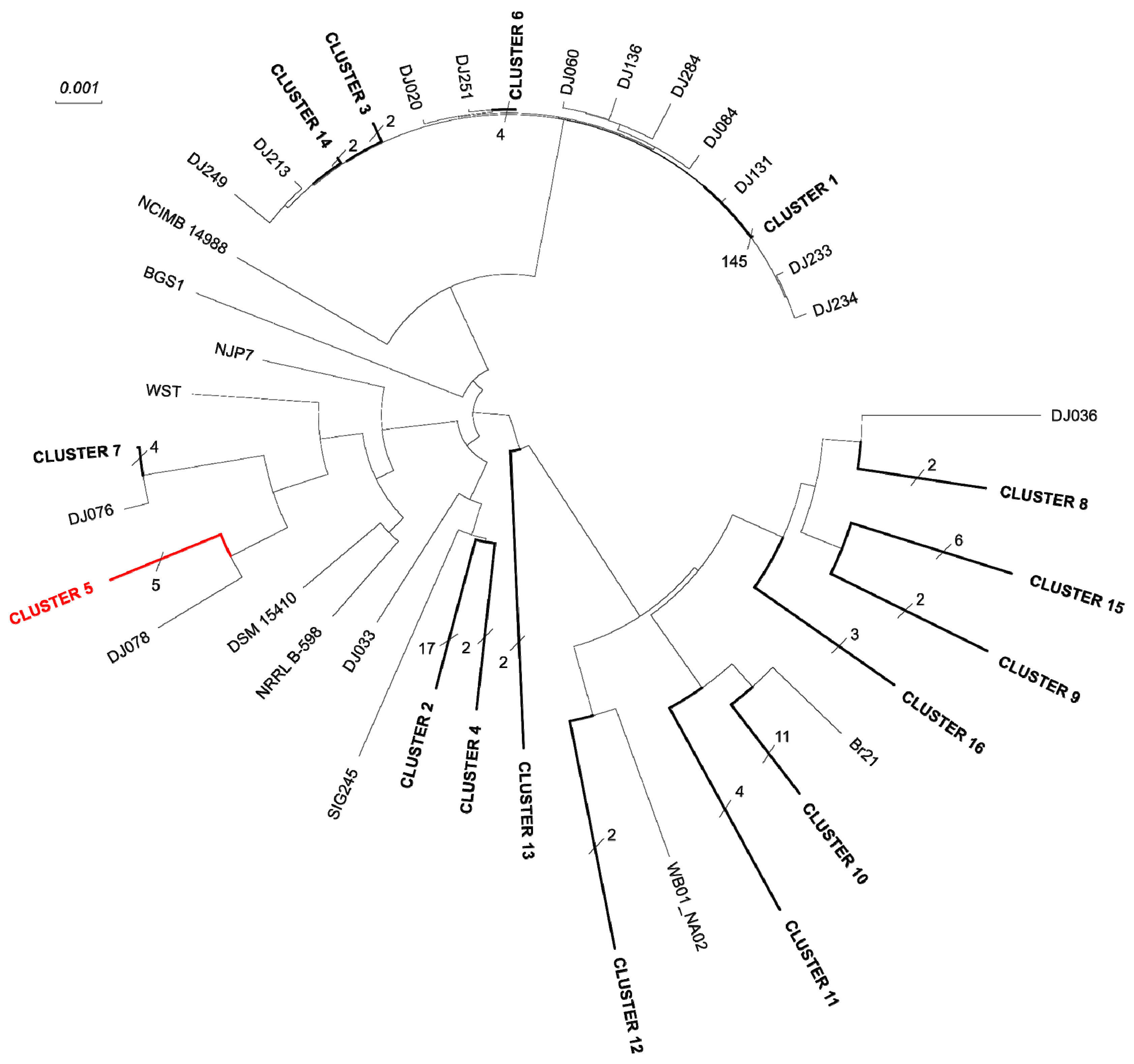
3.5. Phylogeny
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Cruz-Morales, P.; Orellana, C.; Moutafis, G.; Moonen, G.; Rincon, G.; Nielsen, L.; Marcellin, E. Revisiting the Evolution and Taxonomy of Clostridia, a Phylogenomic Update. Genome Biol. Evol. 2019, 11, 2035–2044. [Google Scholar] [CrossRef]
- Schiel-Bengelsdorf, B.; Montoya, J.; Linder, S.; Dürre, P. Butanol fermentation. Environ. Technol. 2013, 34, 1691–1710. [Google Scholar] [CrossRef] [PubMed]
- Vieira, C.F.D.S.; Filho, F.M.; Filho, R.M.; Mariano, A.P. Acetone-free biobutanol production: Past and recent advances in the Isopropanol-Butanol-Ethanol (IBE) fermentation. Bioresour. Technol. 2019, 287, 121425. [Google Scholar] [CrossRef]
- Patakova, P.; Kolek, J.; Sedlar, K.; Koscova, P.; Branská, B.; Kupkova, K.; Paulova, L.; Provaznik, I. Comparative analysis of high butanol tolerance and production in clostridia. Biotechnol. Adv. 2018, 36, 721–738. [Google Scholar] [CrossRef]
- Charubin, K.; Bennett, R.K.; Fast, A.G.; Papoutsakis, E.T. Engineering Clostridium organisms as microbial cell-factories: Challenges & opportunities. Metab. Eng. 2018, 50, 173–191. [Google Scholar] [CrossRef] [PubMed]
- Finegold, S.M.; Song, Y.; Liu, C. Taxonomy—General Comments and Update on Taxonomy of Clostridia and Anaerobic cocci. Anaerobe 2002, 8, 283–285. [Google Scholar] [CrossRef] [PubMed]
- Keis, S.; Bennett, C.F.; Ward, V.K.; Jones, D.T. Taxonomy and Phylogeny of Industrial Solvent-Producing Clostridia. Int. J. Syst. Bacteriol. 1995, 45, 693–705. [Google Scholar] [CrossRef]
- Johnson, J.L.; Francis, B.S. Taxonomy of the Clostridia: Ribosomal Ribonucleic Acid Homologies among the Species. J. Gen. Microbiol. 1975, 88, 229–244. [Google Scholar] [CrossRef]
- Sedlar, K.; Kolek, J.; Provaznik, I.; Patakova, P. Reclassification of non-type strain Clostridium pasteurianum NRRL B-598 as Clostridium beijerinckii NRRL B-598. J. Biotechnol. 2017, 244, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Lawson, P.A.; Rainey, F.A. Proposal to restrict the genus Clostridium Prazmowski to Clostridium butyricum and related species. Int. J. Syst. Evol. Microbiol. 2016, 66, 1009–1016. [Google Scholar] [CrossRef]
- Moon, C.D.; Pacheco, D.M.; Kelly, W.J.; Leahy, S.; Li, D.; Kopečný, J.; Attwood, G. Reclassification of Clostridium proteoclasticum as Butyrivibrio proteoclasticus comb. nov., a butyrate-producing ruminal bacterium. Int. J. Syst. Evol. Microbiol. 2008, 58, 2041–2045. [Google Scholar] [CrossRef]
- Kobayashi, H.; Tanizawa, Y.; Sakamoto, M.; Nakamura, Y.; Ohkuma, M.; Tohno, M. Reclassification of Clostridium diolis Biebl and Spröer 2003 as a later heterotypic synonym of Clostridium beijerinckii Donker 1926 (Approved Lists 1980) emend. Keis et al. 2001. Int. J. Syst. Evol. Microbiol. 2020, 70, 2463–2466. [Google Scholar] [CrossRef]
- Ackermann, M. A functional perspective on phenotypic heterogeneity in microorganisms. Nat. Rev. Genet. 2015, 13, 497–508. [Google Scholar] [CrossRef]
