Abstract
Infections after bone reconstructive surgery become an authentic therapeutic and economic issue when it comes to a modern health care system. In general; infected bone defects are regarded as contraindications for bone grafting. Since the pathogens develop a biofilm on the inner surface of the bone; local delivery of antibiotics becomes more important. The present work focuses on the synthesis of Mesoporous Hydroxyapatite (MPHAP) loaded with drug Vancomycin (Van) and to investigate its loading and leaching ability in phosphate buffer solution (PBS), to be used for post-operative infections. The effect of pore size on MPHAP was analyzed using different fatty acids as organic modifiers. The impacts of various fatty acids chain length on the morphology and pore size were studied. A simple impregnation technique with optimized conditions ensured a high antibiotic loading (up to 0.476 + 0.0135 mg/mL), with a complete in vitro release obtained within 50 h.
1. Introduction
The reconstruction of bone defects emanating due to diseases such as osteoporosis, tumour or due to injury exposes tremendous challenges for orthopedic surgeons. Undeniably, the idyllic material meant for bone substitute other than exhibiting biocompatibility, osteointegration, and biodegradability, must also possess anti-infectious characteristics. Implant-correlated pathogens are among the frequently faced post-operative challenges, where the pathogens form a biofilm within 24 h of surgery over the implant [1]. Consequently, the adhered microorganisms get deeply encapsulated, thereby shielding them from either the host immune system or the internal antibiotic medications, and complete evacuation of implant-correlated infection becomes tricky [2,3]. An implant prosthesis or the bone filling material loaded with the antibiotic can act as a remedy, by efficiently eradicating and preventing the formation of pathogens on the implant. Ceramic materials like Ca/P is used in most of the orthopedic and dental implants. Calcium phosphate (CaP) bioceramics are widely used in the field of bone regeneration, both in orthopedics and in dentistry, due to their good biocompatibility, osseointegration, and osteoconduction. It is also present in the pathological atherosclerotic lesions, urinary calculus and calcification of tissues [4,5]. Hydroxyapatite is one of the ceramic biomaterials considered to be ideal materials for coating on metallic biomaterials as it possesses almost the closest similarity in chemical composition and excellent biocompatibility with natural bone tissue. The Hydroxyapatite (HAP) is non-toxic and biocompatible [6,7,8,9] exhibiting excellent osteointegration and osteoconductivity characteristics [10,11]. In this contest, the bioceramic hydroxyapatite (HAP) with similar chemical constituents of bones and hard tissues is often used as implants in orthopedic, dental or maxillofacial use can approve bone development and osteointegration [12]. In comparison with conventional HAP nanoparticles, mesoporous HAP also remained to improve the capacity of drug adsorption to stabilize drug release [13]. One of the essential properties of HAP filling materials is their porosity that affects drug delivery characteristics as well as their osteoconduction. These pores serve as the drug reservoir, and the drug release kinetic will depend on their size [14]. The fatty acids play an important role in shape-controlled synthesis by allowing simplistic separation between nucleation and growth stages. The surfactant’s role in defining morphology requires a clear indication of its ability to follow specific crystal faces and direct growth of crystals [15].
The literature effectively proved the use of oleic acid as a surfactant; however, it continues unknown how it could cause healing of the bone fracture. Polycaprolactone (PCL)/Osteoarthritis (OA) scaffolds were manufactured using mixed methods to test the theory that fatty acid is a bone induction factor: solvent casting and particulate leaching [16]. It was concluded that the size-controlled synthesis of nanoparticles with appropriate crystallinity could be synthesized using fatty acid [17]. Recently, the HAP based coating has increasingly drawn attention improving the adhesion quality in metallic biomaterials. The ease of synthesis using natural biomacromolecules such as gums, polymers and surfactants enable this soft template to be fabricated using wet chemical route [18], sol-gel [19], mechanochemical [20], hydrothermal [21], microwave heating [22], microemulsion [23] and template addition [24,25]. The control in size with minimal agglomeration made them the most flexible and convenient method for the effective synthesis of nano-level hydroxyapatite particles [26,27]. Besides, the conventional methods adopted for synthesizing nano HAP, sintering aided polymer encapsulated core-shell hydroxyapatite nanoparticles are forecasted to be used as drug delivery vehicles. This research follows on from our earlier work investigating a novel route of synthesizing mesoporous hydroxyapatite nanoparticles using the sol-gel technique in the presence of stearic acid (SA), a biocompatible medium-chain fatty acid that would act as an organic modifier. Structural, functional, and morphological characterization was studied to the as-prepared HAP particles. Mesoporous in HAP were noted by removing the organic template for samples synthesized at pH 11 [15]. Therefore, the aim of this present work is to compare the structural and morphological characteristics of HAP synthesized using lauric acid (LA) and stearic acid (SA) as an organic modifier. The Vancomycin (Van) drug loading/leaching behavior loaded with Mesoporous Hydroxyapatite nanoparticles was also investigated. Furthermore, the study aims to examine the ability of mesoporous HAP to adsorb and control the release of the antibiotic vancomycin and further analyze the effect of pore size with the drug loading/leaching studies.
2. Materials and Methodology
2.1. Materials
The calcium and phosphate precursors include analytical grade calcium nitrate tetrahydrate (Ca(NO3)24H2O) and diammonium hydrogen phosphate ((NH4)2HPO4). The organic modifiers lauric acid (LA) and stearic acid (SA) were purchased from Sigma-Aldrich.
2.2. Synthesis of MPHAP
Briefly, 1M of calcium nitrate tetrahydrate and 1mM of SA was mixed with 100 mL of ethanol using a magnetic stirrer at 60 °C for 4 h to form a uniformly dispersed homogeneous solution. Now, in order to form a milky white solution, 100 mL of 0.6 M aqueous diammonium hydrogen phosphate was added dropwise to the calcium solution while maintaining pH at 11 using liquid ammonia. The solution was aged for 24 h at 80 °C. The precipitates were centrifuged, washed with double distilled water, and dried at 100 °C and further calcinated in a muffle furnace at 800 °C for 2 h. The experiment was repeated with LA and without any fatty acid for reference. The samples were labeled as HAP, HAP-LA, and HAP-SA for those prepared using without fatty acid and using LA and SA respectively.
2.3. Drug Loading and Leaching Studies
Drug Vancomycin (Van) was selected as a drug model to demonstrate the drug up taking capacity and drug leaching behavior between prepared HAP and conventional HAP (HAP-LA and HAP-SA). A calibration graph for the standard drug concentration (vancomycin) was plotted. Using this calibration curve, it is possible to obtain the unknown concentration of Van by knowing the drug’s absorbance value for a given period. 10 mg of HAP nanoparticles suspended in Vancomycin 10 mL (0.1 mg/mL) phosphate buffer solution (PBS, pH = 7.4) with 220 rpm (taken in a shaker) for 2 h at 37 °C. Using ultraviolet-visible spectroscopy at a fixed wavelength of 280 nm, the remaining concentration of vancomycin present in the PSB for a period of 2 days was investigated to calculate the loaded amount of vancomycin in HAP, HAP-LA, HAP-SA (mg/mL).
3. Characterization Techniques
3.1. Structural Analysis
The measurements of X-ray diffraction (X’Pert-MPD system with a CuKα wavelength of 1.5418 Å (Phillips Analytical, Almelo Netherlands) operated at 40 kV and 30 mA at 2 ranges from 20° to 70° using a step size of 0.02 s−1) were used to determine the phase purity. Based on previous studies [9], the crystallite size Dhkl and the degree of crystallinity (Xc) were calculated using the Debye Scherrer equation as follows,
Where K = 0.94 is a constant, λ is the wavelength of monochromatic radiation (λ = 1.5418 Å), β is diffraction peak for full width half maximum.
3.2. Functional Analysis
Using the KBr pellet technique in the spectral wavelength range from 4000 to 650 cm−1, Fourier transforms infrared spectrometer (Cary 630, Agilent, BeiJing, China) was analyzed.
3.3. Porosity Measurement
Brunauer–Emmett–Teller (BET) (Asap 2020 Porosimeter, Micromeritics, Manchester, UK) were used to determine the surface area and pore size distribution of the prepared samples.
3.4. Morphological Characterization
Field Emission Scanning Electron Microscopy (FESEM: Supra VP35 Carl Zeiss, Germany) and High-Resolution Transmission Electron Microscope (HRTEM: FEI-TECNAI G2-200 KV TWIN) performed the morphological characterization. A standard unit (Oxford Instruments, Abingdon, UK) attached to the FESEM obtained the energy dispersive X-ray (EDX: X-Max, MEE, Minneapolis, MN 55441, USA) spectra for elemental composition analysis.
