Synthesis and Characterization of New Fluoro/Trifluoromethyl-Substituted Acylthiourea Derivatives with Promising Activity against Planktonic and Biofilm-Embedded Microbial Cells



 , ,
, ,  , ,
, ,  ,
, 
 and
and
Abstract
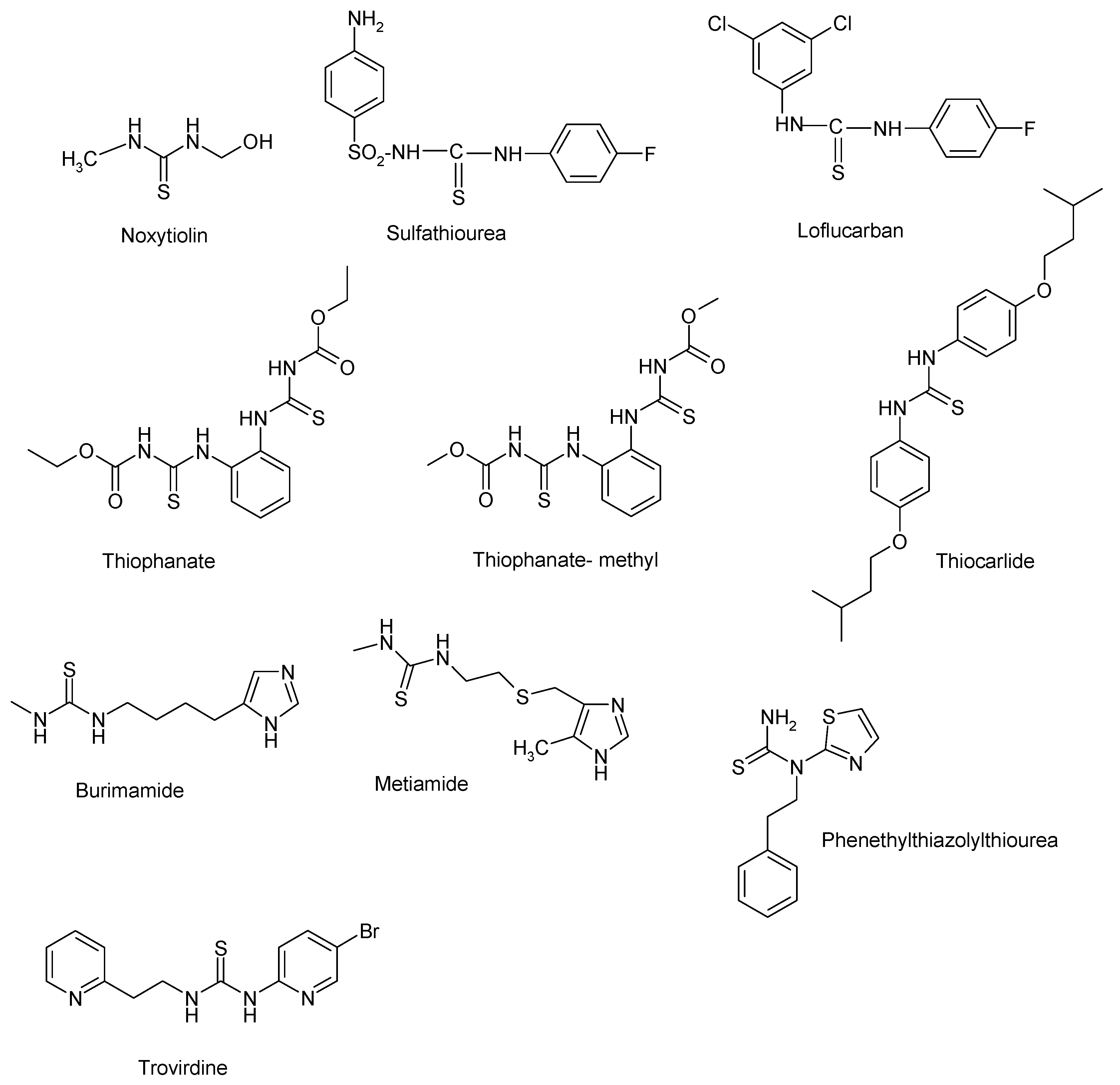
1. Introduction
2. Materials and Methods
2.1. Chemistry
2.1.1. General Synthesis Method of the New 2-((4-Chlorophenoxy)methyl)-N-(Arylcarbamothioyl) Benzamides
2.1.2. Spectral data
2.1.3. In Silico Drug-Likeness Analysis and Molecular Docking Screening
Properties Computations
Molecular Docking Simulations
2.2. Biological Assay
2.2.1. Qualitative Assessment of the Antimicrobial Activity
2.2.2. Quantitative Assessment of the Antimicrobial Activity
2.2.3. The Influence of the Tested Compounds on the Biofilm Development
2.3. Antioxidant Activity of the New Compounds
3. Results
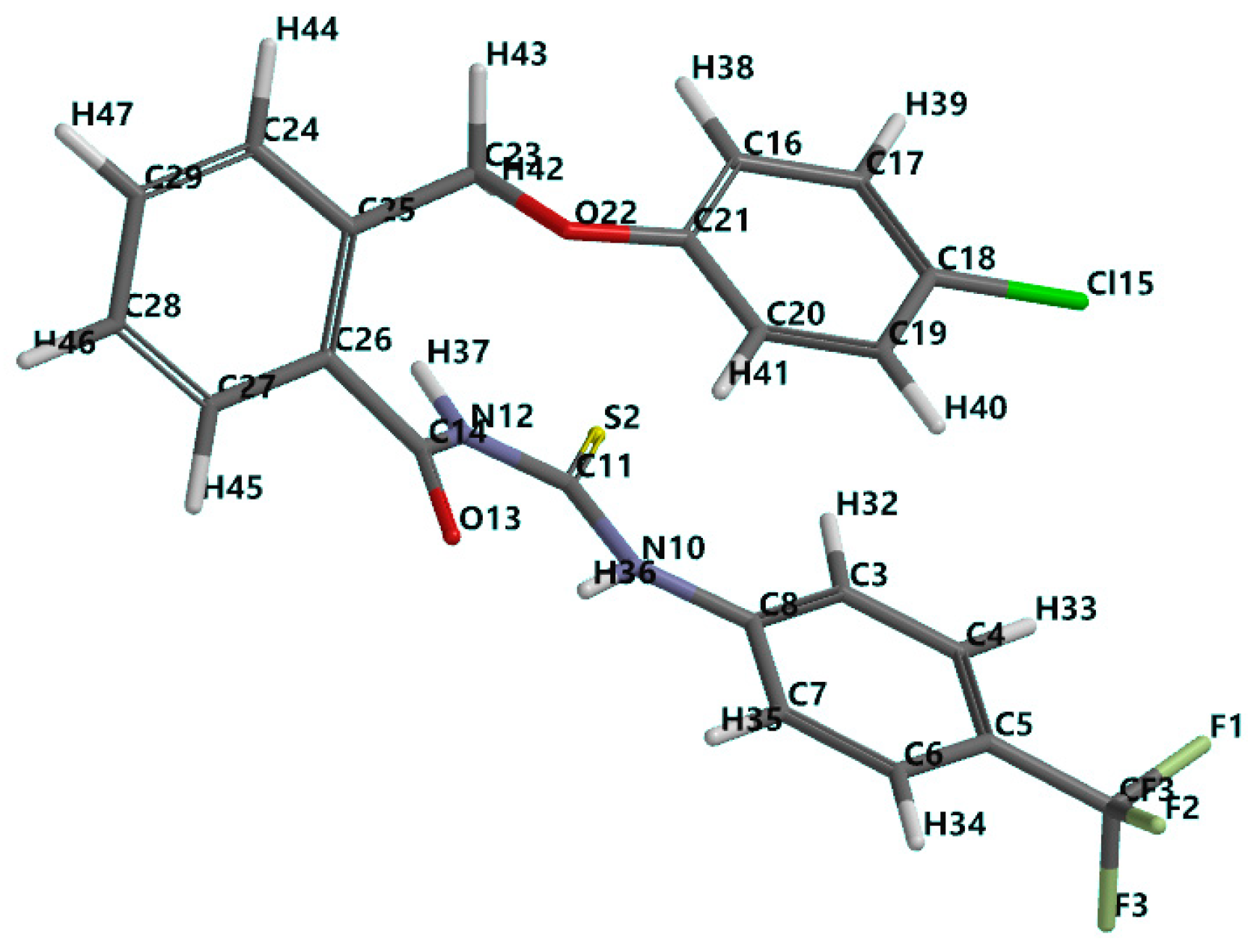
3.1. Chemistry
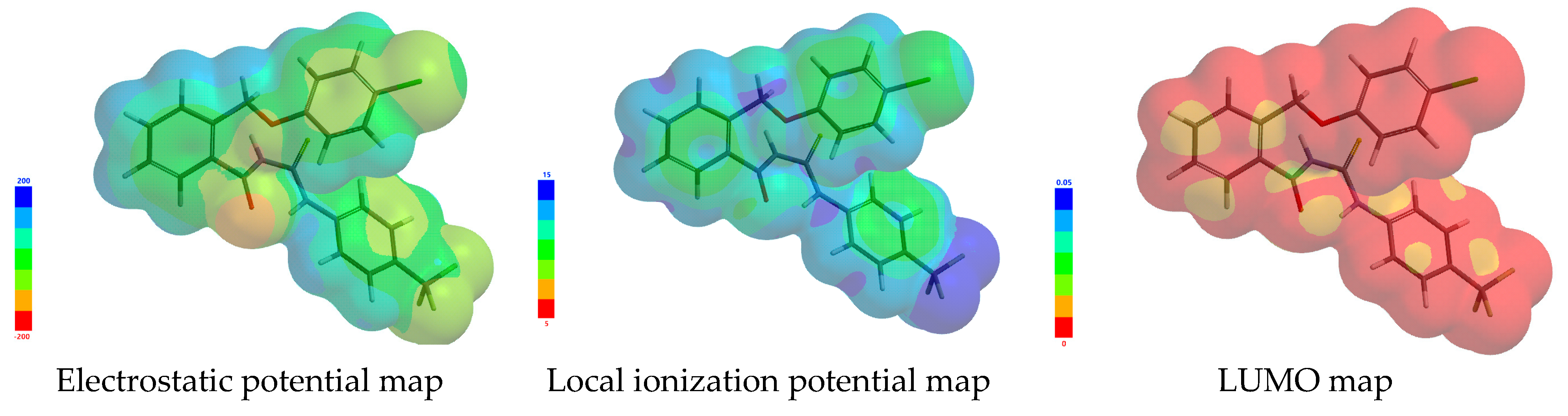
3.2. In Silico Drug-Like and Molecular Docking Screening
3.2.1. Ligand Preparation
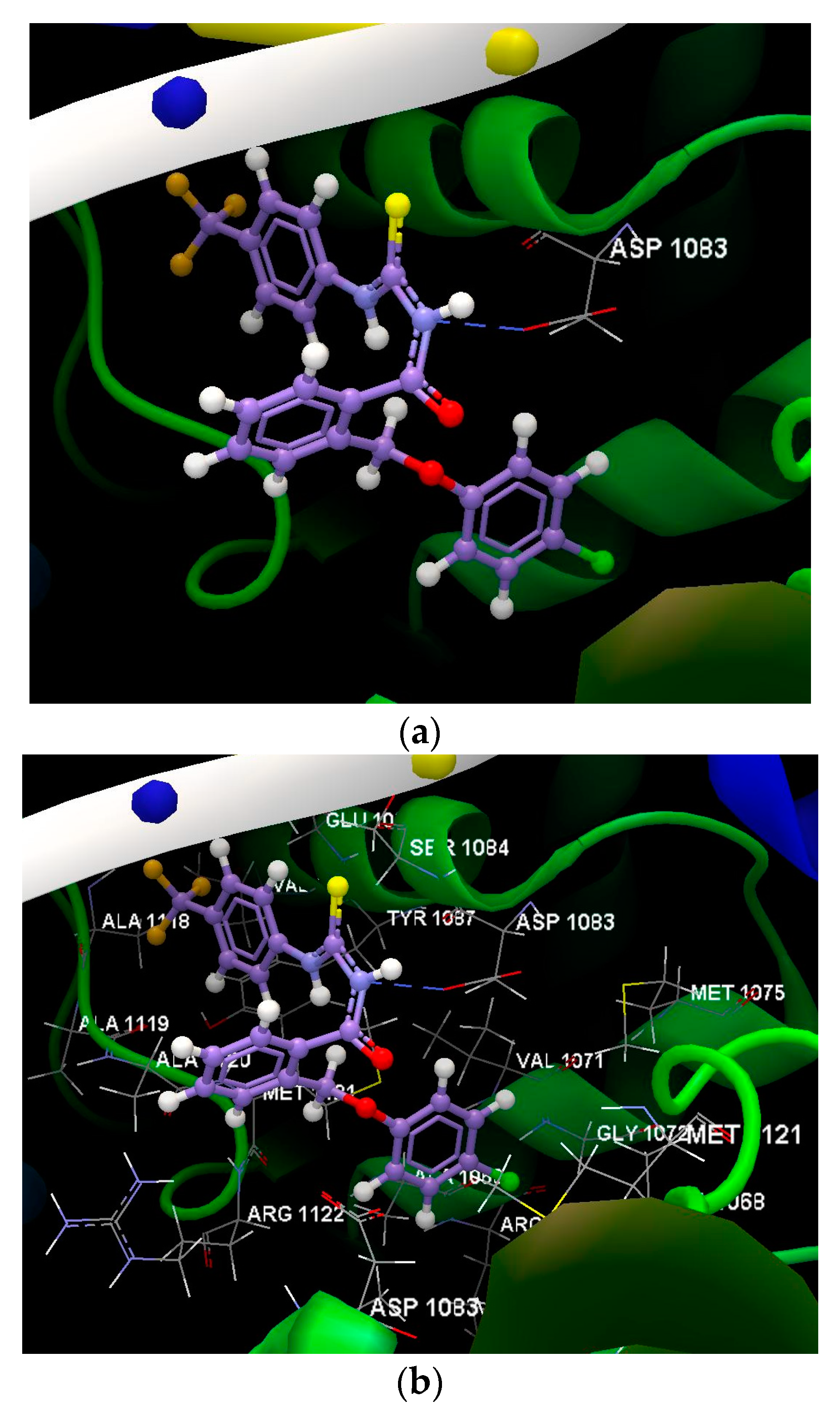
3.2.2. Docking Studies
3.3. Antimicrobial Activity Against Planktonic Cells
3.3.1. Qualitative Assay
3.3.2. Quantitative Assay
3.4. Anti-Biofilm Activity
3.5. Antioxidant Activity
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- DrugBank. Available online: https://www.drugbank.ca/drugs/DB13838 (accessed on 18 February 2020).
- DrugBank. Available online: https://www.drugbank.ca/drugs/DB13699 (accessed on 18 February 2020).
- PubChem. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Loflucarban (accessed on 18 February 2020).
- PubChem. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Thiophanate (accessed on 18 February 2020).
- PubChem. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Thiophanate-methyl (accessed on 18 February 2020).
- DrugBank. Available online: https://www.drugbank.ca/drugs/DB13608 (accessed on 18 February 2020).
- PubChem. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Burimamide (accessed on 18 February 2020).
- DrugBank. Available online: https://www.drugbank.ca/drugs/DB08805 (accessed on 18 February 2020).
- PubChem. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Phenethylthiazolylthiourea (accessed on 18 February 2020).
- Pedersen, O.S.; Pedersen, E.B. Non-nucleoside reverse transcriptase inhibitors: The NNRTI boom. Antivir. Chem. Chemother. 1999, 10, 285–314. [Google Scholar] [CrossRef]
- Maalik, A.; Rahim, H.; Saleem, M.; Fatima, N.; Rauf, A.; Wadood, A.; Malik, M.I.; Ahmed, A.; Rafique, H.; Zafar, M.N.; et al. Synthesis, antimicrobial, antioxidant, cytotoxic, antiurease and molecular docking studies of N-(3-trifluoromethyl)benzoyl-N′-aryl thiourea derivatives. Bioorg. Chem. 2019, 88. [Google Scholar] [CrossRef] [PubMed]