- Sedlar, K.; Vasylkivska, M.; Musilova, J.; Branska, B.; Provaznik, I.; Patakova, P. Phenotypic and genomic analysis of isopropanol and 1,3-propanediol producer Clostridium diolis DSM 15410. Genomics 2021, 113, 1109–1119. [Google Scholar] [CrossRef]
- Biebl, H.; Spröer, C. Taxonomy of the Glycerol Fermenting Clostridia and Description of Clostridium diolis sp. nov. Syst. Appl. Microbiol. 2002, 25, 491–497. [Google Scholar] [CrossRef]
- Wu, J.; Dong, L.; Zhou, C.; Liu, B.; Xing, D.; Feng, L.; Wu, X.; Wang, Q.; Cao, G. Enhanced butanol-hydrogen coproduction by Clostridium beijerinckii with biochar as cell’s carrier. Bioresour. Technol. 2019, 294, 122141. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Dong, L.; Zhou, C.; Liu, B.; Feng, L.; Wu, C.; Qi, Z.; Cao, G. Developing a coculture for enhanced butanol production by Clostridium beijerinckii and Saccharomyces cerevisiae. Bioresour. Technol. Rep. 2019, 6, 223–228. [Google Scholar] [CrossRef]
- Skerman, V.B.D.; Sneath, P.H.A.; McGowan, V. Approved Lists of Bacterial Names. Int. J. Syst. Evol. Microbiol. 1980, 30, 225–420. [Google Scholar] [CrossRef]
- Wang, Y.; Li, X.; Mao, Y.; Blaschek, H.P. Genome-wide dynamic transcriptional profiling in Clostridium beijerinckii NCIMB 8052 using single-nucleotide resolution RNA-Seq. BMC Genom. 2012, 13, 102. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, X.; Mao, Y.; Blaschek, H.P. Single-nucleotide resolution analysis of the transcriptome structure of Clostridium beijerinckii NCIMB 8052 using RNA-Seq. BMC Genom. 2011, 12, 479. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ezeji, T.C. Transcriptional analysis of Clostridium beijerinckii NCIMB 8052 to elucidate role of furfural stress during acetone butanol ethanol fermentation. Biotechnol. Biofuels 2013, 6, 66. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.; Blaschek, H.P. Transcriptional Analysis of Clostridium beijerinckii NCIMB 8052 and the Hyper-Butanol-Producing Mutant BA101 during the Shift from Acidogenesis to Solventogenesis. Appl. Environ. Microbiol. 2008, 74, 7709–7714. [Google Scholar] [CrossRef]
- Wilkinson, S.R.; Young, M. Physical map of the Clostridium beijerinckii (formerly Clostridium acetobutylicum) NCIMB 8052 chromosome. J. Bacteriol. 1995, 177, 439–448. [Google Scholar] [CrossRef][Green Version]
- Kolek, J.; Sedlar, K.; Provaznik, I.; Patakova, P. Dam and Dcm methylations prevent gene transfer into Clostridium pasteurianum NRRL B-598: Development of methods for electrotransformation, conjugation, and sonoporation. Biotechnol. Biofuels 2016, 9, 1–14. [Google Scholar] [CrossRef]
- Sedlar, K.; Kolek, J.; Gruber, M.; Jureckova, K.; Branska, B.; Csaba, G.; Vasylkivska, M.; Zimmer, R.; Patakova, P.; Provaznik, I. A transcriptional response of Clostridium beijerinckii NRRL B-598 to a butanol shock. Biotechnol. Biofuels 2019, 12, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Vasylkivska, M.; Branska, B.; Sedlar, K.; Jureckova, K.; Provaznik, I.; Patakova, P. Phenotypic and Genomic Analysis of Clostridium beijerinckii NRRL B-598 Mutants With Increased Butanol Tolerance. Front. Bioeng. Biotechnol. 2020, 8, 598392. [Google Scholar] [CrossRef]