3.5. Drug Loading
The competition of nanomedicine over other predictable instruments in the diagnosis, treatment, and therapy of illnesses for biomedical disciplines improved our understanding of refining human health and hygiene. In evaluating the drug’s release frequency from nanoparticles in a suitable medium, a calibration curve for the average drug concentration is essential. The samples were uniformly mixed in water with 0.5 mg of Vancomycin drug and then pressed into pellets. A tabulating machine recorded the average weight for each sample as all the tests were conducted in triplicate. The pellet samples loaded with 0.5 mg Vancomycin was transferred to a 100 mL of dissolution medium consisting of a freshly prepared phosphate buffer saline, maintained at 37 °C and stirred at 100 rpm. Three-milliliter aliquots were removed for all batches and their absorbance (280 nm) was measured at predetermined time intervals. To ensure ideal sink conditions, an equal amount of phosphate buffer was added to the dissolution medium for each sample removed. All the batches were evaluated for the release profiles and the absorbance was recorded by a spectrophotometer. The drug release kinetics of samples were analyzed at 280 nm using UV-visible spectrophotometer. One gram of this was spread over 100 mL of physiological PBS and placed in an incubator shaker. The temperature and shaking velocity were maintained at 37 °C and 100 rpm respectively. Every 20 min, 1 mL of PBS solution was removed and substituted with a new medium. The calibration graph was used to assess the concentration released in the buffer solution of HAP, HAP-SA, and HAP-LA and the experiment was repeated three times. The amount of vancomycin released was calculated using Equation (3).
Therefore, using a UV-visible spectrophotometer, a known concentration of (2–10 mg/mL) vancomycin was scanned.
4. Results and Discussions
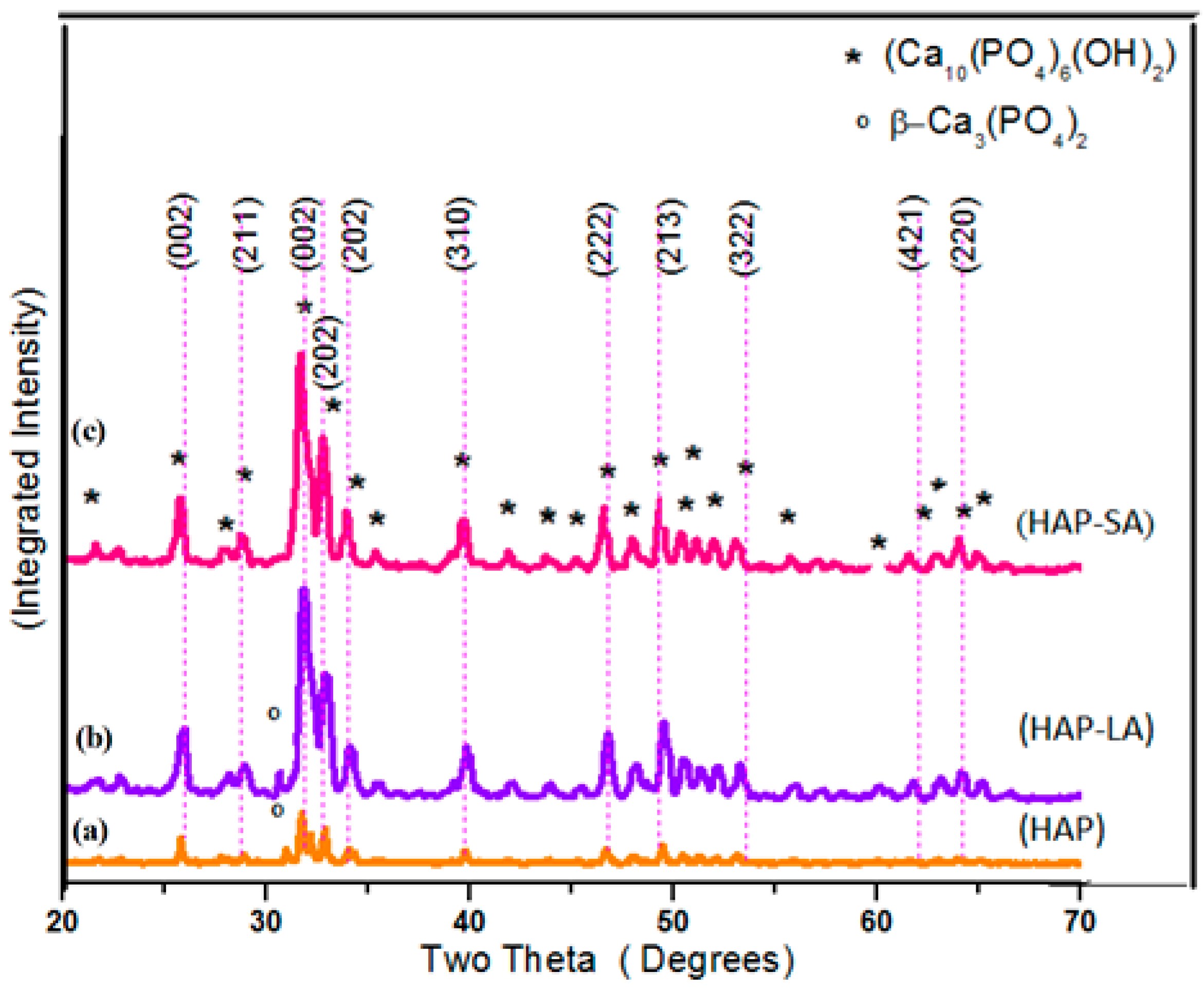
Figure 1a–c shows the X-ray diffraction (XRD) patterns of the sintered samples HAP, HAP-LA, and HAP-SA. The diffraction patterns of the synthesized samples are in good agreement with those of the standard HA crystallites, showing the characteristic peaks at 2θ = 25.9° (002), 2θ = 31.7° (211), 2θ = 32.1° (112), 2θ = 32.8° (300), 2θ = 46.6° (222) and 2θ = 49.4° (213) well compatible with (JCPDS No.9-0432) [10] for all samples, except for a small additional peak present due to β–TCP in HAP and HAP-LA respectively, which could have been introduced during the process of sintering. Table 1 shows the size of crystallite and the percentage of HAP, HAP-LA and HAP-SA crystallinity along with stoichiometry. The presence of traces of β-TCP is further reflected in the EDAX analysis, where the stoichiometric ratio of Ca/P for HAP and HAP-LA samples is slightly lesser than the Stoichiometry of pure HAP samples. These signs indicate that adding fatty acids does not affect as-synthesized HAP’s phase of purity. The crystal size and percentage of crystallinity were calculated to be 46.1 nm, 20.8 nm, 10.32 nm, and 2.392%, 1.83%, and 1.779% corresponding to HAP, HAP-LA, and HAP-SA, respectively. Table 1 clearly shows the decrease in as-synthesized HAP crystallinity and crystallite size using LA and SA. This was authenticated according to Scherrer’s equation by the calculated crystallite size and crystallinity. Also, from the XRD pattern, it is seen that the broadening of peak occurs for HAP samples (HAP-LA and HAP-SA) synthesized using fatty acids. This broadening of peak occurs when the particle size falls in the nano range, suggesting that the synthesis of HAP using fatty acids can positively modify the crystal growth of HAP.
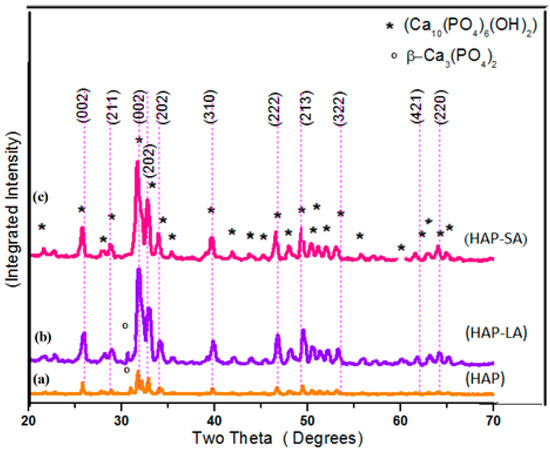
Figure 1.
XRD pattern of (a) HAP (b) HAP-LA (c) HAP-SA.

Table 1.
Crystallite size percentage of crystallinity and stochiometric ratio of HAP.