- Alimohammadi, A.; Mostafavi, H.; Mahdavi, M. Thiourea derivatives based on the dapsone-naphthoquinone hybrid as anticancer and antimicrobial agents: In vitro screening and molecular docking studies. Chem. Select. 2020, 5, 847–852. [Google Scholar] [CrossRef]
- Bielenica, A.; Kedzierska, E.; Fidecka, S.; Maluszynska, H.; Miroslaw, B.; Koziol, A.E.; Stefanska, J.; Madeddu, S.; Giliberti, G.; Sanna, G.; et al. Synthesis, antimicrobial and pharmacological evaluation of thiourea derivatives of 4H-1,2,4-triazole. Lett. Drug. Des. Discov. 2015, 12, 251–252. [Google Scholar] [CrossRef]
- Naz, S.; Zahoor, M.; Umar, M.N.; Ali, B.; Ullah, R.; Shahat, A.A.; Mahmood, H.M.; Sahibzada, M.U.K. Enzyme inhibitory, antioxidant and antibacterial potentials of synthetic symmetrical and unsymmetrical thioureas. Drug Des. Dev. Ther. 2019, 13, 3485–3495. [Google Scholar] [CrossRef]
- Tatar, E.; Karakuş, S.; Küçükgüzel, Ș.G.; Okullu, S.O.; Ünübol, N.; Kocagöz, T.; De Clercq, E.; Andrei, G.; Snoeck, R.; Pannecouque, C.; et al. Design, synthesis, and molecular docking studies of a conjugated thiadiazole–thiourea scaffold as antituberculosis agents. Biol. Pharm. Bull. 2016, 39, 502–515. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhai, Z.-W.; Shi, Y.-X.; Tan, C.-X.; Weng, J.-Q.; Han, L.; Li, B.-J.; Liu, X.-H. Novel trifluoromethylpyrazole acyl thiourea derivatives: Synthesis, antifungal activity and docking study. Lett. Drug. Des. Discov. 2019, 16. [Google Scholar] [CrossRef]
- Larik, A.F.; Saeed, A.; Faisal, M.; Channar, P.A.; Azam, S.S.; Ismail, H.; Dilshad, E.; Mirza, B. Synthesis, molecular docking and comparative efficacy of various alkyl/aryl thioureas as antibacterial, antifungal and α-amylase inhibitors. Comput. Biol. Chem. 2018, 77, 193–198. [Google Scholar] [CrossRef]
- Larik, F.A.; Saeed, A.; Channar, P.A.; Ismail, H.; Dilshad, E.; Mirza, B. New 1-octanoyl-3-aryl thiourea derivatives: Solvent-free synthesis, characterization and multi-target biological activities. Bangladesh J. Pharmacol. 2016, 11, 894–902. [Google Scholar] [CrossRef]
- Pingaew, R.; Sinthupoom, N.; Mandi, P.; Prachayasittikul, V.; Cherdtrakulkiat, R.; Prachayasittikul, S.; Ruchirawat, S.; Prachayasittikul, V. Synthesis, biological evaluation and in silico study of bis-thiourea derivatives as anticancer, antimalarial and antimicrobial agents. Med. Chem. Res. 2017, 26, 3136–3148. [Google Scholar] [CrossRef]
- El Bissati, K.; Redel, H.; Ting, L.-M.; Lykins, J.D.; McPhillie, M.J.; Upadhya, R.; Woster, P.M.; Yarlett, N.; Kim, K.; Weiss, L.M. Novel synthetic polyamines have potent antimalarial activities in vitro and in vivo by decreasing intracellular spermidine and spermine concentrations. Front. Cell. Infect. Microbiol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Vajragupta, O.; Pananookooln, S.; Tuntiwachwuttikul, P.; Foye, W. Ovicidal activity of 6-substituted-2- and -4-aminoquinolines and their mono and bis(thiourea)derivatives against human hookworm. J. Sci. Soc. Thailand 1998, 14, 51–59. [Google Scholar] [CrossRef]