- Sedlar, K.; Kolek, J.; Skutkova, H.; Branska, B.; Provaznik, I.; Patakova, P. Complete genome sequence of Clostridium pasteurianum NRRL B-598, a non-type strain producing butanol. J. Biotechnol. 2015, 214, 113–114. [Google Scholar] [CrossRef]
- Diallo, M.; Hocq, R.; Collas, F.; Chartier, G.; Wasels, F.; Wijaya, H.S.; Werten, M.W.; Wolbert, E.J.; Kengen, S.W.; van der Oost, J.; et al. Adaptation and application of a two-plasmid inducible CRISPR-Cas9 system in Clostridium beijerinckii. Methods 2020, 172, 51–60. [Google Scholar] [CrossRef] [PubMed]
- De Gérando, H.M.; Wasels, F.; Bisson, A.; Clement, B.; Bidard, F.; Jourdier, E.; López-Contreras, A.M.; Ferreira, N.L. Genome and transcriptome of the natural isopropanol producer Clostridium beijerinckii DSM6423. BMC Genom. 2018, 19, 242. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Li, T.; He, J. Characterization and genome analysis of a butanol–isopropanol-producing Clostridium beijerinckii strain BGS1. Biotechnol. Biofuels 2018, 11, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Lanfear, R.; Schalamun, M.; Kainer, D.; Wang, W.; Schwessinger, B. MinIONQC: Fast and simple quality control for MinION sequencing data. Bioinformatics 2019, 35, 523–525. [Google Scholar] [CrossRef]
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Kolmogorov, M.; Yuan, J.; Lin, Y.; Pevzner, P.A. Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 2019, 37, 540–546. [Google Scholar] [CrossRef] [PubMed]
- Antipov, D.; Hartwick, N.; Shen, M.; Rayko, M.; Lapidus, A.; Pevzner, P.A. plasmidSPAdes: Assembling plasmids from whole genome sequencing data. Bioinformatics 2016, 32, 3380–3387. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, S.; Phillippy, A.; Delcher, A.L.; Smoot, M.; Shumway, M.; Antonescu, C.; Salzberg, S.L. Versatile and open software for comparing large genomes. Genome Biol. 2004, 5, R12. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef] [PubMed]
- Vaser, R.; Sović, I.; Nagarajan, N.; Šikić, M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 2017, 27, 737–746. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An Integrated Tool for Comprehensive Microbial Variant Detection and Genome Assembly Improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Gao, F.; Zhang, C.-T. Ori-Finder: A web-based system for finding oriC s in unannotated bacterial genomes. BMC Bioinforma. 2008, 9, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Luo, H.; Gao, F. DoriC 10.0: An updated database of replication origins in prokaryotic genomes including chromosomes and plasmids. Nucleic Acids Res. 2019, 47, D74–D77. [Google Scholar] [CrossRef]
- Tatusova, T.; DiCuccio, M.; Badretdin, A.; Chetvernin, V.; Nawrocki, P.; Zaslavsky, L.; Lomsadze, A.; Pruitt, K.D.; Borodovsky, M.; Ostell, J. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 2016, 44, 6614–6624. [Google Scholar] [CrossRef] [PubMed]