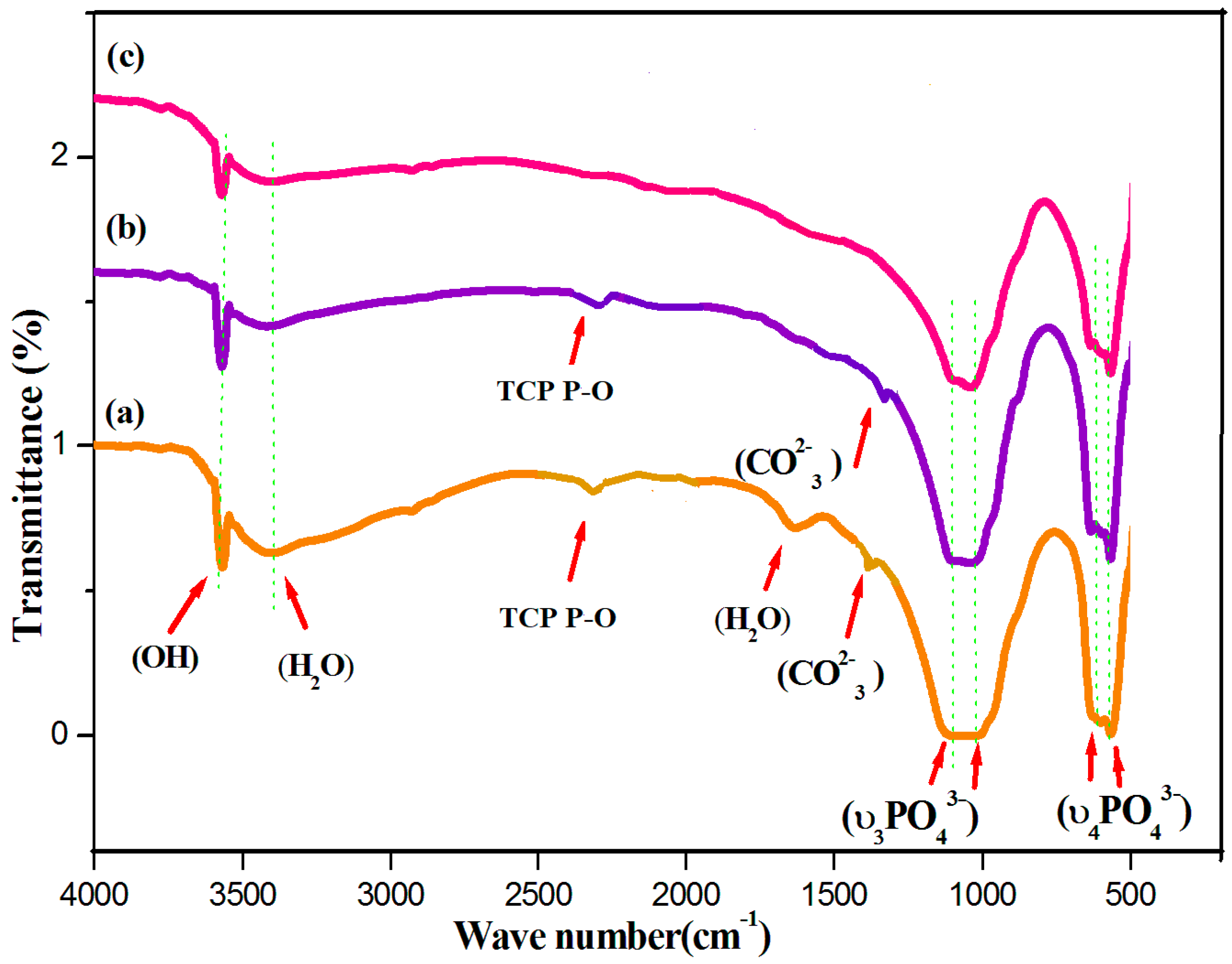
Fourier-transform infrared (FTIR) spectroscopy is an analytical technique generally used to describe the functional groups existing in biomaterials such as HAP, HAP-LA, and HAP-SA. The wide FTIR band centered around 1000–1100 cm−1 is the primary symbol of HA development [28] and represents the υ3 phosphate modes (Figure 2). The peaks found at 604 and 564 cm-1, resulting from the P-O band’s doubly degenerate bending mode (υ4). The broadband at 3550–3200 cm−1 implies the water absorbed by the lattice. The OH− is stretching, and vibrational bending modes appear at 3569 and 631 cm−1 [29]. The weak absorption bands at 1408, 1456 cm−1 were observed for samples HAP and HAP-LA as reported for carbonated HAP, which could result from the presence of atmospheric carbon dioxide during calcination processes [30,31]. The presence of TCP is also shown by the stretching frequency of the phosphate group at 2360 cm-1 [15] for the HAP and HAP-LA samples. Furthermore, the existence of TCP is also weakened, which is in coincidence with the XRD results. For HAP synthesized in the presence of SA, the HAP peaks were only detected without any other Ca-P impurities, as shown in Figure 1 and Figure 2, whereas, for samples HAP and HAP-LA, traces of TCP were confirmed. Thus, the conventionally prepared HAP was carbonated compared to those to HAP-SA. Also, the absence of C=O bands at 1760 cm−1 specifies the non-appearance of fatty acids in HA after sintering; thus the functional groups of LA or either SA are not incorporated in the sintered HA-HAP-LA or HAP-SA samples.
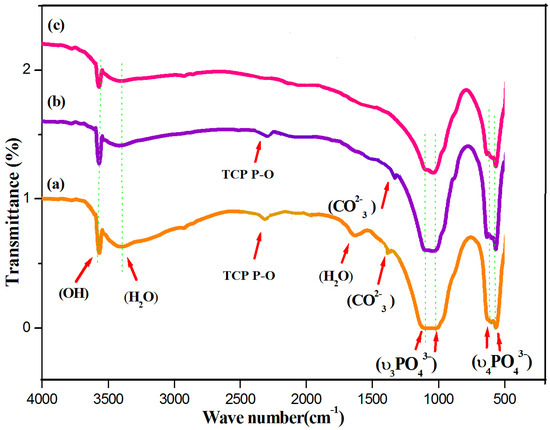
Figure 2.
FTIR spectra of (a) HAP (b) HAP-LA (c) HAP-SA.
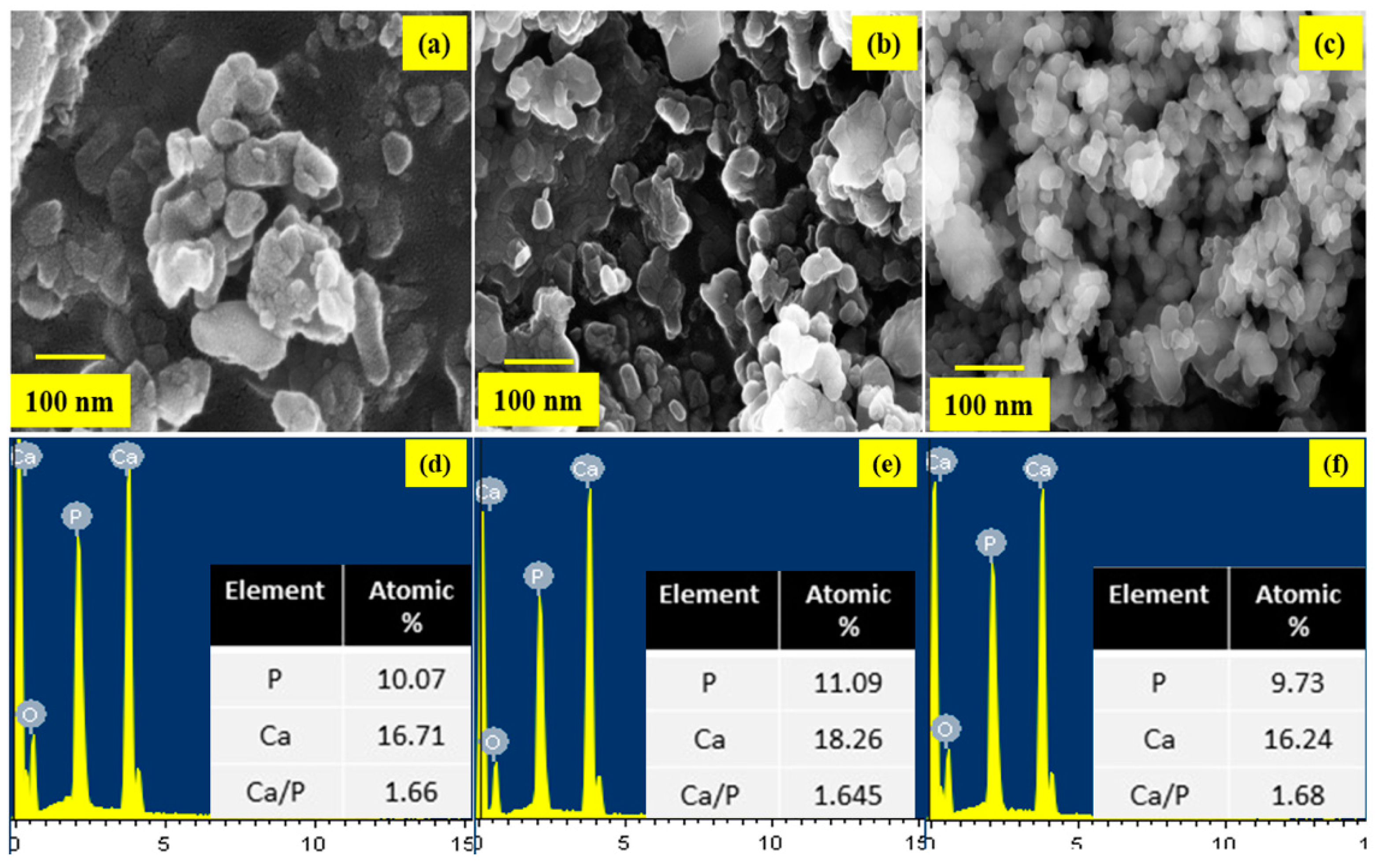
The size and morphology and of the prepared HAP nanostructures were observed using Field Emission Scanning Microscopy (FESEM) and its corresponding Energy Dispersive X-ray (EDX) analysis for the samples HAP, HAP-LA, and HAP-SA are shown in Figure 3a–c and Figure 3d–f respectively. HAP samples are larger and aggregated, while those synthesized in the presence of LA/SA show a significant reduction in particle size and uniform rod-shaped morphology for HAP-SA. Also, for example, precursors, temperature, time, pH, solvent, pressure, and organic modifiers, the compositional changes and response conditions play an essential role in stabilizing the particles [32,33]. It is admirable to note that the HA composition is the same as the standard bone composition. EDX is used to examine the various components present in the sample and their composition. The spectra highlight the occurrence of Ca, P, and O, together with the elemental spectra as the major elements. There are no traces in the spectrum of other elements that confirm that the samples prepared are pure in form. However, the EDX analysis shows that HAP of Ca/P is 1.68 obtained stoichiometry HAP highly for HAP-SA whereas the stoichiometric ratio was found to be slightly lower for these samples HAP and HAP-LA, which could be due to the presence of β-TCP traces present in the sample during the synthesis process.