- Ansari, M.M.; Deshmukh, S.P.; Khan, R.; Musaddiq, M. Synthesis antimicrobial and anticancer evaluation of 1-aryl-5-(o-methoxyphenyl)-2-S-benzyl Isothiobiurets. Int. J. Med. Chem. 2014. [Google Scholar] [CrossRef]
- Viswas, R.S.; Pundir, S.; Lee, H. Design and synthesis of 4-piperazinyl quinoline derived urea/thioureas for anti-breast cancer activity by a hybrid pharmacophore approach. J. Enzyme Inhib. Med. Chem. 2019, 34, 620–630. [Google Scholar] [CrossRef] [PubMed]
- Al-Harbi, R.A.K.; El-Sharief, M.A.M.S.; Abbas, Y. Synthesis and anticancer activity of bis-benzo[d][1,3]dioxol-5-yl thiourea derivatives with molecular docking study. Bioorg. Chem. 2019, 90, 103088. [Google Scholar] [CrossRef]
- Zhang, R.-Z.H.B.; Huang, X.-C.; Liang, G.-B.; Qin, J.-M.; Pan, Y.-M.; Liao, Z.-X.; Wang, H.-S. Synthesis and biological evaluation of terminal functionalized thiourea-containing dipeptides as antitumor agents. RSC Adv. 2017, 7, 8866. [Google Scholar]
- Çelen, A.O.; Kaymakçioùlu, B.; Gümrü, S.; Toklu, H.Z.; Aricioùlu, F. Synthesis and anticonvulsant activity of substituted thiourea derivatives. Marmara Pharm. J. 2011, 15, 43–47. [Google Scholar] [CrossRef]
- Kumar, P.; Sharma, A.; Gupta, S.K. Synthesis, characterization and anticonvulsant activity on novel thiourea derivatives. Int. J. Pharm. Med. Res. 2014, 2, 116–122. [Google Scholar]
- Thakur, A.S.; Deshmukh, R.; Jha, A.K.; Kumar, P.S. Molecular docking study and anticonvulsantactivity of synthesized 4-((4,6-dimethyl-6H-1,3-thiazin-2-yl)phenylsulfonyl)urea/ thiourea derivatives. J. King. Saud. Univ. Sci. 2018, 30, 330–336. [Google Scholar] [CrossRef]
- Alagarsamy, V.; Meena, S.; Ramseshu, K.V.; Solomon, V.R.; Thirumurugan, K.; Dhanabal, K.; Murugan, M. Synthesis, analgesic, anti-inflammatory, ulcerogenic index and antibacterial activities of novel 2-methylthio-3-substituted-5,6,7,8-tetrahydrobenzo(b)thieno[2,3-d]pyrimidin-4(3H)-ones. Eur. J. Med. Chem. 2006, 41, 1293–1300. [Google Scholar] [CrossRef]
- Sheorey, R.V.; Thangathiruppathy, A.; Alagarsamy, V. Synthesis, analgesic and anti-inflammatory activities of 3-ethyl-2-substituted amino-3H-quinazolin-4-ones. Trop. J. Pharm. Res. 2013, 12, 583–589. [Google Scholar] [CrossRef]
- Sudzhaev, A.R.; Rzaeva, I.A.; Nadzhafova, R.A.; Safarov, Y.A.; Allakhverdiev, M.A. Antioxidant properties of some thiourea derivatives. Russ. J. Appl. Chem. 2011, 84, 1394. [Google Scholar]
- Hameed, A.; Jafri, L.; Sheikh, M.A. Effect of thiourea on proteins, catalase, guaiacol-peroxidase and protease activities in wheat leaves under H2O2 induced oxidative stress. Plant Physiol. 2013, 4, 857–864. [Google Scholar]
- Kalaiyarasi, A.; Haribabu, J.; Gayathri, D.; Gomathi, K.; Bhuvanesh, N.S.P.; Karvembu, R.; Biju, V.M. Chemosensing, molecular docking and antioxidant studies of 8-aminoquinoline appended acylthiourea derivatives. J. Mol. Struct. 2019, 1185, 450–460. [Google Scholar] [CrossRef]