- Taboada, B.; Estrada, K.; Ciria, R.; Merino, E. Operon-mapper: A web server for precise operon identification in bacterial and archaeal genomes. Bioinformatics 2018, 34, 4118–4120. [Google Scholar] [CrossRef] [PubMed]
- Huerta-Cepas, J.; Szklarczyk, D.; Heller, D.; Hernández-Plaza, A.; Forslund, S.K.; Cook, H.V.; Mende, D.R.; Letunic, I.; Rattei, T.; Jensen, L.J.; et al. eggNOG 5.0: A hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2019, 47, D309–D314. [Google Scholar] [CrossRef] [PubMed]
- Carver, T.; Thomson, N.; Bleasby, A.; Berriman, M.; Parkhill, J. DNAPlotter: Circular and linear interactive genome visualization. Bioinform. 2008, 25, 119–120. [Google Scholar] [CrossRef]
- Rutherford, K.; Parkhill, J.; Crook, J.; Horsnell, T.; Rice, P.; Rajandream, M.-A.; Barrell, B. Artemis: Sequence visualization and annotation. Bioinform. 2000, 16, 944–945. [Google Scholar] [CrossRef]
- Akhter, S.; Aziz, R.; Edwards, R.A. PhiSpy: A novel algorithm for finding prophages in bacterial genomes that combines similarity- and composition-based strategies. Nucleic Acids Res. 2012, 40, e126. [Google Scholar] [CrossRef]
- Biswas, A.; Staals, R.; Morales, S.; Fineran, P.; Brown, C.M. CRISPRDetect: A flexible algorithm to define CRISPR arrays. BMC Genom. 2016, 17, 1–14. [Google Scholar] [CrossRef]
- Sayers, E.W.; Cavanaugh, M.; Clark, K.; Ostell, J.; Pruitt, K.D.; Mizrachi, I.K. GenBank. Nucleic Acids Res. 2019, 48, D84–D86. [Google Scholar] [CrossRef]
- Alikhan, N.-F.; Petty, N.K.; Ben Zakour, N.L.; Beatson, S.A. BLAST Ring Image Generator (BRIG): Simple prokaryote genome comparisons. BMC Genom. 2011, 12, 402. [Google Scholar] [CrossRef]
- Murrell, P. R Graphics. Wiley Interdiscip. Rev. Comput. Stat. 2009, 1, 216–220. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Göker, M. TYGS is an automated high-throughput platform for state-of-the-art genome-based taxonomy. Nat. Commun. 2019, 10, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Chaudhari, N.M.; Gupta, V.K.; Dutta, C. BPGA- an ultra-fast pan-genome analysis pipeline. Sci. Rep. 2016, 6, 24373. [Google Scholar] [CrossRef] [PubMed]
- Wolstencroft, K.; Krebs, O.; Snoep, J.L.; Stanford, N.J.; Bacall, F.; Golebiewski, M.; Kuzyakiv, R.; Nguyen, Q.; Owen, S.; Soiland-Reyes, S.; et al. FAIRDOMHub: A repository and collaboration environment for sharing systems biology research. Nucleic Acids Res. 2017, 45, D404–D407. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, B.; Gao, S.; Lercher, M.J.; Hu, S.; Chen, W.-H. Evolview v3: A webserver for visualization, annotation, and management of phylogenetic trees. Nucleic Acids Res. 2019, 47, W270–W275. [Google Scholar] [CrossRef]
- Cornillot, E.; Nair, R.V.; Papoutsakis, E.T.; Soucaille, P. The genes for butanol and acetone formation in Clostridium acetobutylicum ATCC 824 reside on a large plasmid whose loss leads to degeneration of the strain. J. Bacteriol. 1997, 179, 5442–5447. [Google Scholar] [CrossRef]
- Kemperman, R.; Jonker, M.; Nauta, A.; Kuipers, O.P.; Kok, J. Functional Analysis of the Gene Cluster Involved in Production of the Bacteriocin Circularin A by Clostridium beijerinckii ATCC 25752. Appl. Environ. Microbiol. 2003, 69, 6174–6178. [Google Scholar] [CrossRef]
