Improvement of the Mechanical Properties of Thermosetting-Binding-System-Based Composites by Means of Kneading Procedure Modification and Composite Formulation

Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
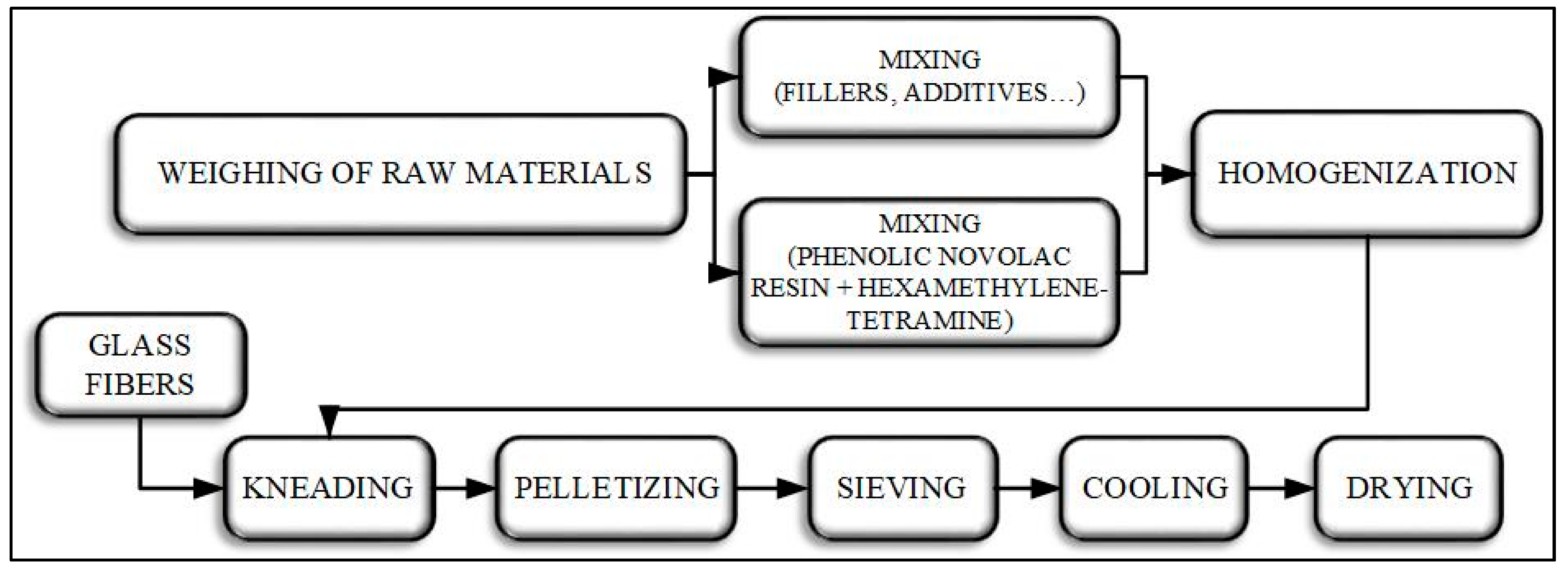
2.2. Preparation of Phenolic Molding Compounds
2.3. Preparation of Phenolic Molding Compound Standard Test Specimens
2.4. Testing Methods
- Differential scanning calorimetry (DSC): The test samples were heated in a thermal analyzer apparatus (DSC823e Module STARe System, Mettler Toledo, Greifensee, Switzerland), in order to determine heat flow profiles of the PFs before the compounding process and of the binding system before the compounding process. Aluminum pans (40 µL) with pierced lids, containing 12.0 mg ± 2.0 mg of test samples for PF resins and 10.0 mg ± 1.0 mg for binding system, were analyzed at the temperature range from 25 °C to 300 °C, with a heating rate of 10 K/min under air atmosphere.
- Melt viscosity analysis: Melt viscosities of PF resins were measured using a cone-plate rotating springless viscometer (RM100 CP2405, Lamy Rheology, Champagne au Mont d’Or, France). Samples of 80.0 mg ± 0.2 mg of the tested PF resins were weighed and used for determination of melt viscosity of PF resins. Preset time of measurement was 60 s, temperature was 150 °C, and shear rate was 600 s−1.