- Wang, B.; Ma, Y.; Xiong, L.; Li, Z. Synthesis and insecticidal activity of novel N-pyridylpyrazole carbonyl thioureas. Chin. J. Chem. 2012, 30, 815–821. [Google Scholar] [CrossRef]
- Yunyun, G.; Wei, H.; Xinghai, L.; Jianquan, V.; Chengxia, T. Synthesis and herbicidal activities of 1-methylcyclohexyl-formayl thioureas. Chin. J. Org. Chem. 2013, 33, 2396–2401. [Google Scholar]
- Wua, A.-B.; Duana, L.-P. Transition metal complexes of N-(4,6-dimethoxypyrimidin-2-ylcarbamothioyl)benzamide: Design, synthesis and herbicidal activity. J. Chin. Chem. Soc. 2009, 56, 539–542. [Google Scholar] [CrossRef]
- Lin, Q.; Yao, H.; Wei, T.-B.; Zhang, Y.-M. Synthesis and plant growth regulatory activity of o-nitrobenzoylthiourea derivatives. Indian J. Chem. 2009, 48, 124–127. [Google Scholar]
- Kachhadia, V.V.; Patel, M.R.; Joshi, H.S. Heterocyclic systems containing S/N regioselective nucleophilic competition: Facile synthesis, antitubercular and antimicrobial activity of thiohydantoins and iminothiazolidinones containing the benzo[b]thiophene moiety. J. Serb. Chem. Soc. 2005, 70, 153–161. [Google Scholar] [CrossRef]
- Sun, C.; Huang, H.; Feng, M.; Shi, X.; Zhang, X.; Zhou, P. A novel class of potent influenza virus inhibitors: Polysubstituted acylthiourea and its fused heterocycle derivatives. Bioorg. Med. Chem. Lett. 2005, 16, 162–166. [Google Scholar] [CrossRef]
- Chandrasekhar, M.; Syam Prasad, G.; Venkataramaiah, C.; Umapriya, K.; Raju, C.N.; Seshaiah, K.; Rajendra, W. In silico and in vitro antioxidant and anticancer activity profiles of urea and thiourea derivatives of 2,3-dihydro-1H-inden-1-amine. J. Recept. Signal. Transduct. Res. 2020, 40, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Sudhamani, H.; Basha, S.T.; Venkateswarlu, N.; Vijaya, T.; Raju, C.N. Synthesis and characterization of new thiourea and urea derivatives of 6-fluoro-3-(piperidin-4-yl)benzo[d]isoxazole: In vitro antimicrobial and antioxidant activity. J. Chem. Sci. 2015, 127, 1739–1746. [Google Scholar] [CrossRef]
- Firdausiah, S.; Hasbullah, S.A.; Yamin, B.M. Synthesis, structurale elucidation and antioxidant study of ortho-substituted N,N’-bis(benzamidothiocarbonyl)hydrazine derivatives. J. Phys. Conf. Ser. 2018, 979, 012010. [Google Scholar] [CrossRef]
- Yeşilkaynak, T. Synthesis and characterization of N-((6-methylpyridin-2-yl)carbamothioyl)thiophene-2-carboxamide and its Co(II), Ni(II) and Cu(II) complexes: Calculation of the molecular orbitals and antioxidant and antitumor activities. J. Turkish Chem. Soc. Sect. Chem. 2016, 3, 1–14. [Google Scholar] [CrossRef]
- Da Silva, T.L.; Forain Miolo, L.M.; Sousa, F.S.S.; Brod, L.M.P.; Savegnago, L.; Schneider, P.H. New thioureas based on thiazolidines with antioxidant potential. Tetrahedron Lett. 2015, 56, 6674–6680. [Google Scholar] [CrossRef]