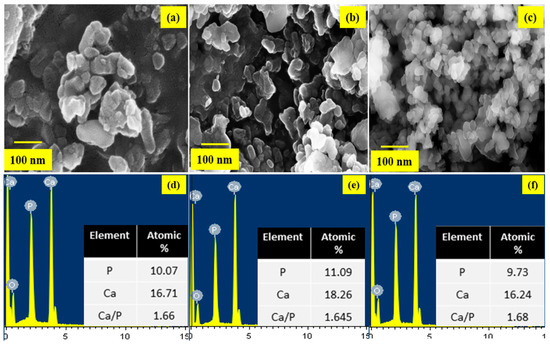
Figure 3.
FESEM images of (a) HAP (b) HAP-LA (c) HAP-SA and EDX spectrum (d) HAP (e) HAP-LA (f) HAP-SA.
The HRTEM images of HAP synthesized without fatty acid shows larger particles of a 150–200 nm size and exhibit spherical particles as shown in Figure 4a–c. Whereas for HAP-LA, the particles were agglomerated and those of HAP-SA showed elongated particles. The shape, size, and length of nanorods of the sample HAP-SA are much enhanced compared to HAP and HAP-LA. Thus, it is found that the HAP-SA sample has a rod-like morphology without agglomeration, and a close look at the morphology of HAP-LA and HAP-SA samples shows poorly crystalline nanometer pores in nature. Many organic modifiers, such as citric acid, ethylenediamine tetraacetic acid, trisodium citrates, and vitamin C, were shown to control the size, shape, morphology, and pores of HAP particles during synthesis.

Figure 4.
The HRTEM images of (a) HAP (b) HAP-LA (c) HAP-SA.
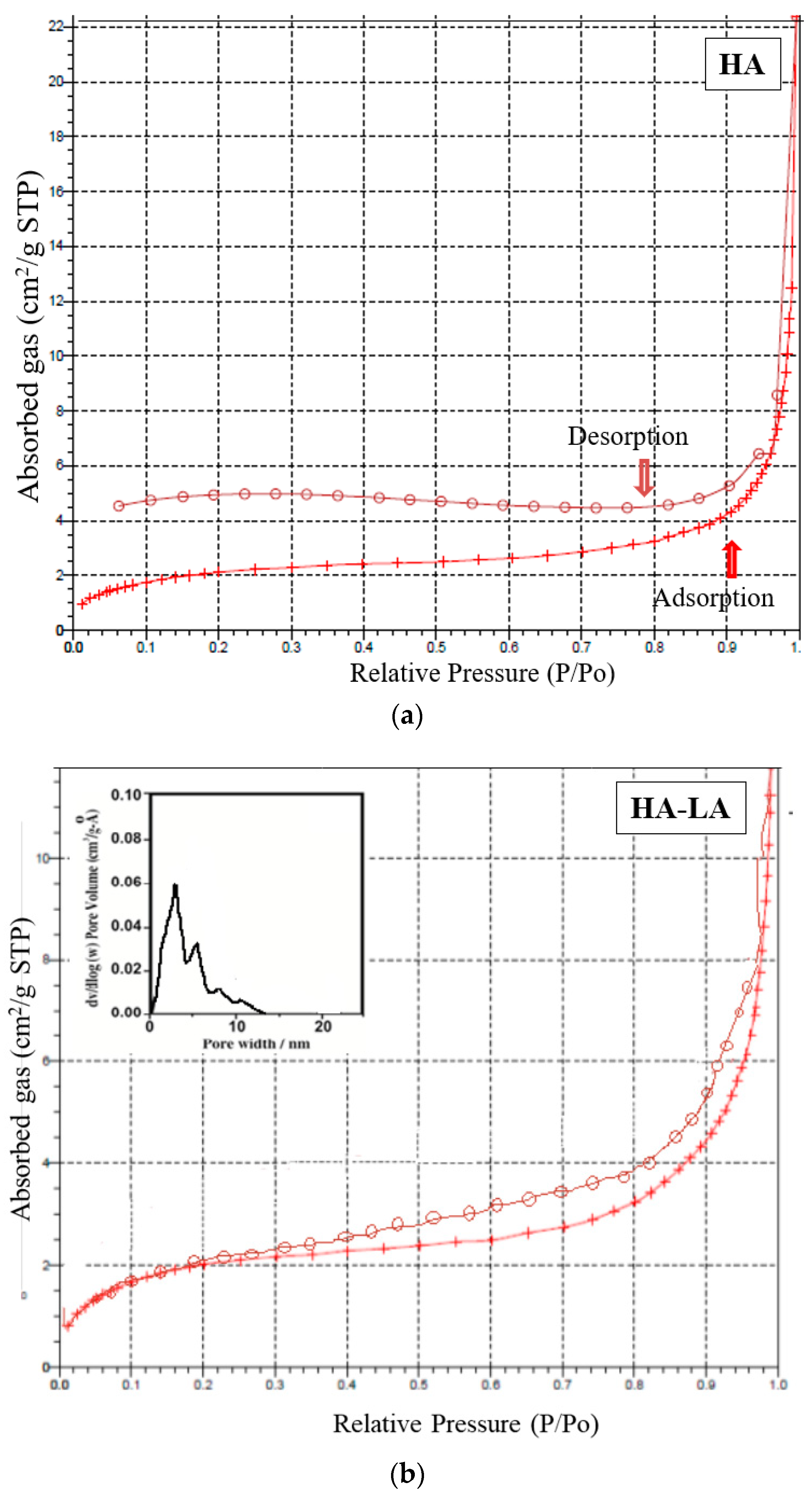
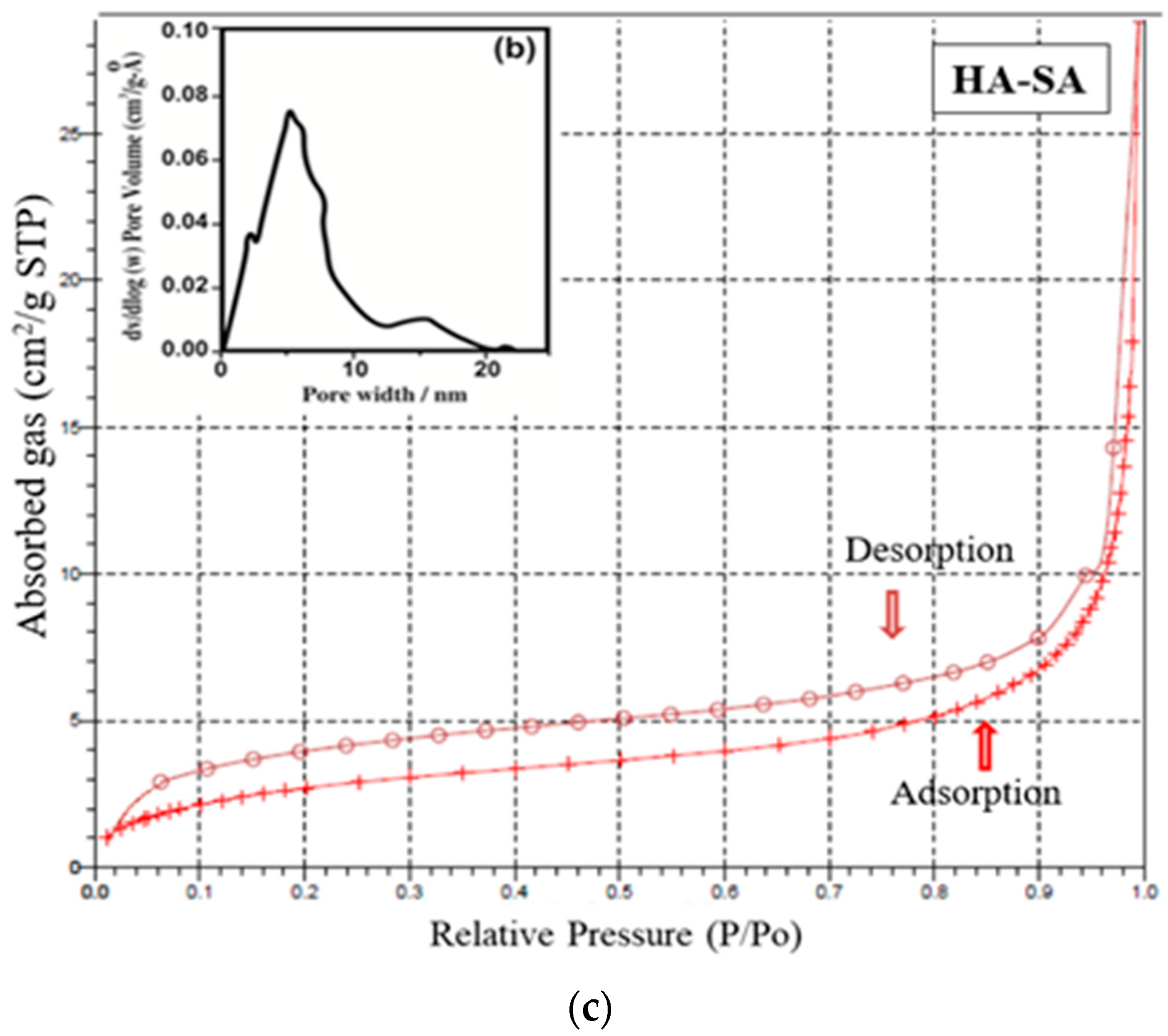
The mesoporous and microporous nature of samples HAP, HAP-LA and HAP-SA were analyzed based on the adsorption-desorption mechanism of nitrogen gas by BET analysis. Since nitrogen gas has the capacity to be adsorbed on the surface, it is used for the analysis of the surface area and porosity of pure HAP and HAP-LA and HAP-SA. The depth profile of pores was examined by the N2 adsorption and desorption isotherm method. The nature of the adsorption and desorption curves implies that they were not reversible, which leads to the appearance of hysteresis between the adsorption and desorption curves. Both the samples exhibited a type IV curve with a