- Phenolic resin flow distance test: Phenolic resin flow distance is a standardized test (ISO 8619) used to check the reactivity and viscosity of resin. We applied two modifications to the standardized method: the pellet size was 14.50 mm ± 0.3 mm in diameter and 5.0 mm ± 0.2 mm in height, and the inclining apparatus was made out of stainless steel. Twenty grams of PF and HMTA were weighed in a weight ratio of 9:1 and homogenized in a coffee grinder (Coffee grinder SMK150B, Gorenje, Velenje, Slovenia) for 60 s. Pellets were crafted by weighing 0.50 g ± 0.01 g of powered homogeneous mixture of PF and HMTA, followed by pressing to specified dimensions. The stainless steel inclining apparatus was tempered in a horizontal position at 125 °C ± 1 °C for 60 min in an oven (Performance laboratory oven SP-45C, Kambič, Semič, Slovenia), while we placed the previously crafted pellets on a cold smooth glass plate—the distance from the edges of the smooth glass plate was 1 cm. After placing the cold smooth glass plate, carrying test pellets, on the inclining apparatus, we kept the plate in a horizontal position for 180 s ± 3 s, then lowered the shelf and glass plate to 60° and continued heating for 20 min. Finally, we removed the glass plate from the oven and cooled to room temperature before measuring the distance from the upper point in the original position of the pellets to the point of most extreme flow. We expressed the results as the arithmetic mean of the two flow distances.