- Kemperman, R.; Kuipers, A.; Karsens, H.; Nauta, A.; Kuipers, O.; Kok, J. Identification and Characterization of Two Novel Clostridial Bacteriocins, Circularin A and Closticin 574. Appl. Environ. Microbiol. 2003, 69, 1589–1597. [Google Scholar] [CrossRef]
- Thieme, N.; Panitz, J.C.; Held, C.; Lewandowski, B.; Schwarz, W.H.; Liebl, W.; Zverlov, V. Milling byproducts are an economically viable substrate for butanol production using clostridial ABE fermentation. Appl. Microbiol. Biotechnol. 2020, 104, 8679–8689. [Google Scholar] [CrossRef]
- Wischral, D.; Zhang, J.; Cheng, C.; Lin, M.; de Souza, L.M.G.; Pessoa, F.L.P.; Pereira, N.; Yang, S.-T. Production of 1,3-propanediol by Clostridium beijerinckii DSM 791 from crude glycerol and corn steep liquor: Process optimization and metabolic engineering. Bioresour. Technol. 2016, 212, 100–110. [Google Scholar] [CrossRef]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef]
- Song, W.; Sun, H.-X.; Zhang, C.; Cheng, L.; Peng, Y.; Deng, Z.; Wang, D.; Wang, Y.; Hu, M.; Liu, W.; et al. Prophage Hunter: An integrative hunting tool for active prophages. Nucleic Acids Res. 2019, 47, W74–W80. [Google Scholar] [CrossRef] [PubMed]
- Pyne, M.E.; Liu, X.; Moo-Young, M.; Chung, D.A.; Chou, C.P. Genome-directed analysis of prophage excision, host defence systems, and central fermentative metabolism in Clostridium pasteurianum. Sci. Rep. 2016, 6, 26228. [Google Scholar] [CrossRef]
- Schüler, M.; Stegmann, B.; Poehlein, A.; Daniel, R.; Dürre, P. Genome sequence analysis of the temperate bacteriophage TBP2 of the solvent producer Clostridium saccharoperbutylacetonicum N1-4 (HMT, ATCC 27021). FEMS Microbiol. Lett. 2020, 367. [Google Scholar] [CrossRef] [PubMed]
- Sorek, R.; Lawrence, C.M.; Wiedenheft, B. CRISPR-Mediated Adaptive Immune Systems in Bacteria and Archaea. Annu. Rev. Biochem. 2013, 82, 237–266. [Google Scholar] [CrossRef] [PubMed]
- Anderson, E.L.; Jang, J.; Venterea, R.T.; Feyereisen, G.W.; Ishii, S. Isolation and characterization of denitrifiers from woodchip bioreactors for bioaugmentation application. J. Appl. Microbiol. 2020, 129, 590–600. [Google Scholar] [CrossRef]
- Wayne, L.G.; Brenner, D.J.; Colwell, R.R.; Grimont, P.A.D.; Kandler, O.; Krichevsky, M.I.; Moore, L.H.; Moore, W.E.C.; Murray, R.G.E.; Stackebrandt, E.; et al. Report of the Ad Hoc Committee on Reconciliation of Approaches to Bacterial Systematics. Int. J. Syst. Bacteriol. 1987, 37, 463–464. [Google Scholar] [CrossRef]
- Chen, Z.; Erickson, D.L.; Meng, J. Polishing the Oxford Nanopore long-read assemblies of bacterial pathogens with Illumina short reads to improve genomic analyses. Genomics 2021, 113, 1366–1377. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Auch, A.F.; Klenk, H.-P.; Göker, M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinforma. 2013, 14, 60. [Google Scholar] [CrossRef] [PubMed]