- Nacea, V.; Boscenco, R.; Missir, A.V.; Limban, C.; Bărbuceanu, S. Sinteza şi caracterizarea unor noi combinaţii complexe ale Cu (I) şi Cu (II) cu noi tioureide N,N’-disubstituite. Rev. Chim. Buchar. 2005, 56, 68–71. [Google Scholar]
- Limban, C.; Missir, A.V.; Chiriţă, I.C.; Bădiceanu, C.D.; Drăghici, C.; Balotescu, M.C.; Stamatoiu, O. New thioureides of 2-(4-methyl-phenoxymethyl)-benzoic and 2-(4-methoxy-phenoxymethyl)-benzoic acids with biological activity. Rev. Roum. Chim. 2008, 53, 595–602. [Google Scholar]
- Limban, C.; Missir, A.V.; Grumezescu, A.M.; Oprea, A.E.; Grumezescu, V.; Vasile, B.S.; Socol, G.; Trușcă, R.; Caproiu, M.T.; Chifiriuc, M.C.; et al. Bioevaluation of novel anti-biofilm coatings based on PVP/Fe3O4 nanostructures and 2-((4-ethylphenoxy)methyl)-N-(arylcarbamothioyl)benzamides. Molecules 2014, 19, 12011–12030. [Google Scholar] [CrossRef]
- Limban, C.; Missir, A.V.; Nuţă, D.C.; Căproiu, M.T.; Papacocea, M.T.; Chiriţă, C. Synthesis of some new 2-((4-chlorophenoxy)methyl)-N-(arylcarbamothioyl)benzamides as potential antifungal agents. Farmacia 2016, 64, 775–779. [Google Scholar]
- Limban, C.; Balotescu Chifiriuc, M.C.; Missir, A.V.; Chiriţă, I.C.; Bleotu, C. Antimicrobial activity of some new thioureides derived from 2-(4-chlorophenoxymethyl)benzoic acid. Molecules 2008, 13, 567–580. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle- Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Hehre, W.J. A Guide to Molecular Mechanics and Quantum Chemical Calculations; Wavefunction, Inc.: Irvine, CA, USA, 2003. [Google Scholar]
- Halgren, T.A. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J. Comput. Chem. 1996, 17, 490–519. [Google Scholar] [CrossRef]
- Bax, D.B.; Chan, P.F.; Eggleston, D.S.; Fosberry, A.; Gentry, D.R.; Gorrec, F.; Giordano, I.; Hann, M.; Hennessy, A.; Hibbs, M.; et al. Type IIA topoisomerase inhibition by a new class of antibacterial agents. Nature 2010, 466, 935–940. [Google Scholar] [CrossRef] [PubMed]
- Calu, L.; Badea, M.; Chifiriuc, M.C.; Bleotu, C.; David, G.-I.; Ioniţă, C.; Măruţescu, L.; Lazăr, V.; Stanică, N.; Soponaru, I.; et al. Synthesis, spectral, thermal, magnetic and biological characterization of Co(II), Ni(II), Cu(II) and Zn(II) complexes with a Schiff base bearing a 1,2,4-triazole pharmacophore. J. Therm. Anal. Calorim. 2015, 120, 375–386. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Misral, H.; Sapari, S.; Rahman, T.; Ibrahim, N.; Yamin, B.M.; Hasbulla, S.A. Evaluation of novel N-(dibenzylcarbamothioyl)benzamide derivatives as antibacterial agents by using DFT and drug-likeness assessment. J. Chem. 2018. [Google Scholar] [CrossRef]
- Korb, O.; Stützle, T.; Exner, T.E. Empirical scoring functions for advanced protein-ligand docking with plants. J. Chem. Inf. Model. 2009, 49, 84–96. [Google Scholar] [CrossRef]