- Volatile organic compounds: The test samples were heated in a halogen moisture analyzer apparatus (HG63 Halogen Moisture, Mettler Toledo, Greifensee, Switzerland). We determined volatile organic compounds of PFs before the compounding process and of PMC granulates by heating (and maintaining at the temperature 168 °C for 12 min) aluminum sample pans containing 13.00 g ± 0.10 g of test samples.
- Chemical rheology analysis: The flow-curing behavior of the PMC granulates was determined using a torque rheometer (Brabender Plastograph EC plus equipped with MB30 mixer, Brabender GmbH & Co. KG, Duisburg, Germany), obtaining typical plastograms, which show the relationship between torque (corresponding to the melt viscosity of the tested sample) and the temperature–time in consideration of structural changes of the material. Twenty-five grams of test sample were loaded into the mixer bowl, which was heated and kept at a temperature of 140 °C. The speed of rotation of the triangle-shaped mixing blades was 30 rpm and the duration of the analysis was 130 s.
- Mechanical properties: The mechanical characterization of the injection-molded PMC standard test specimens was carried out using a universal tensile testing machine (AG-x plus 10 kN, Shimadzu Corporation, Kyoto, Japan) to determine tensile strength and flexural strength (according to ISO 527-1 and ISO 178, respectively). To determine the Charpy impact strength, we used an impact strength measuring machine (Charpy LY-XJJDSM; according to ISO 179-1). For all methods