Density Functional Theory Analysis of the Adsorption Interactions of Carbon Impurities in Coal-associated Kaolinite



Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. SEM-EDS Analysis
2.3. XPS Analysis
2.4. DFT Simulation Method
3. Results and Discussion
3.1. SEM-EDS and XPS Analyses of Carbon Impurities in Coal-Associated Kaolinite
3.2. Occurrence of C Element in Coal-Associated Kaolinite
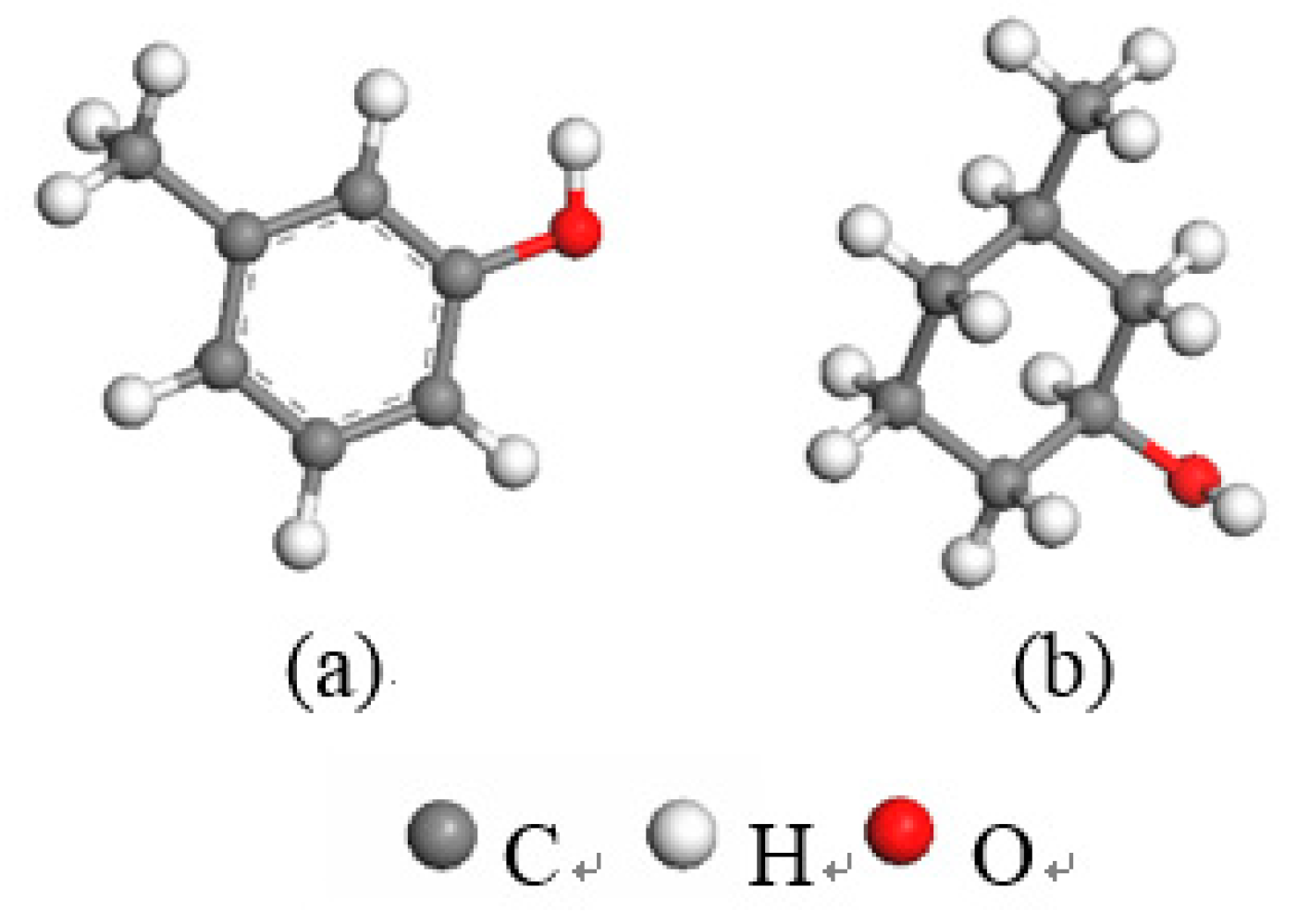