- Saha, J.; Saha, B.K.; Sarkar, M.P.; Roy, V.; Mandal, P.; Pal, A. Comparative Genomic Analysis of Soil Dwelling Bacteria Utilizing a Combinational Codon Usage and Molecular Phylogenetic Approach Accentuating on Key Housekeeping Genes. Front. Microbiol. 2019, 10, 2896. [Google Scholar] [CrossRef]
- Chung, M.; Munro, J.B.; Tettelin, H.; Hotopp, J.C.D. Using Core Genome Alignments to Assign Bacterial Species. mSystems 2018, 3, e00236-18. [Google Scholar] [CrossRef] [PubMed]
- Seo, S.; Janssen, H.; Magis, A.; Wang, Y.; Lu, T.; Price, N.D.; Jin, Y.; Blaschek, H.P. Genomic, Transcriptional, and Phenotypic Analysis of the Glucose Derepressed Clostridium beijerinckii Mutant Exhibiting Acid Crash Phenotype. Biotechnol. J. 2017, 12, 1700182. [Google Scholar] [CrossRef]
- Patakova, P.; Branská, B.; Sedlar, K.; Vasylkivska, M.; Jureckova, K.; Kolek, J.; Koscova, P.; Provaznik, I. Acidogenesis, solventogenesis, metabolic stress response and life cycle changes in Clostridium beijerinckii NRRL B-598 at the transcriptomic level. Sci. Rep. 2019, 9, 1–21. [Google Scholar] [CrossRef]
- Branska, B.; Vasylkivska, M.; Raschmanova, H.; Jureckova, K.; Sedlar, K.; Provaznik, I.; Patakova, P. Changes in efflux pump activity of Clostridium beijerinckii throughout ABE fermentation. Appl. Microbiol. Biotechnol. 2021, 105, 877–889. [Google Scholar] [CrossRef] [PubMed]
- Jureckova, K.; Raschmanova, H.; Kolek, J.; Vasylkivska, M.; Branska, B.; Patakova, P.; Provaznik, I.; Sedlar, K. Identification and Validation of Reference Genes in Clostridium beijerinckii NRRL B-598 for RT-qPCR Using RNA-Seq Data. Front. Microbiol. 2021, 12, 640054. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Feature | Chromosome | Plasmid |
|---|---|---|
| Length (bp) | 5,876,902 | 73,345 |
| GC content (%) | 29.90 | 27.84 |
| Total number of ORF | 5209 | 70 |
| Total number of operons | 3237 | 54 |
| Protein coding genes | 4929 | 68 |
| Pseudogenes | 132 | 2 |
| rRNA genes (5S, 16S, 23S) | 17, 16, 16 | 0, 0, 0 |
| tRNA | 93 | 0 |
| ncRNA | 6 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sedlar, K.; Nykrynova, M.; Bezdicek, M.; Branska, B.; Lengerova, M.; Patakova, P.; Skutkova, H. Diversity and Evolution of Clostridium beijerinckii and Complete Genome of the Type Strain DSM 791T. Processes 2021, 9, 1196. https://doi.org/10.3390/pr9071196
Sedlar K, Nykrynova M, Bezdicek M, Branska B, Lengerova M, Patakova P, Skutkova H. Diversity and Evolution of Clostridium beijerinckii and Complete Genome of the Type Strain DSM 791T. Processes. 2021; 9(7):1196. https://doi.org/10.3390/pr9071196
Chicago/Turabian StyleSedlar, Karel, Marketa Nykrynova, Matej Bezdicek, Barbora Branska, Martina Lengerova, Petra Patakova, and Helena Skutkova. 2021. "Diversity and Evolution of Clostridium beijerinckii and Complete Genome of the Type Strain DSM 791T" Processes 9, no. 7: 1196. https://doi.org/10.3390/pr9071196
APA StyleSedlar, K., Nykrynova, M., Bezdicek, M., Branska, B., Lengerova, M., Patakova, P., & Skutkova, H. (2021). Diversity and Evolution of Clostridium beijerinckii and Complete Genome of the Type Strain DSM 791T. Processes, 9(7), 1196. https://doi.org/10.3390/pr9071196