applied, we used standard injection-molded PMC standard test specimens. For the Charpy impact strength measurements, we modified the PMC standard test specimens by cutting them to dimensions 120.0 mm × 20.0 mm × 4.0 mm (L × W × H).
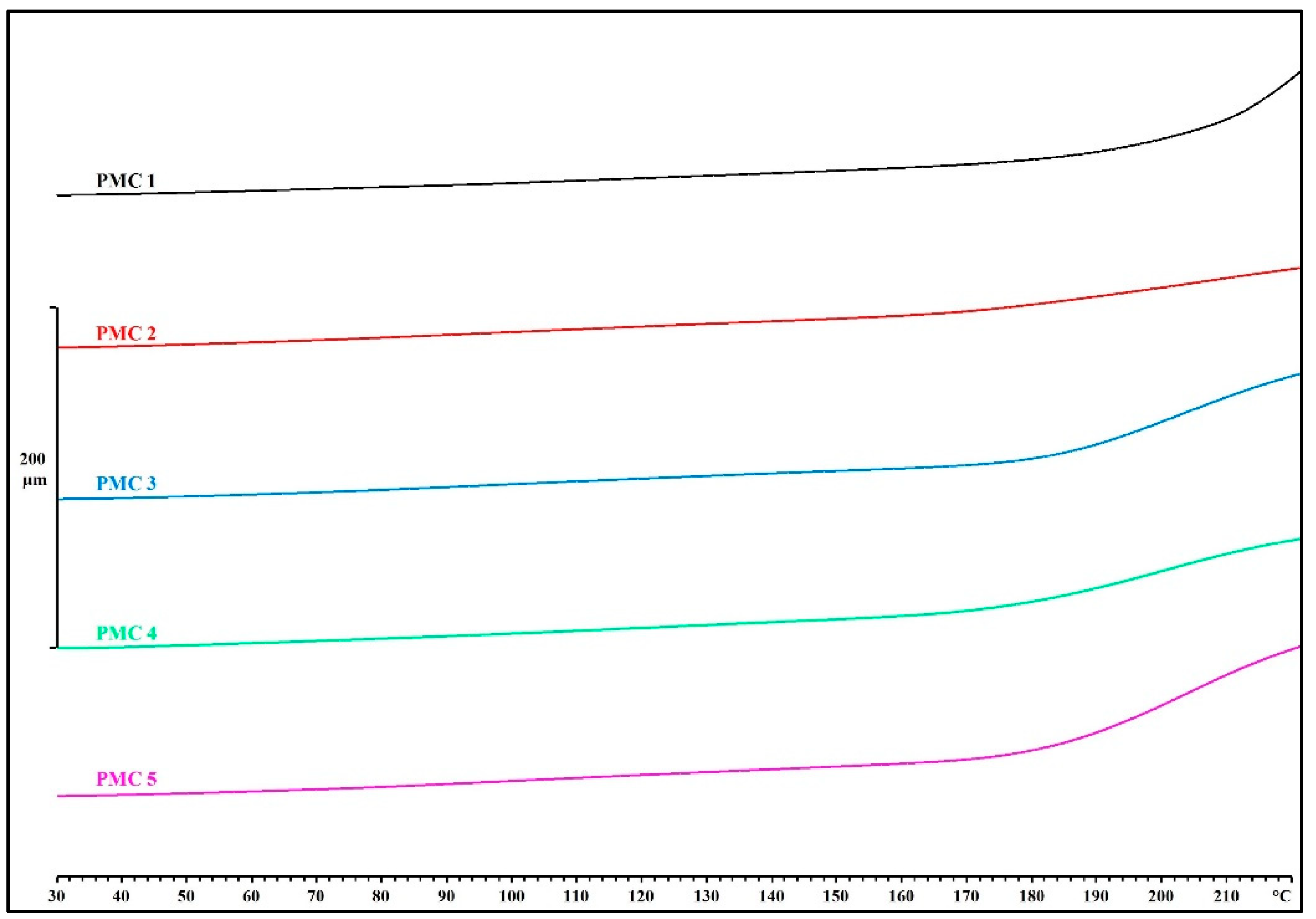
- Thermogravimetric analysis (TMA): We determined thermal expansion profiles of PMC standard test specimens using a thermomechanical analyzer apparatus (TMA/SDTA840, Mettler Toledo, Greifensee, Switzerland). Standard test specimens were cut to dimensions 10 mm × 10 mm × (3.5 mm ± 0.5 mm) and analyzed at a heating rate of 10 K/min from 30 °C to 400 °C, using a 3.0 mm probe with a flat end (0.05 N force) under air atmosphere.
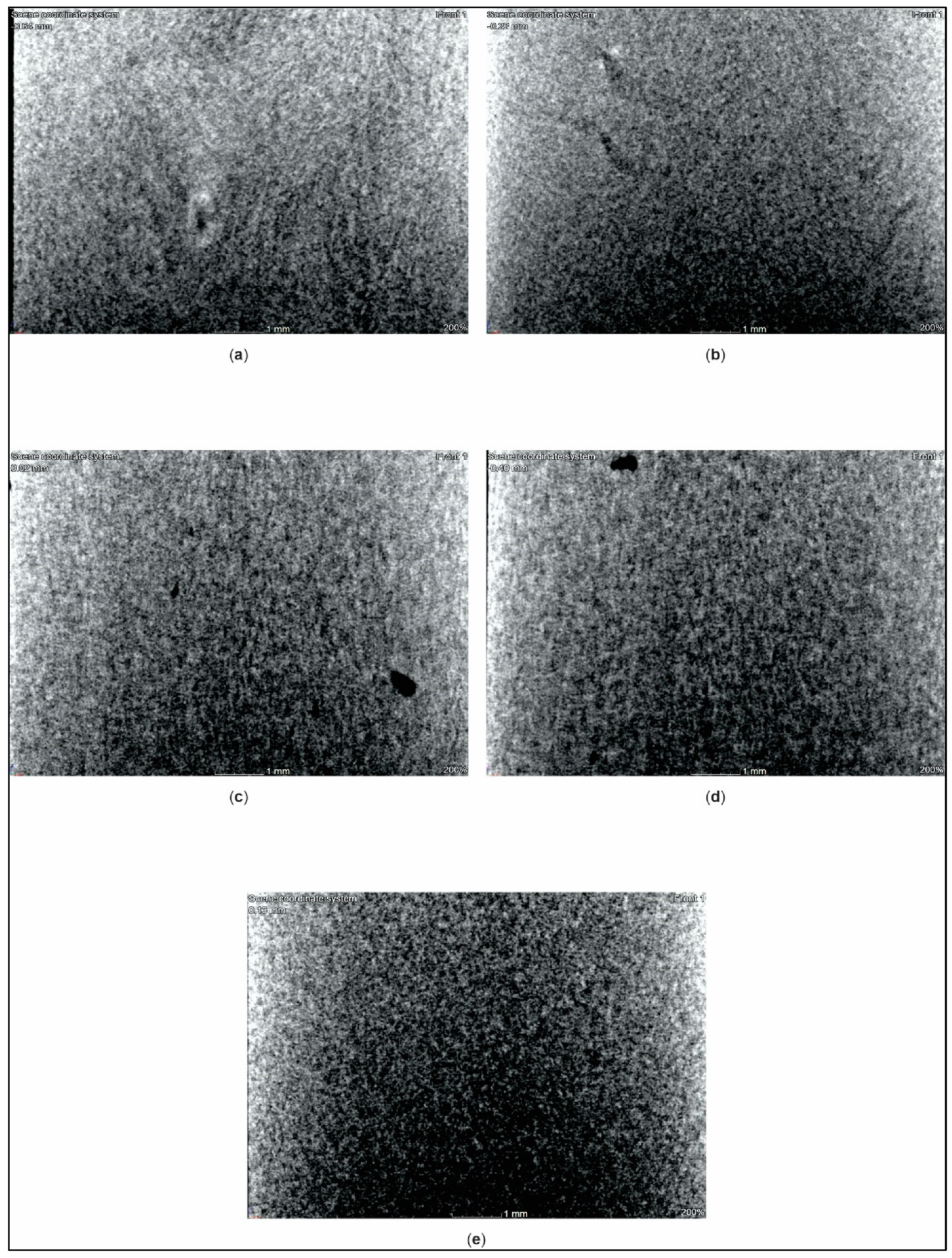
- X-ray computed tomography (XCT): We determined manufacturing defects in the PMC standard test specimens using an X-ray computed tomography system (phoenix v|tome|x s240 system, General Electric, Boston, United States of America). Standard test specimens were cut to dimensions of 16.0 mm × 10.0 mm × 4.0 mm and scanned at a nano-focus tube voltage of 180 kV with a measurement current of 200 µA.
3. Results and Discussion
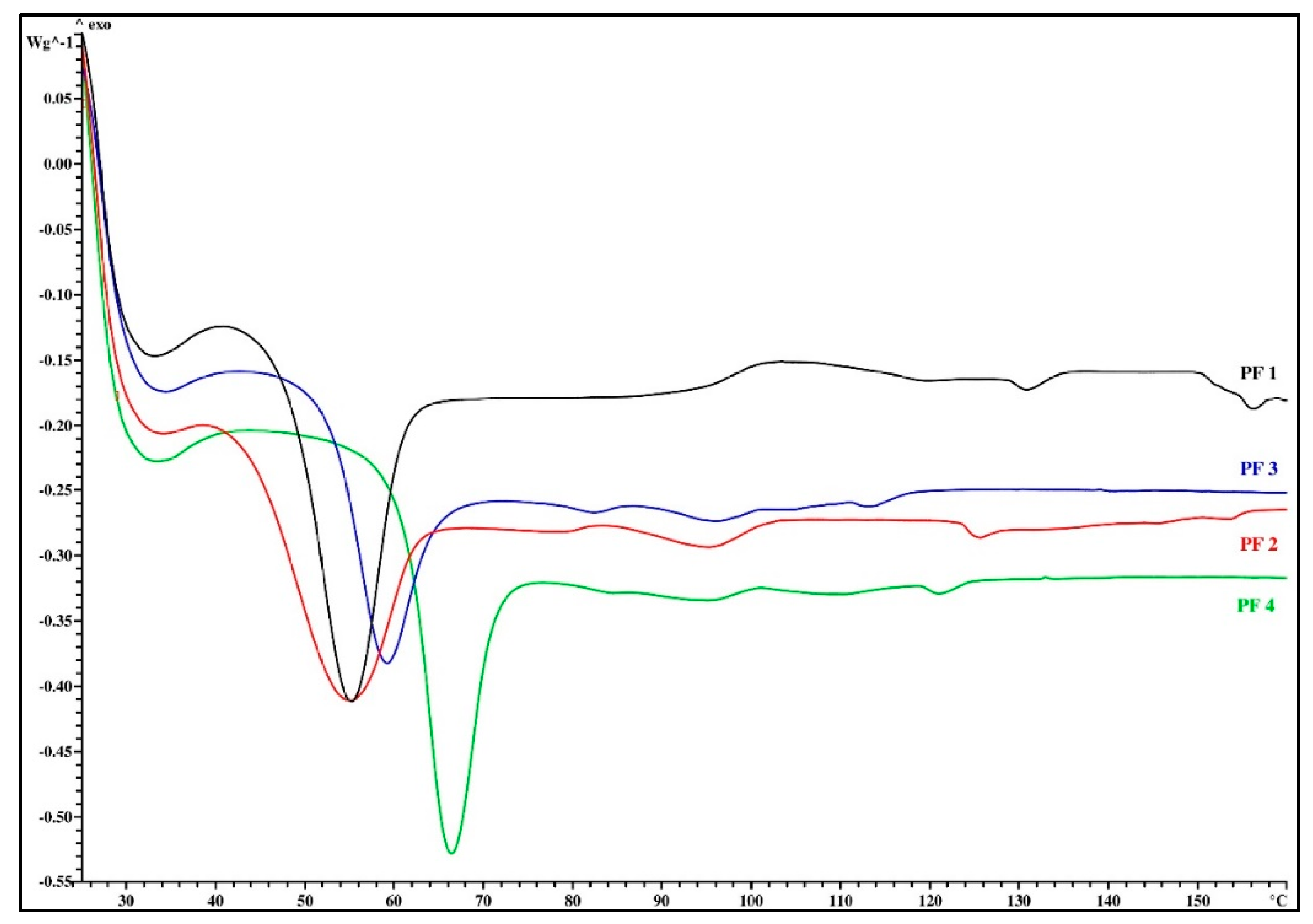
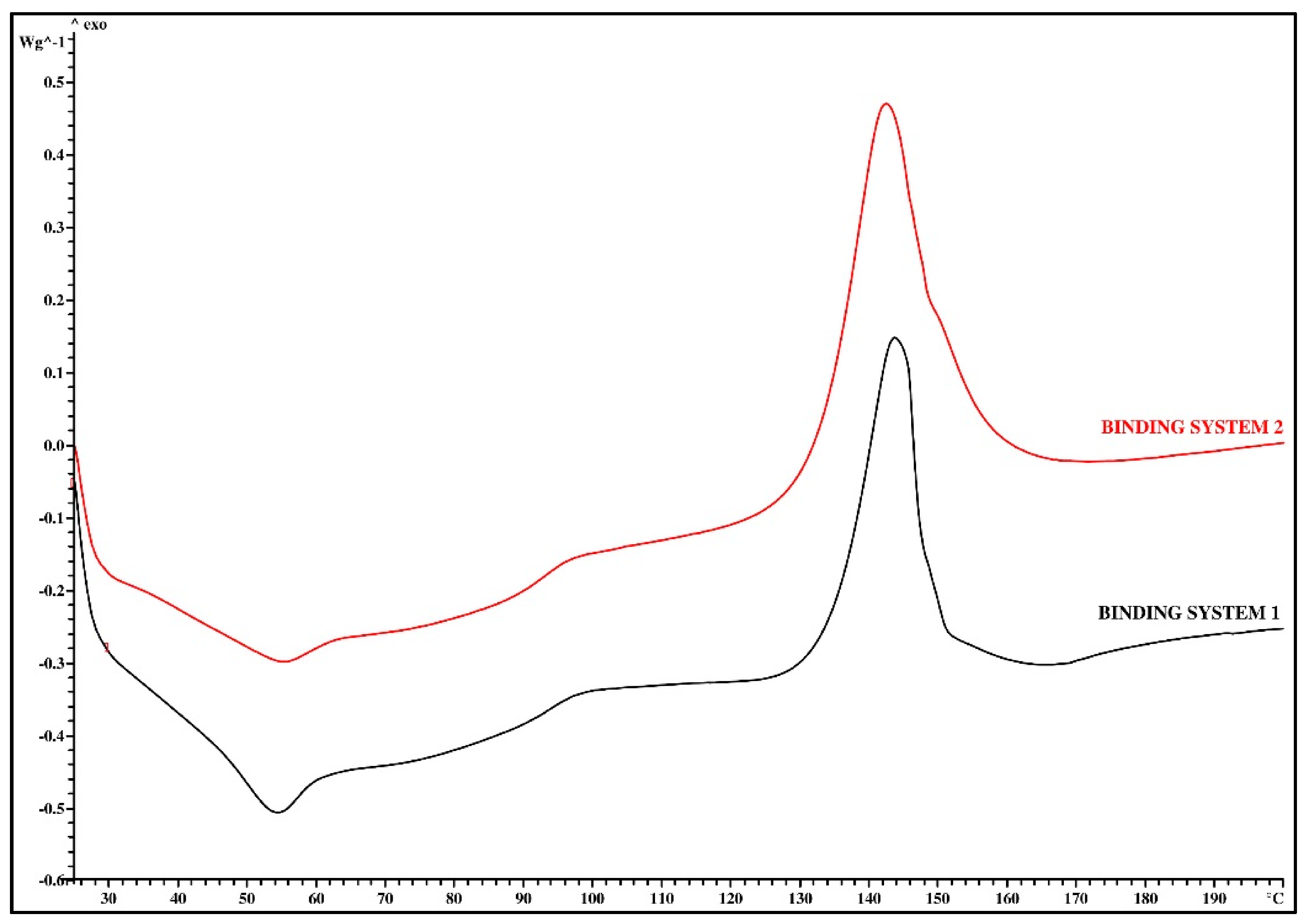
3.1. Thermal Analysis
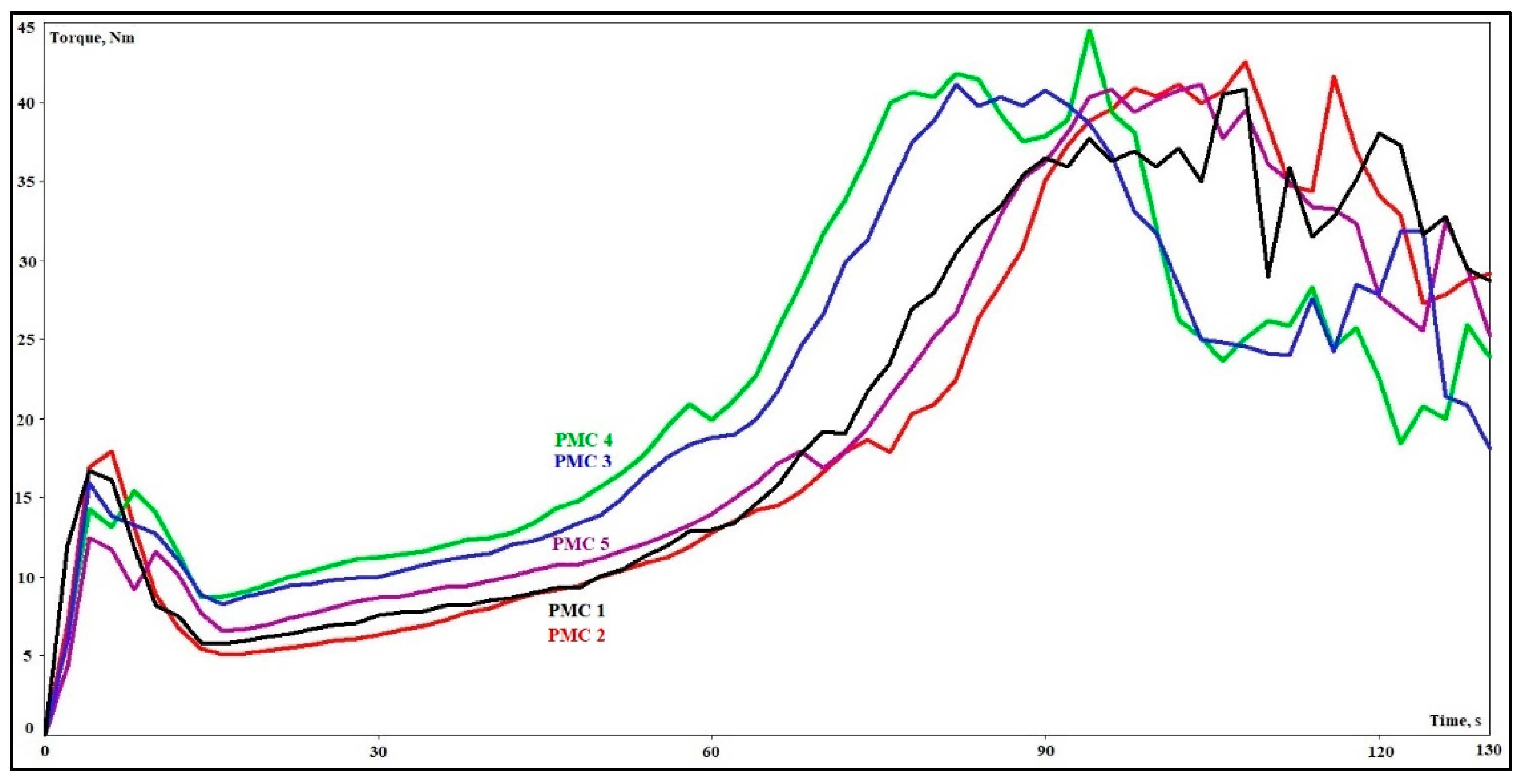
3.2. Chemical Rheological Analysis
3.3. Mechanical and Thermomechanical Properties
3.4. X-ray Computed Tomography Analysis