- Ghose, A.K.; Pritchett, A.; Crippenic, G.M. Atomic physicochemical parameters for three dimensional structure directed quantitative structure-activity relationships III: Modeling hydrophobic interactions. J. Comput. Chem. 1988, 9, 80–90. [Google Scholar] [CrossRef]
- Cheng, T.; Zhao, Y.; Li, X.; Lin, F.; Xu, Y.; Zhang, X.; Li, Y.; Wang, R. Computation of octanol-water partition coefficients by guiding an additive model with knowledge. J. Chem. Inf. Model. 2007, 47, 2140–2148. [Google Scholar] [CrossRef]
- Prior, R.L.; Wu, X.; Schaich, K. Standardized methods for the determination of antioxidant capacity and phenolics in food and dietary supplements. J. Agric. Food Chem. 2005, 53, 4290–4302. [Google Scholar] [CrossRef]
- Shah, P.; Westwell, A.D. The role of fluorine in medicinal chemistry. J. Enzyme Inhib. Med. Chem. 2007, 22, 527–540. [Google Scholar] [CrossRef] [PubMed]
- Algburi, A.; Comito, N.; Kashtanov, D.; Dicks, L.M.T.; Chikindas, M.L. Control of biofilm formation: Antibiotics and beyond. Appl. Environ. Microbiol. 2017, 83, e02508–e02516. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
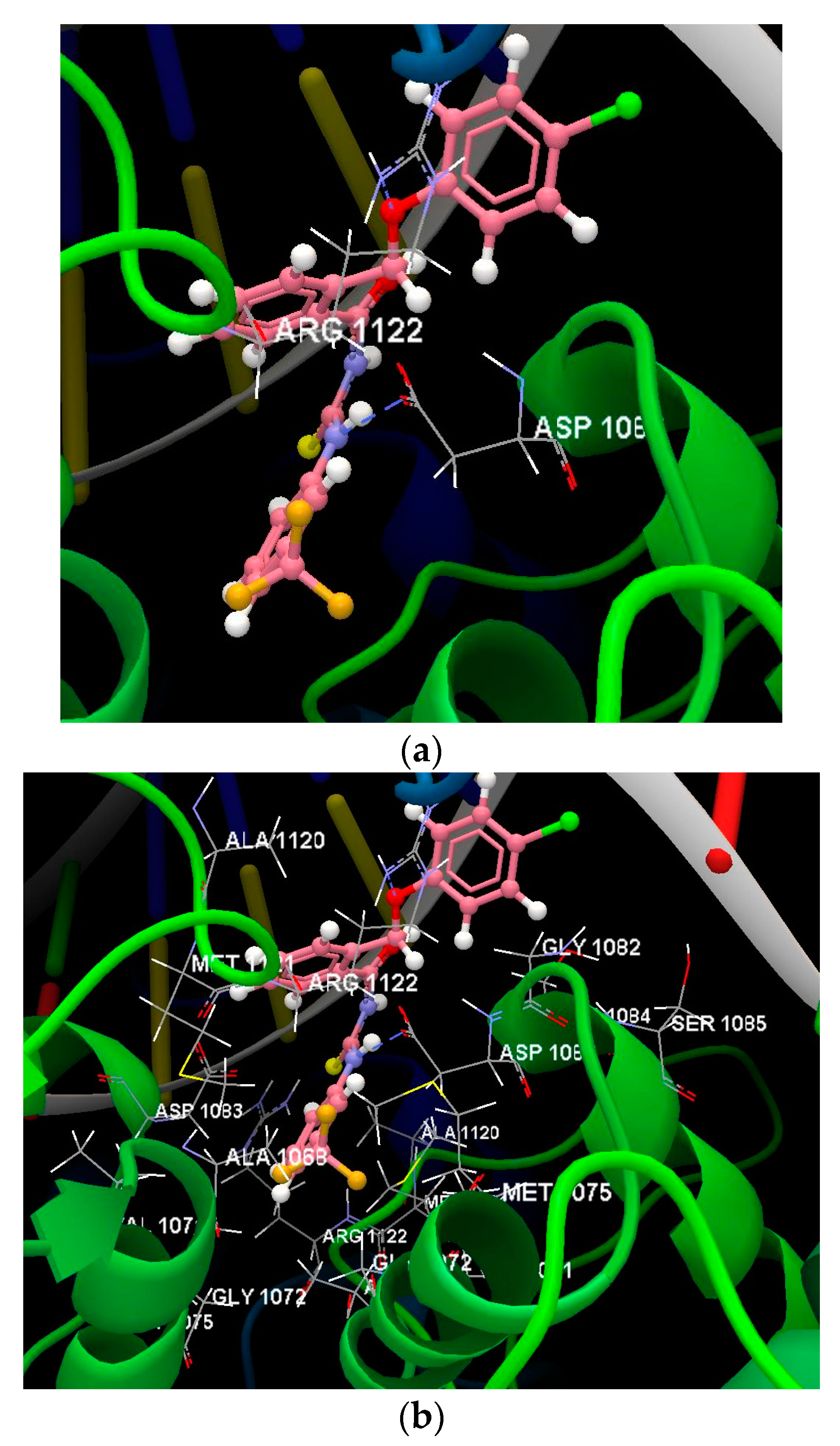{kind=link}
{kind=link}
| Property/Descriptor | Compound | ||||||
|---|---|---|---|---|---|---|---|
| 1a | 1b | 1c | 1d | 1e | 1f | 1g | |
| Weight (g.mol−1) | 414.89 | 414.89 | 450.87 | 450.87 | 464.89 | 464.89 | 464.89 |
| Energy (au) | 2028.06424 | 2028.06386 | 2226.51347 | 2226.52502 | 2265.87000 | 2265.86835 | 2265.86900 |
| Area (Å2) | 408.89 | 408.87 | 419.32 | 416.83 | 435.03 | 438.72 | 438.79 |
| Volume (Å3) | 387.96 | 388.00 | 397.10 | 396.30 | 414.66 | 415.36 | 415.41 |
| Ovality | 1.59 | 1.59 | 1.60 | 1.60 | 1.62 | 1.63 | 1.63 |
| Polarizability (10−30.m3) | 71.85 | 71.88 | 72.58 | 72.59 | 74.09 | 74.06 | 74.08 |
| logP | 0.61 | 0.61 | -0.46 | −0.46 | 1.75 | 1.75 | 1.75 |
| PSA (Å2) | 32.84 | 32.80 | 32.24 | 28.87 | 28.66 | 32.69 | 32.66 |
| Dipole moment (debye) | 6.37 | 6.28 | 6.23 | 6.88 | 4.15 | 6.21 | 7.79 |
| E HOMO (eV) | −6.04 | −5.92 | −6.16 | −6.04 | −5.82 | −6.17 | −6.19 |
| E LUMO (eV) | −1.84 | −1.81 | −1.90 | −2.08 | −1.94 | −1.91 | −1.99 |
| ΔE (eV) | 4.20 | 4.11 | 4.26 | 3.96 | 3.88 | 4.26 | 4.20 |
| Ligand | Score */RMSD | Group Interaction | Hydrogenv Bond | Bond Length |
|---|---|---|---|---|
| Co-crystallized | −61.50/2.49 Å | SER 1084(B), ASP 1083(B), MET 1075(B), VAL 1071(B), MET 1121(B), GLY 1072(B), ALA 1068(B), ARG 1069(B), ARG 1122(D), MET 1121(D), ALA 1068(D), GLY 1072(D), MET 1075(D), VAL 1071(D), ASP 1083(D) | N sp2 (N21)- O sp2 from ASP 1083(B) | 2.902 Å |
| 1a | −44.96/0.15 Å | ARG 1122(B), ASP 1083(D), SER 1084(D), MET 1121(B0, ALA 1120(B), ALA 1119, (B) ALA 118(B), MET 1121(D), ALA 1068(D), MEET 1075(B), ARG 1122(D), PHE 1123(D), ASP 1083(B), TYR 1087(B), ALA 1120(D), SER 1084(B) | N sp2 (N12)- O sp2 from ASP 1083(D) N sp2 (N10)- O sp2 from ASP 1083(D) | 3.077 Å 3.154 Å |