hysteresis loop that corresponds to a mesoporous material. The associated nitrogen adsorption–desorption isotherms and the resultant pore size distribution of HAP, HAP-LA, and HAP-SA are presented in Figure 5a–c. It is found that HAP synthesized by the conventional method does not show any mesoporous structure as the isotherm curve does not show closed hysteresis loop as shown in Figure 5a. While those synthesized with LA and SA (HAP-LA and HAP-SA) exhibit an average pore size of the order of 5.86 ± 0.0256 nm and 3.87 ± 0.0171 nm with the corresponding BET surface area as high as 66.265 ± 0.0345 cm2/g for HAP-SA and 47.567 ± 0.0595 cm2/g for HAP-LA respectively. The BET surface area of HAP sample synthesized without fatty acid showed the least area of 7.7138 ± 0.0474 cm2/g due to nonporous nature, thus with the least surface area. A lower porosity of 47.567 ± 0.0595 cm2/g was observed for HAP-LA using a short length fatty acid whereas an enhanced porosity of 66.265 ± 0.0345 cm2/g was observed using SA a medium-chain fatty acid. Table 2 represents the average pore size and BET surface area of the prepared mesoporous HAP using fatty acid. Thus, it is convincible to predict that the porosity and hence the surface area are connected with the length of the fatty acid used for the synthesis of HAP.
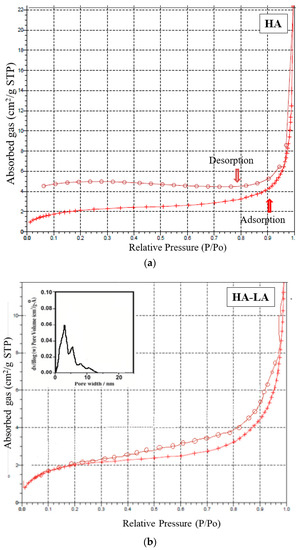
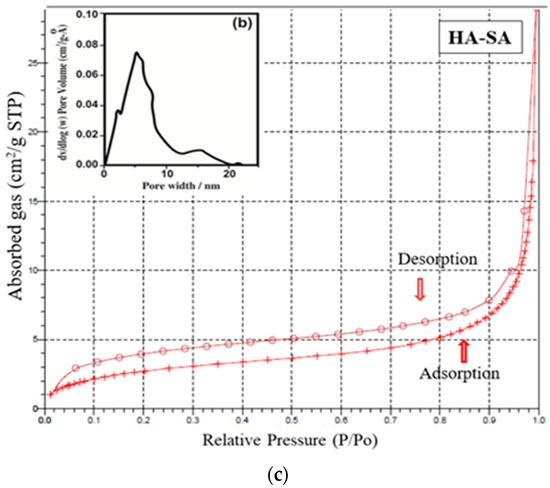
Figure 5.
(a) Nitrogen adsorption–desorption isotherm for sample HAP. (b) Nitrogen adsorption–desorption isotherm and pore size distribution of HAP-LA. (c) Nitrogen adsorption–desorption isotherm and pore size distribution of HAP-SA.
Table 2.
Average pore size and BET surface area of the prepared mesoporous HAP using fatty acid.