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sapuan, S.M. (Ed.) Composite materials. In Composite Materials: Concurrent Engineering Approach, 1st ed.; Butterworth-Heinemann: Oxford, UK, 2017; pp. 57–72. [Google Scholar]
- Hirano, K.; Asami, M. Phenolic resins—100 years of progress and their future. React. Funct. Polym. 2013, 73, 256–269. [Google Scholar] [CrossRef]
- Gardziella, A.; Pilato, L.A.; Knop, A. Phenolic Resins, 2nd ed.; Springer: Berlin/Heidelberg, Germany; New York, NY, USA, 2000; Volume 2, pp. 24–79. [Google Scholar]
- Wan, J.; Wang, S.; Li, C.; Zhou, D.; Chen, J.; Liu, Z.; Yu, L.; Fan, H.; Li, B.-G. Effect of molecular weight and molecular weight distribution on cure reaction of novolac with hexamethylenetetramine and properties of related composites. Thermochimica Acta 2012, 530, 32–41. [Google Scholar] [CrossRef]
- Mikhailin, Y.A.; Kerber, M.L.; Gorbunova, I.Y. Binders for polymer composites. P. Curtis (translation). Int. Polym. Sci. Technol. 2002, 29, 49–61. [Google Scholar] [CrossRef]
- Kalyon, D.M.; Hallouch, M. Compounding of Thermosets in Continuous Kneaders. Adv. Polym. Technol. 1986, 3, 237–249. [Google Scholar] [CrossRef]
- Števulová, N.; Schwarzova, I.; Hospodarova, V.; Junak, J.; Ondova, M.; Vaclavik, V. Possibilities of Using Alternative Materials as a Substitute Binder or Filler into Composites. Adv. Mater. Res. 2015, 1105, 31–35. [Google Scholar] [CrossRef]