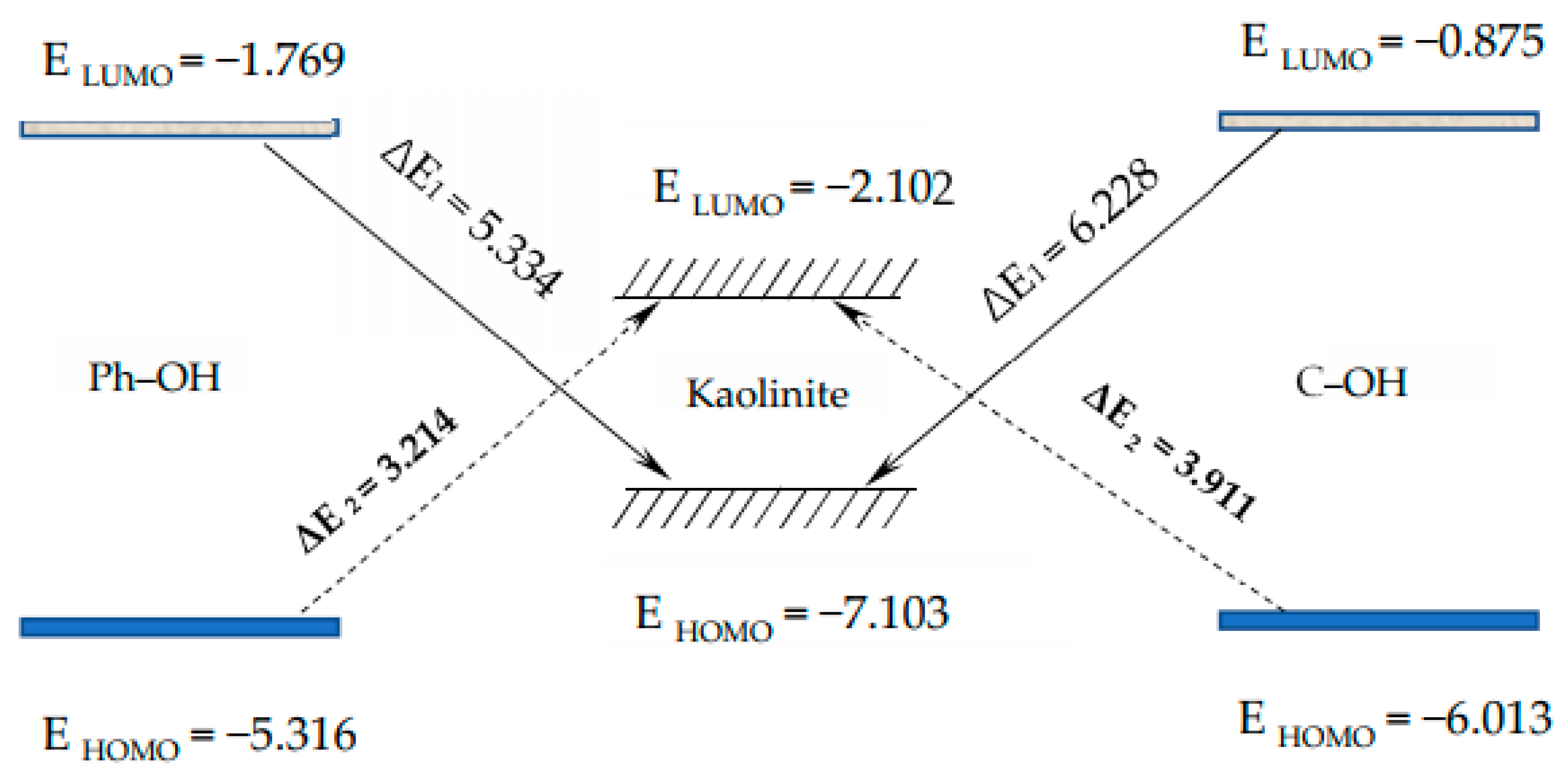
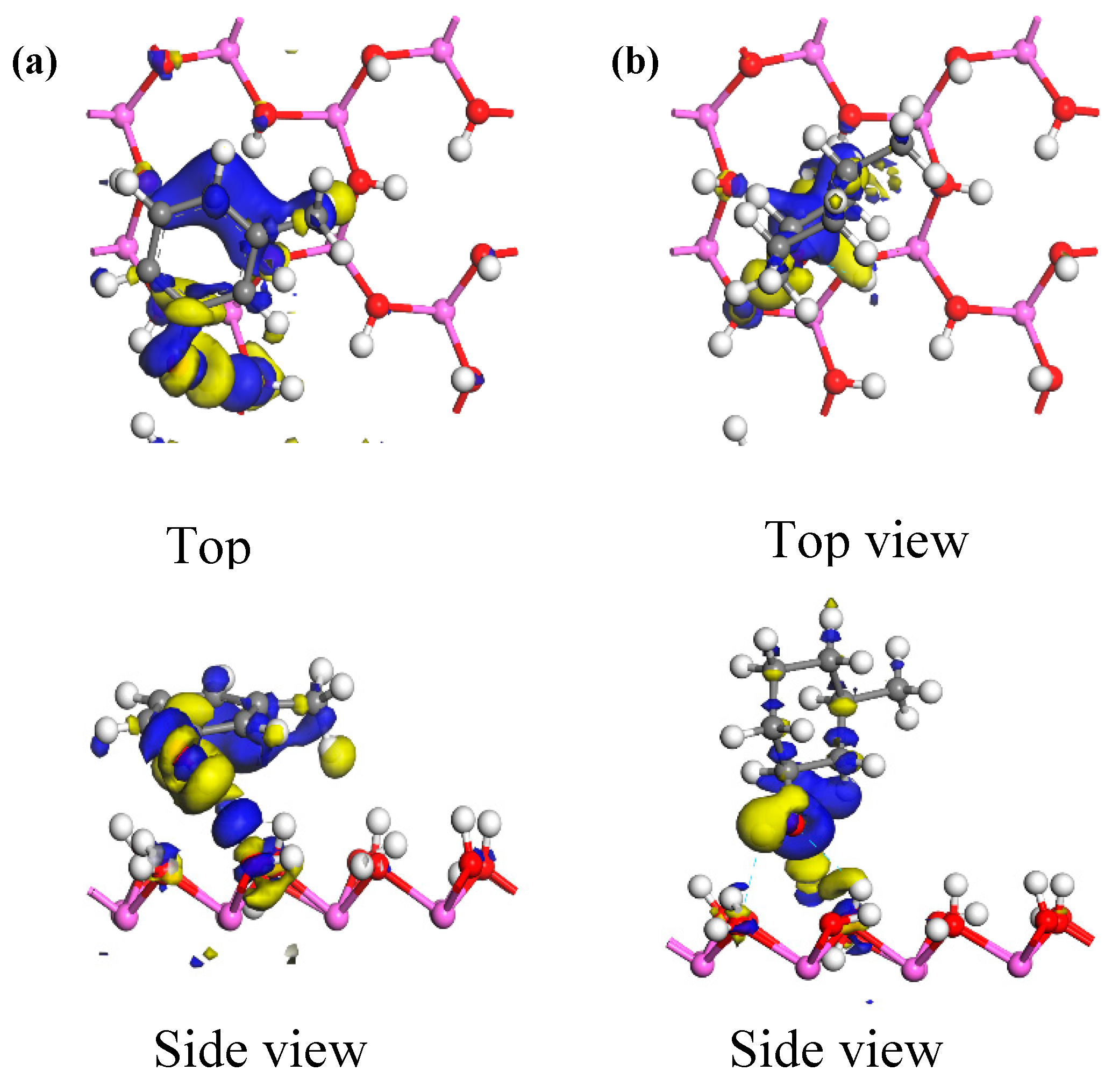
3.2.1. Frontier Orbital Analysis
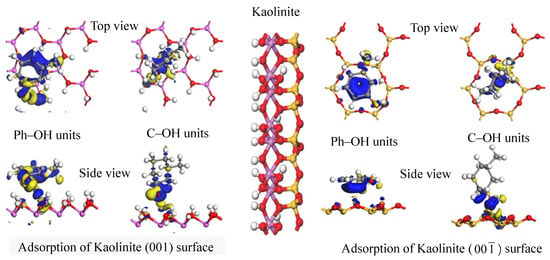