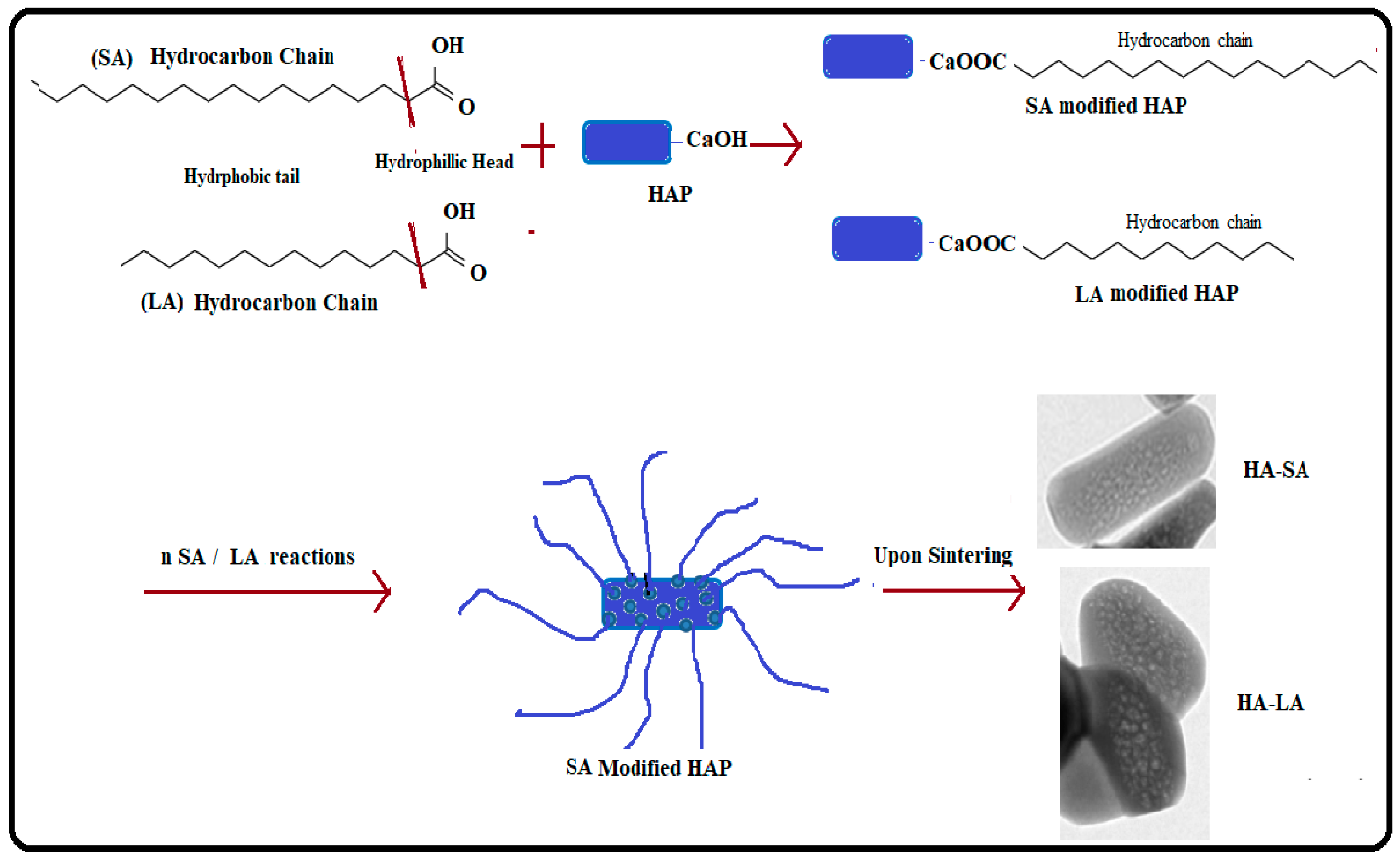
On the other hand, the relevance of fatty acids as an agent for controlling material morphology is seldom reported. Nevertheless, numerous water-soluble organic compounds and aqueous extract from various plants were studied to modify the surface of the HAP nanoparticles as an organic modifier. We used SA and LA (fatty acid series) for the first time as a water-insoluble compound to produce nanorods of MPHAP.
Figure 6 shows the mechanism for the formation of MPHAP using LA and HAP. In the present work, fatty acid molecules were first adsorbed to calcium phosphate precursors via its –OH group. After that, due to the hydrophobic part of molecules, the growth of HAP crystals in the docked area of LA and SA was hampered. A structure surrounding fatty acid molecules continued to form the growth of HAP in another area. The thermal motion of water-insoluble fatty acids was intensified in the sol-gel environment. This triggered the removal of HAP molecules, ensuring that mesopores were established. Thus, the overall functional carboxyl group in fatty acid can promote calcium ion binding (Ca2+) from the carboxylate ion solution for HAP formation. This initiates the growth and nucleation of crystals and therefore helps HAP to improve size-controlled formations [34,35].
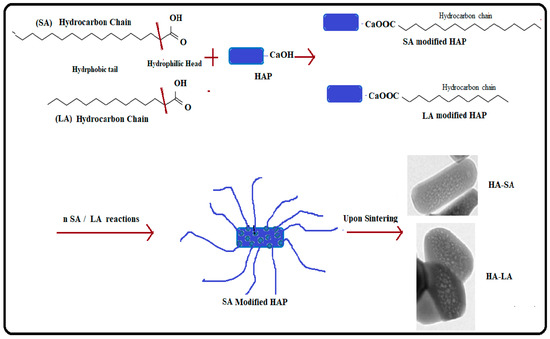
Figure 6.
Mechanism of formation of MPHAP using LA and HAP.
Vancomycin is considered a last-resort antibiotic and tends to be effective in treating bacteria that is highly resistant to other drugs. It is a glycopeptide, derived from Amycolatopsis Orientalis cultures. One of the most well-known bacteria that vancomycin is effective against is methicillin-resistant Staphylococcus aureus (MRSA). Overall, vancomycin is successful against various Gram-positive bacteria, including highly resistant streptococci, enterococci, and staphylococci strains.
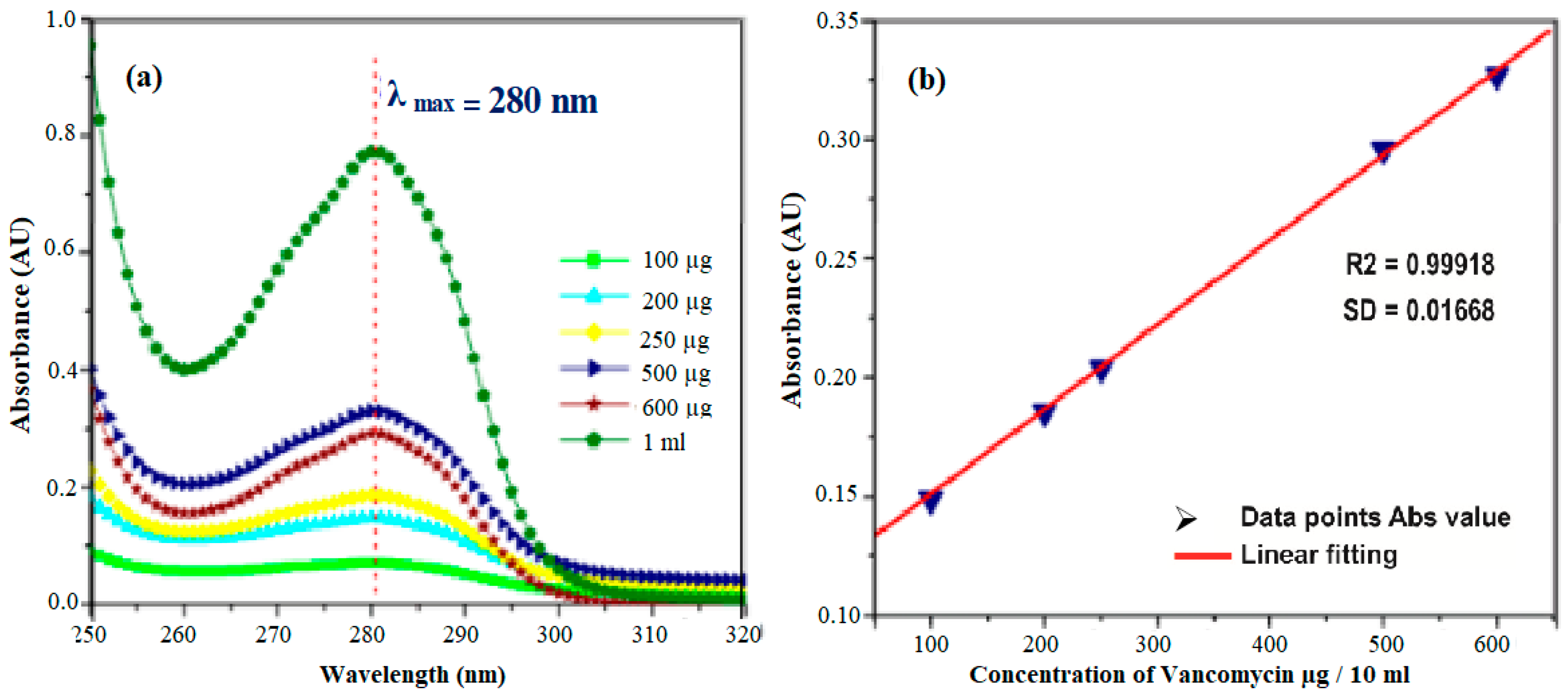
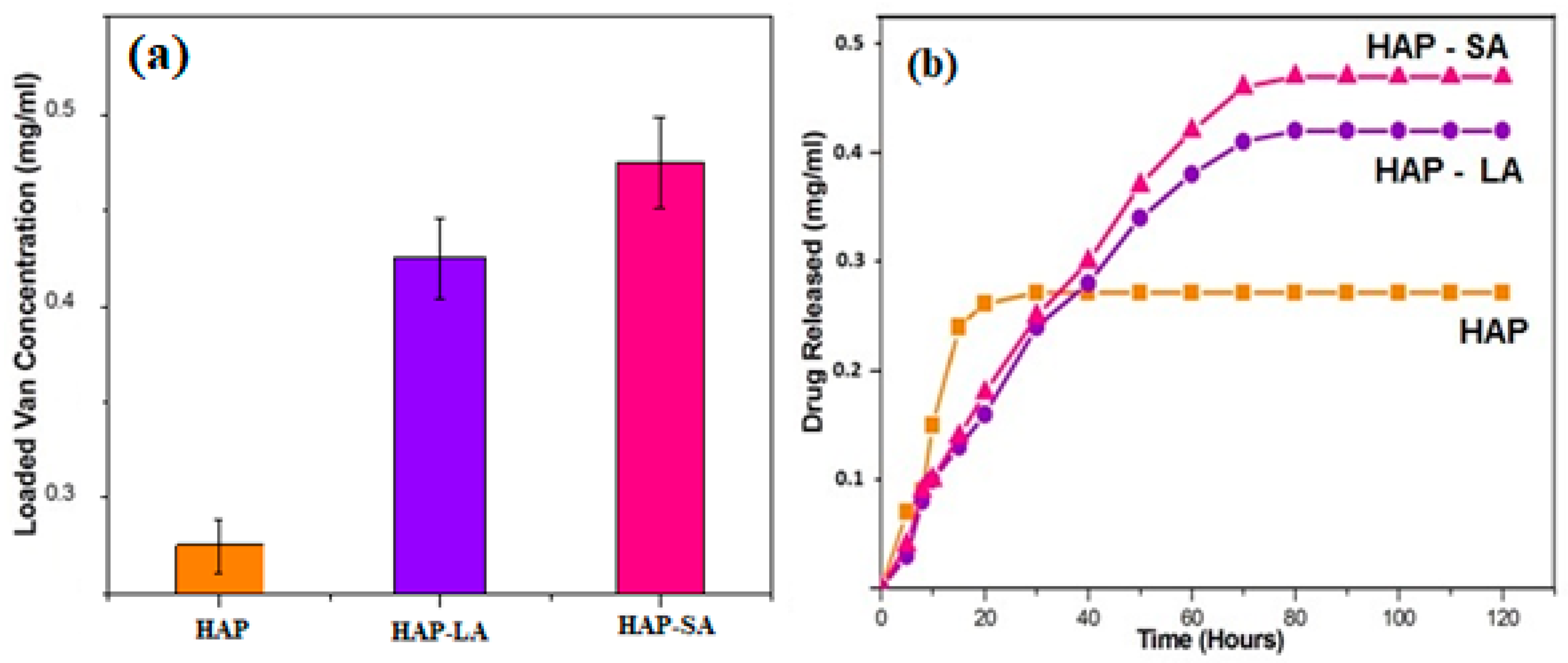
To estimate the unknown concentration of the drug vancomycin in the leaching studies a standard calibration graph is plotted for various concentrations of the drug such as 100 µg, 200 µg, 250 µg, 500 µg, 600 µg, and 1 mg/10mL. A prominent peak at 280 nm was noticed for vancomycin with a range concentration of 2–10 mg/mL. With the respective concentration, the absorbance values at 280 nm were noted, and a standard calibration graph was plotted in Figure 7a. Thus, the unknown concentration of the drug (vancomycin) leached in SBF can be calculated from the standard calibration graph (Figure 7b). The Quantity of drug vancomycin loaded on the samples was found to be 0.272 ± 0.0235, 0.424 ± 0.21 and 0.476 ± 0.0135 mg/mL for samples labeled HAP, HAP-LA, and HAP-SA respectively as shown in Figure 7a,b. The leaching studies were evaluated as a function of time in PBS at pH 7.4.
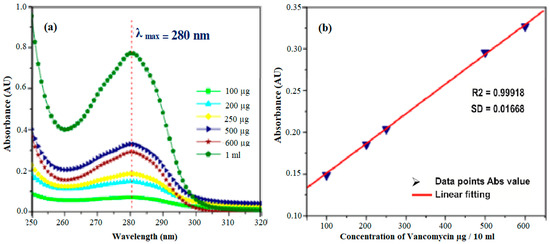
Figure 7.
(a) STD absorption curves for drug Vancomycin and (b) its calibration graph.