- Ribeiro, M.C.S.; Meira-Castro, A.C.; Silva, F.G.; Santos, J.; Meixedo, J.P.; Fiúza, A.; Dinis, M.L.; Alvim, M.R. Re-use assessment of thermoset composite wastes as aggregate and filler replacement for concrete-polymer composite materials: A case study regarding GFRP pultrusion wastes. Resour. Conserv. Recycl. 2015, 104 Pt B, 417–426. [Google Scholar] [CrossRef]
- Liaw, J.H.; Hsueh, T.Y.; Tan, T.-S.; Wang, Y.; Chiao, S.-M. Twin-screw compounding of poly (methyl methacrylate)/clay nanocomposites: Effects of compounding temperature and matrix molecular weight. Polym. Int. 2007, 56, 1045–1052. [Google Scholar] [CrossRef]
- Williams, C.K.; Hillmyer, M.C. Polymers from Renewable Resources: A Perspective for a Special Issue of Polymer Reviews. Polym. Rev. 2008, 48, 1–10. [Google Scholar] [CrossRef]
- Elemans, P.H.M. Continuous equipment simulation—Co-kneader. In Mixing and Compounding of Polymers, 2nd ed.; Manas-Zloczower, I., Ed.; Carl Hanser Verlag: Munich, Germany, 2009; Volume 1, pp. 419–450. [Google Scholar]
- Saba, N.; Jawaid, M.; Sultan, M.T.H. An overview of mechanical and physical testing of composite materials. In Mechanical and Physical Testing of Biocomposites, Fibre-Reinforced Composites and Hybrid Composites, 1st ed.; Jawaid, M., Thariq, M., Saba, N., Eds.; Woodhead Publishing: Duxford, UK, 2019; Volume 1, pp. 1–12. [Google Scholar]
- Kastner, J.; Plank, B.; Salaberger, D.; Sekelja, J. Defect and porosity determination of fibre reinforced polymers by X-ray computed tomography. In Proceedings of the 2nd International Symposium on NDT in Aerospace, Hamburg, Germany, 22–24 November 2010. [Google Scholar]