| 1b | −39.91/1.76 Å | MET 1121(B), ASP 1083(B0, VAL 1071(B), MET 1075(B), ALA 1120(D), PHE 1123 (D), ARG 1122(D), MET 1121(D), ALA 1068(D), TYR 1087(D), GLU 1088(D), SER 1084(D), ASP 1083(D), ARG 1122(B) | O sp3 (O22)- O sp2 from ARG 1122(D) | 3.250 Å |
| 1c | −42.89/0.12 Å | MET 1121(B), MET 1075(B), ALA 1068(D), ARG 1122(B), MET 1121(D), ALA 1120(D), ALA 1119(D), ALA 1118(D), VAL 1091(D), TYR 1087(D), GLU 1088(D), GLY 1117(D), ASP 1083(D), SER 1084(D). | - | - |
| 1d | −44.28/0.06 Å | ALA 1118(D), GLU 1088(D), TYR 1087(D), SER 1084(D), ARG 1069(D), ALA 1068(B), ALA 1068(D), GLY 1072(B), VAL 1071(B), MET 1075(B), MET 1121(B), ARG 1122(D), ALA 1120(D), MET 1121(D), ARG 1069(B). | - | - |
| 1e | −47.64/0.05 Å | GLY 1117(B), SER 438(D), ALA 1119(B), ALA 1118(B), ALA 1120(B), SER 1084(B), TYR 1087(B), ASP 1083(B), MET 1121(B), ARG 1122(B), ASP 1023(D), MET 1075(D), MET 1121(D), VAL 1071(D), GLY 1072(D), ALA 1068(D), ARG 1069(D), MET 1075(B), VAL 1071(B), ALA 1068(B), GLY 1072(B) | - | - |
| 1f | −45.02/0.40 Å | ALA 1120(B), MET 1121(B), ARG 1122(B), GLY 1082(D), SER 1085(D), SER 1084(D), ASP 1083(D), MET 121(D), MET 1075(D), ALA 1120(D), VAL 1071(D), GLY 1072(D), ALA 1068(D), ARG 1122(D), GLY 1072(B), MET 1075(B), VAL 1071(B), ALA 1068(B), ASP 083(B), VAL 1071(B), ALA 1068(B), ASP 1083(B), | N sp2 (N10)- O sp2 from ASP 1083(D) O sp3 (O22)- O sp2 from ARG 1122(B) | 3.115 Å 3.191 Å |
| 1g | −45.93/0.75Å | ALA 1119(D), ALA 1118(D), ALA 1120(D), ARG 1122(D), MET 1121(D), VAL 1091(D), GLU 1088(D), TYR 1087(D), SER 1084(D), ASP 1083(D), GLY 1072(D), VAL 1071(D), MET 1121(B), ALA 1068(B), VAL 1071(B), GLY 1072(B), ARG 1069(D), ASP 1083(B) | N sp2 (N12)- O sp2 from ASP 1083(D) | 2.917 Å |
| Ligand | Atoms | Weight [Daltons] | Flexible Bonds | Lipinski Violations | Hydrogen Donors | Hydrogen Acceptors | Log P |
|---|---|---|---|---|---|---|---|
| Co-crystallized | 59 | 460.57 | 8 | 0 | 1 | 7 | 3.67 |
| 1a | 44 | 414.88 | 5 | 1 | 2 | 4 | 5.35 |
| 1b | 44 | 414.88 | 5 | 1 | 2 | 4 | 5.35 |
| 1c | 44 | 450.86 | 5 | 1 | 2 | 4 | 5.56 |
| 1d | 44 | 450.86 | 5 | 1 | 2 | 4 | 5.56 |
| 1e | 47 | 464.89 | 6 | 1 | 2 | 4 | 6.14 |
| 1f | 47 | 464.89 | 6 | 1 | 2 | 4 | 6.14 |
| 1g | 47 | 464.89 | 6 | 1 | 2 | 4 | 6.14 |
| Chemical Compound | 1a | 1b | 1c | 1d | 1e | 1f | 1g | Tobramycin | Fluconazole | |
|---|---|---|---|---|---|---|---|---|---|---|
| Microbial Strain | ||||||||||
| Staphylococcus aureus ATCC 25923 | - | - | - | - | - | - | + | + | ||
| Enterococcus faecalis ATCC 29212 | - | - | - | - | - | + | + | + | ||
| Escherichia coli ATCC 25922 | - | - | - | - | - | - | + | + | ||
| Pseudomonas aeruginosa ATCC 27853 | - | + | + | - | + | + | + | + | ||
| Candida albicans ATCC 10231 | - | + | - | - | - | - | + | + | ||
| Compound | Staphylococcus aureus ATCC 25923 | Enterococcus faecalis ATCC 29212 | Escherichia coli ATCC 25922 | Pseudomonas aeruginosa ATCC 27853 | Candida albicans ATCC 10231 |
|---|---|---|---|---|---|
| 1a | 0.31 | 0.15 | 0.31 | 0.62 | 0.62 |
| 1b | 1.25 | 0.31 | 2.5 | 1.25 | 0.31 |