Drug loading and leaching studies for HAP, HAP-LA, and HAP-SA are shown in Figure 8a,b. For HAP samples, a rapid burst of nearly 80% of vancomycin was released within the first 4 h and gradually decreased, and almost 98% of drugs released at the end of 12 h. The porous HAP-LA and HAP-SA recorded a rapid release of the drug of 50% of the drug during the first 20 h and prolonged release that extended up to 50 h. This delayed-release was due to the release of drugs trapped in the pores. This drug explosive release is useful for quick recovery [28,29]. The initial speedy release is accredited to the initial high difference in the drug concentration gradient between the pellet and the release medium. This observed difference in drug release behavior as a function of time might be due to the difference in the extent of porosity generated. For HAP, HAP-SA, and HAP-LA, the generation of the mesoporous structure with a higher pore volume and pore diameter helps to control the drug release behavior. On the whole, no statistical variations were observed for the samples ccrystallite size with respect to its average pore size and its corresponding loading and leaching capability. Among the fatty acids used HAP-SA samples showed non-agglomerated particles with larger mesopores and hence larger loading and leaching behavior. Further by variating the concentration of LA and SA, the mesopores can be modified and the degree of drug loading and release from HAP-SA and HAP-LA could be effectively changed. Thus, for appropriate application the pore size can be controlled and drug-loaded MHAP can be used as a carrier for drug delivery applications.
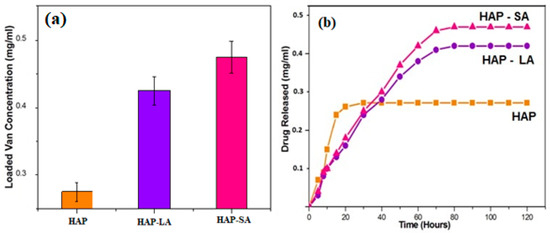
Figure 8.
(a) Drug loading (b) leaching studies for HAP, HAP-LA, and HAP-SA.
5. Conclusions
Mesoporous HAP nanoparticles were successfully synthesized by the sol-gel method using fatty acids, lauric acid, and stearic acid as an organic modifier. The HRTEM images reveal un-agglomerated rod-shaped MPHAP nanoparticles with an average pore size of 5.86 ± 0.0256 nm for HA-SA and 3.87 ± 0.0171 nm for HAP-LA that appears agglomerated with more substantial sized particles. Thus, mesoporous rod-like HAP nanoparticles can be obtained with an average pore size of 5.86 nm corresponding to the BET surface area of 66.265 ± 0.0345 m2/g. Also, the MPHAP revealed improved adsorption of drug vancomycin in comparison to HAP synthesized without fatty acid. The drug vancomycin loading capacity was proportional to the pore size of the samples with a sustained drug release attainable up to 50 h. Further studies are needed to compare the strength and durability of MPHA with traditional HAP to be able to be used as bone substitute.
Author Contributions
J.A.L.—data curation, format analysis, investigation, writing—original draft; S.S.—Writing—review & editing; J.J.P.—data curation; N.A.H.—data curation; I.A.B.—Funding acquisition; M.R.J.—data curation; A.R.M.—data curation; Y.A.W.—data curation; T.M.Y.K.—Funding acquisition; S.K.—Funding acquisition.