- Garcea, S.C.; Wang, Y.; Withers, P.J. X-ray computed tomography for polymer composites. Compos. Sci. Technol. 2018, 156, 305–319. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PF Resin Mark | Mw (Da) | Mn (Da) | Mw/Mn | Free Phenol Content |
|---|---|---|---|---|
| PF 1 | n.a. | n.a. | n.a. | n.a. |
| PF 2 | n.a. | n.a. | n.a. | 0.44 |
| PF 3 1 | 1900 | 1000 | 1.9 | 0.20–0.50 |
| PF 4 | 1900 | 1000 | 1.9 | 0.20–0.50 |
| Phenolic Molding Compound | Compounding Process Temperature (°C) | Material Throughput (kg/h) | Screw Speed (rpm) |
|---|---|---|---|
| PMC 1 | 86 | 25 | 55 |
| PMC 2 | 80 | 20 | 44 |
| PMC 3 | 80 | 20 | 53 |
| PMC 4 | 83 | 25 | 55 |
| PMC 5 | 83 | 25 | 66 |
| PF Resin Mark | Tg (°C) | ΔTg (°C) | Δcp (J/g∙K) | Melt Viscosity (mPa∙s) | Flow Length (mm) | VOC (%) |
|---|---|---|---|---|---|---|
| PF 1 | 48.5 | 1.2 | 0.33 | 2834 | 87.0 | 1.1 |
| PF 2 | 45.4 | 3.2 | 0.43 | 1070 | 78.0 | 1.6 |
| PF 3 | 53.8 | 2.4 | 0.56 | 1130 | 88.5 | 1.0 |
| PF 4 | 61.2 | 1.6 | 0.65 | 1717 | 100.5 | 0.8 |
| Binding System Mark | Tg (°C) | ΔTg (°C) | Δcp (J/g∙K) | Tc (°C) | ΔTc (°C) | ΔHc (J/g) |
|---|---|---|---|---|---|---|
| Binding System 1 | 48.2 | 13.0 | 0.18 | 143.6 | 13.8 | −28.40 |
| Binding System 2 | 43.6 | 3.2 | 0.32 | 142.4 | 19.3 | −45.06 |
| Phenolic Molding Composite | Residence Time (s) | Reaction Time (s) | Torque Minimum (Nm) | VOC (%) |
|---|---|---|---|---|
| PMC 1 | 37 | 82 | 5.8 | 3.1 |
| PMC 2 | 33 | 84 | 5.1 | 3.2 |
| PMC 3 | 29 | 72 | 8.3 | 3.0 |
| PMC 4 | 27 | 66 | 8.7 | 3.1 |
| PMC 5 | 30 | 77 | 6.6 | 3.4 |