| 1c | 2.5 | 0.62 | 2.5 | 1.25 | 1.25 |
| 1d | 0.62 | 0.31 | 0.62 | 1.25 | 0.62 |
| 1e | 1.25 | 0.62 | 2.5 | 1.25 | 0.31 |
| 1f | 0.62 | 0.15 | 2.5 | 1.25 | 0.62 |
| 1g | 0.62 | 0.31 | 1.25 | 1.25 | 0.31 |
| DMSO | 1.25 | 2.5 | 1.25 | 1.25 | 0.62 |
| Tobramycin | 0.019 | 0.31 | 0.019 | 0.019 | |
| Fluconazole | 0.019 |
| Compound | Staphylococcus aureus ATCC 25923 | Enterococcus faecalis ATCC 29212 | Escherichia coli ATCC 25922 | Pseudomonas aeruginosa ATCC 27853 | Candida albicans ATCC 10231 |
|---|---|---|---|---|---|
| 1a | 0.62 | 0.62 | 2.5 | 1.25 | 0.31 |
| 1b | 2.5 | 2.5 | 2.5 | 1.25 | 0.019 |
| 1c | 1.25 | 0.62 | 0.62 | 1.25 | 0.31 |
| 1d | 1.25 | 0.31 | 2.5 | 1.25 | 0.15 |
| 1e | 0.62 | 1.25 | 2.5 | 1.25 | 0.62 |
| 1f | 0.31 | 1.25 | 0.62 | 1.25 | 0.31 |
| 1g | 0.15 | 1.25 | 2.5 | 1.25 | 0.019 |
| DMSO | 2.5 | 2.5 | 2.5 | 1.25 | 0.62 |
| Tobramycin | 1.25 | 2.5 | 1.25 | 1.25 | |
| Fluconazole | 1.25 |
| Compound | Abs. 517nm | % Inhibition |
|---|---|---|
| DPPH | 1.357 | |
| 1a | 1.182 | 12.89 |
| 1b | 1.229 | 9.43 |
| 1c | 1.187 | 12.53 |
| 1d | 1.211 | 10.76 |
| 1e | 1.236 | 8.92 |
| 1f | 1.213 | 10.61 |
| 1g | 1.225 | 9.73 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Limban, C.; Nuta, D.C.; Missir, A.V.; Roman, R.; Caproiu, M.T.; Dumitrascu, F.; Pintilie, L.; Stefaniu, A.; Chifiriuc, M.C.; Popa, M.; et al. Synthesis and Characterization of New Fluoro/Trifluoromethyl-Substituted Acylthiourea Derivatives with Promising Activity against Planktonic and Biofilm-Embedded Microbial Cells. Processes 2020, 8, 503. https://doi.org/10.3390/pr8050503
Limban C, Nuta DC, Missir AV, Roman R, Caproiu MT, Dumitrascu F, Pintilie L, Stefaniu A, Chifiriuc MC, Popa M, et al. Synthesis and Characterization of New Fluoro/Trifluoromethyl-Substituted Acylthiourea Derivatives with Promising Activity against Planktonic and Biofilm-Embedded Microbial Cells. Processes. 2020; 8(5):503. https://doi.org/10.3390/pr8050503
Chicago/Turabian StyleLimban, Carmen, Diana Camelia Nuta, Alexandru Vasile Missir, Roxana Roman, Miron Teodor Caproiu, Florea Dumitrascu, Lucia Pintilie, Amalia Stefaniu, Mariana Carmen Chifiriuc, Marcela Popa, and et al. 2020. "Synthesis and Characterization of New Fluoro/Trifluoromethyl-Substituted Acylthiourea Derivatives with Promising Activity against Planktonic and Biofilm-Embedded Microbial Cells" Processes 8, no. 5: 503. https://doi.org/10.3390/pr8050503
APA StyleLimban, C., Nuta, D. C., Missir, A. V., Roman, R., Caproiu, M. T., Dumitrascu, F., Pintilie, L., Stefaniu, A., Chifiriuc, M. C., Popa, M., Zarafu, I., Arsene, A. L., Dinu Pirvu, C. E., Udeanu, D. I., & Papacocea, I. R. (2020). Synthesis and Characterization of New Fluoro/Trifluoromethyl-Substituted Acylthiourea Derivatives with Promising Activity against Planktonic and Biofilm-Embedded Microbial Cells. Processes, 8(5), 503. https://doi.org/10.3390/pr8050503