Acknowledgments
The authors extend their appreciation to the Deanship of Scientific Research at King Khalid University for funding this work through research groups program under grant number (R.G.P. 2/11/39). This work was also financially supported by University of Malaya Research Grant (RU001-2018 and RU001-2019).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Gurjala, A.N.; Geringer, M.R.; Seth, A.K.; Hong, S.J.; Smeltzer, M.S.; Galiano, R.D.; Mustoe, T.A. Development of a novel, highly quantitative in vivo model for the study of biofilm-impaired cutaneous wound healing. Wound Repair Regen. 2011, 19, 400–410. [Google Scholar] [CrossRef] [PubMed]
- Kostenko, V.; Ceri, H.; Martinuzzi, R.J. Increased tolerance of Staphylococcus aureus to vancomycin in viscous media. FEMS Immunomol. Med. Microbiol. 2007, 51, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Mah, T.F.; O’Toole, G.A. Mechanisms of biofilm resistance to antimicrobial agents. Trends Microbiol. 2001, 9, 34–39. [Google Scholar] [CrossRef]
- Dorozhkin, S.V.; Epple, M. Biological and medical significance of calcium phosphates. Angew. Chem. Int. Ed. 2002, 41, 3130–3146. [Google Scholar] [CrossRef]
- Dorozhkin, S.V. Multiphasic calcium orthophosphate (CaPO4) bioceramics and their biomedical applications. Ceram. Int. 2016, 42, 6529–6554. [Google Scholar] [CrossRef]
- Thompson, J.B.; Kindt, J.H.; Drake, B.; Hansma, H.G.; Morse, D.E.; Hansma, P.K. Bone indentation recovery time correlates with bond reforming time. Nature 2001, 414, 773. [Google Scholar] [CrossRef]
- Currey, J. Sacrificial bonds heal bone. Nature 2001, 414, 699. [Google Scholar] [CrossRef]
- Rey, C.; Kim, H.M.; Gerstenfeld, L.; Glimcher, M.J. Structural and chemical characteristics and maturation of the calcium-phosphate crystals formed during the calcification of the organic matrix synthesized by chicken osteoblasts in cell culture. J. Bone Miner. Res. 1995, 10, 1577. [Google Scholar] [CrossRef]
- Krajewski, A.; Ravaglioli, A.; Roncari, E.; Pinasco, P.; Montanari, L. Porous ceramic bodies for drug delivery. J. Mater. Sci. Mater. Med. 2000, 11, 763–771. [Google Scholar] [CrossRef]
- Meskinfam, M.; Bertoldi, S.; Albanese, N.; Cerri, A.; Tanzi, M.C.; Imani, R.; Fare, S. Polyurethane foam/nano hydroxyapatite composite as a suitable scaffold for bone tissue regeneration. Mater. Sci. Eng. C 2018, 82, 130–140. [Google Scholar] [CrossRef]
- Tsetsekou, A.; Brasinika, D.; Vaou, V.; Chatzitheodoridis, E. On the synthesis of tailored biomimetic hydroxyapatite nanoplates through a bioinspired approach in the presence of collagen or chitosan and L-arginine. Mater. Sci. Eng. C 2014, 43, 555–565. [Google Scholar] [CrossRef] [PubMed]
- Vallet-Regi, M.; Gonzalez-Calbet, J.M. Calcium phosphates as substitution of bone tissues. Prog. Solid State Chem. 2004, 32, 1–31. [Google Scholar] [CrossRef]
- Zhang, N.; Gao, T.; Wang, Y.; Wang, Z.; Zhang, P.; Liu, J. Environmental pH-controlled loading and release of protein on mesoporous hydroxyapatite nanoparticles for bone tissue engineering. Mater. Sci. Eng. C 2015, 46, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Padmanabhan, V.P.; Kulandaivelu, R.; Nellaiappan, S.N.T.; Lakshmipathy, M.; Sagadevan, S.; Johan, M.R. Facile fabrication of phase transformed Cerium (IV) doped Hydroxyapatite for biomedical applications—A health care approach. Ceram. Int. 2019. [Google Scholar] [CrossRef]
- Lett, J.A.; Sundareswari, M.; Ravichandran, K.; Latha, M.B.; Sagadevan, S.; Johan, M.R.B. Tailoring the morphological features of sol-gel synthesized mesoporous hydroxyapatite using fatty acids as an organic modifier. RSC Adv. 2019, 9, 6228–6240. [Google Scholar] [CrossRef]
- Cardoso, G.B.; Chacon, E.; Chacon, P.G.; Bordeaux-Rego, P.; Duarte, A.S.; Saad, S.T.O.; Cunha, M.R. Fatty acid is a potential agent for bone tissue induction: In vitro and in vivo approach. Exp. Biol. Med. 2017, 242, 1765–1771. [Google Scholar] [CrossRef]
- Abidi, S.S.A.; Murtaz, Q. Synthesis, and Characterization of Nano-hydroxyapatite Powder Using Wet Chemical Precipitation Reaction. Mater. Sci. Eng. C 2007, 27, 865–869. [Google Scholar] [CrossRef]
- Liu, Y.; Hou, D.; Wang, G. A simple wet chemical synthesis and characterization of hydroxyapatite nanorods. Mater. Chem. Phys. 2004, 86, 69–73. [Google Scholar] [CrossRef]
- Ashuri, M.; Moztarzadeh, F.; Nezafati, N.; Hamedani, A.A.; Tahriri, M. Development of a composite based on hydroxyapatite and magnesium and zinc-containing sol-gel-derived bioactive glass for bone substitute applications. Mater. Sci. Eng. C 2012, 32, 2330–2339. [Google Scholar] [CrossRef]
- Fahami, A.; Tabrizi, B.N.; Kahrizsangi, R.E. Synthesis of calcium phosphate-based composite nanopowders by mechanochemical process and subsequent thermal treatment. Ceram. Inter. 2012, 38, 6729–6738. [Google Scholar] [CrossRef]
- Cardoso, G.B.; Chacon, E.; Chacon, P.G.; Bordeaux-Rego, P.; Duarte, A.S.; Saad, S.T.O.; Cunha, M.R. Controlling morphology of hydroxyapatite nanoparticles through hydrothermal microemulsion chemical synthesis. Inorg. Chem. Commun. 2012, 20, 90–92. [Google Scholar]
- Farzadi, A.; Hashjin, M.S.; Bakhshi, F.; Aminian, A. Synthesis and characterization of hydroxyapatite/β-tricalcium phosphate nanocomposites using microwave irradiation. Ceram. Inter. 2011, 37, 65–71. [Google Scholar] [CrossRef]
- Chen, B.H.; Chen, K.I.; Ho, M.L.; Chen, H.N.; Chen, W.C.; Wang, C.K. Synthesis of calcium phosphates and porous hydroxyapatite beads prepared by emulsion method. Mater. Chem.Phys. 2009, 113, 365–371. [Google Scholar] [CrossRef]
- Shanthi, P.M.S.L.; Mangalaraja, R.V.; Uthirakumar, A.P.; Velmathi, S.; Balasubramanian, T.; Ashok, M. Synthesis and characterization of porous shell-like nanohydroxyapatite using cetrimide as template. J. Colloid Interface Sci. 2010, 350, 39–43. [Google Scholar] [CrossRef]
- Gopi, D.; Bhuvaneshwari, N.; Indira, J.; Kanimozhi, K.; Kavitha, L. novel green template-assisted synthesis of hydroxyapatite nanorods and their spectral characterization. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2013, 107, 196–202. [Google Scholar] [CrossRef]
- Hadavi, M.; Hasannia, S.; Faghihi, S.; Mashayekhi, F.; Zadeh, H.; Mostofi, S. Novel calcified gum Arabic porous nano-composite scaffold for bone tissue regeneration. Biochem. Biophys. Res. Commun. 2017, 488, 671–678. [Google Scholar] [CrossRef]
- Wang, A.; Liu, D.; Yin, H.; Wu, H.; Wada, Y.; Ren, M.; Xu, Y. Size-controlled synthesis of hydroxyapatite nanorods by chemical precipitation in the presence of organic modifiers. Mater. Sci. Eng. C 2007, 27, 865–869. [Google Scholar] [CrossRef]
- Sheikh, Z.; Najeeb, S.; Khurshid, Z.; Verma, V.; Rashid, H.; Glogauer, M. Biodegradable Materials for Bone Repair and Tissue Engineering Applications. Materials 2015, 8, 5744–5794. [Google Scholar] [CrossRef]
- Varma, H.K.; Babu, S.S. Synthesis of calcium phosphate bioceramics by citrate gel pyrolysis method. Ceram. Int. 2005, 31, 109. [Google Scholar] [CrossRef]
- Yan, D.; Lou, Y.; Han, Y.; Wickramaratne, M.N.; Dai, H.; Wang, X. Controllable synthesis of poly(acrylic acid)-stabilized nano-hydroxyapatite suspension by an ultrasound-assisted precipitation method. Mater. Lett. 2018, 227, 9–12. [Google Scholar] [CrossRef]
- Chandra, V.S.; Elayaraja, K.; Arul, K.T.; Ferraris, S.; Spriano, S.; Ferraris, M.; Kalkura, S.N. Synthesis of magnetic hydroxyapatite by hydrothermal–microwave technique: Dielectric, protein adsorption, blood compatibility, and drug release studies. Ceram. Int. 2015, 41, 13153–13163. [Google Scholar] [CrossRef]
- Zhang, Y.; Lu, J. A simple method to tailor spherical nanocrystal hydroxyapatite at low temperature. J. Nanopart. Res. 2007, 9, 589–594. [Google Scholar] [CrossRef]
- Cai, Y.; Liu, Y.; Yan, W.; Hu, Q.; Tao, J.; Zhang, M.; Shi, Z.; Tang, R. Role of hydroxyapatite nanoparticle size in bone cell proliferation. J. Mater. Chem. 2007, 17, 3780–3787. [Google Scholar] [CrossRef]
- Li, Y.; Weng, W. Surface modification of hydroxyapatite by stearic acid: characterization and in vitro behaviors. J. Mater. Sci. Mater. Med. 2008, 19, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Koutsopoulos, S.; Dalas, E. The Crystallization of Hydroxyapatite in the Presence of Lysine. J. Colloid Interface Sci. 2000, 231, 207. [Google Scholar] [CrossRef]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).