| Standard Test Specimen | Td (°C) | Tensile Strength (MPa) | Flexural Strength (MPa) | Charpy Impact Strength (Unnotched) (kJ/m2) |
|---|---|---|---|---|
| PMC 1 | 190.7 | 50.3 ± 2.5 | 118.4 ± 16.0 | 5.9 ± 1.7 |
| PMC 2 | 165.9 | 88.7 ± 7.7 | 181.6 ± 5.9 | 8.3 ± 0.8 |
| PMC 3 | 178.4 | 85.7 ± 4.5 | 175.0 ± 7.5 | 7.3 ± 1.0 |
| PMC 4 | 171.6 | 86.0 ± 2.3 | 173.2 ± 8.1 | 7.8 ± 1.0 |
| PMC 5 | 177.7 | 87.7 ± 5.8 | 174.0 ± 3.6 | 8.2 ± 0.5 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Šahinović, A.; Mušič, B. Improvement of the Mechanical Properties of Thermosetting-Binding-System-Based Composites by Means of Kneading Procedure Modification and Composite Formulation. Processes 2020, 8, 625. https://doi.org/10.3390/pr8050625
Šahinović A, Mušič B. Improvement of the Mechanical Properties of Thermosetting-Binding-System-Based Composites by Means of Kneading Procedure Modification and Composite Formulation. Processes. 2020; 8(5):625. https://doi.org/10.3390/pr8050625
Chicago/Turabian StyleŠahinović, Adis, and Branka Mušič. 2020. "Improvement of the Mechanical Properties of Thermosetting-Binding-System-Based Composites by Means of Kneading Procedure Modification and Composite Formulation" Processes 8, no. 5: 625. https://doi.org/10.3390/pr8050625
APA StyleŠahinović, A., & Mušič, B. (2020). Improvement of the Mechanical Properties of Thermosetting-Binding-System-Based Composites by Means of Kneading Procedure Modification and Composite Formulation. Processes, 8(5), 625. https://doi.org/10.3390/pr8050625