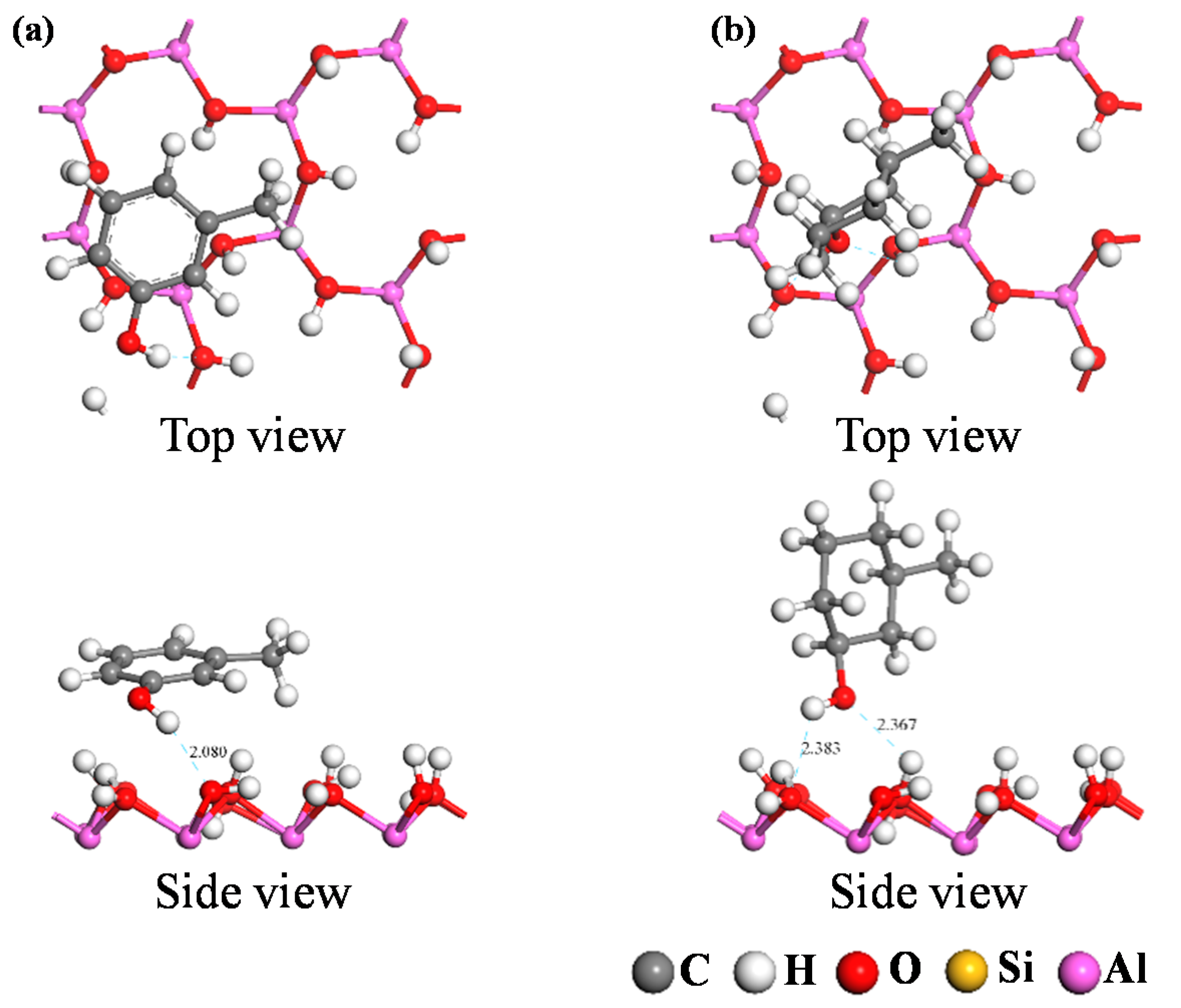
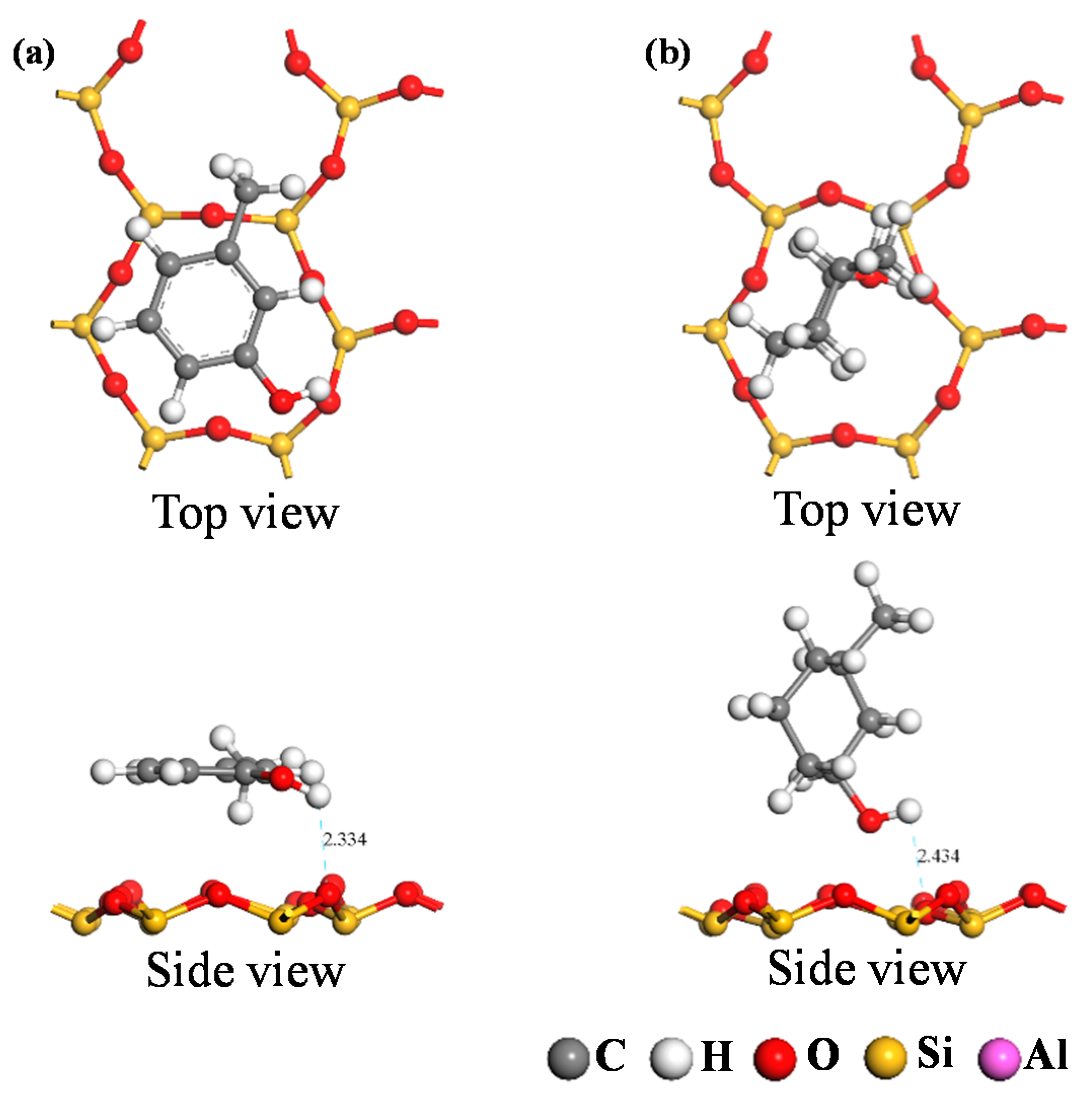
3.2.2. Analysis of Adsorption Configurations and Adsorption Energies
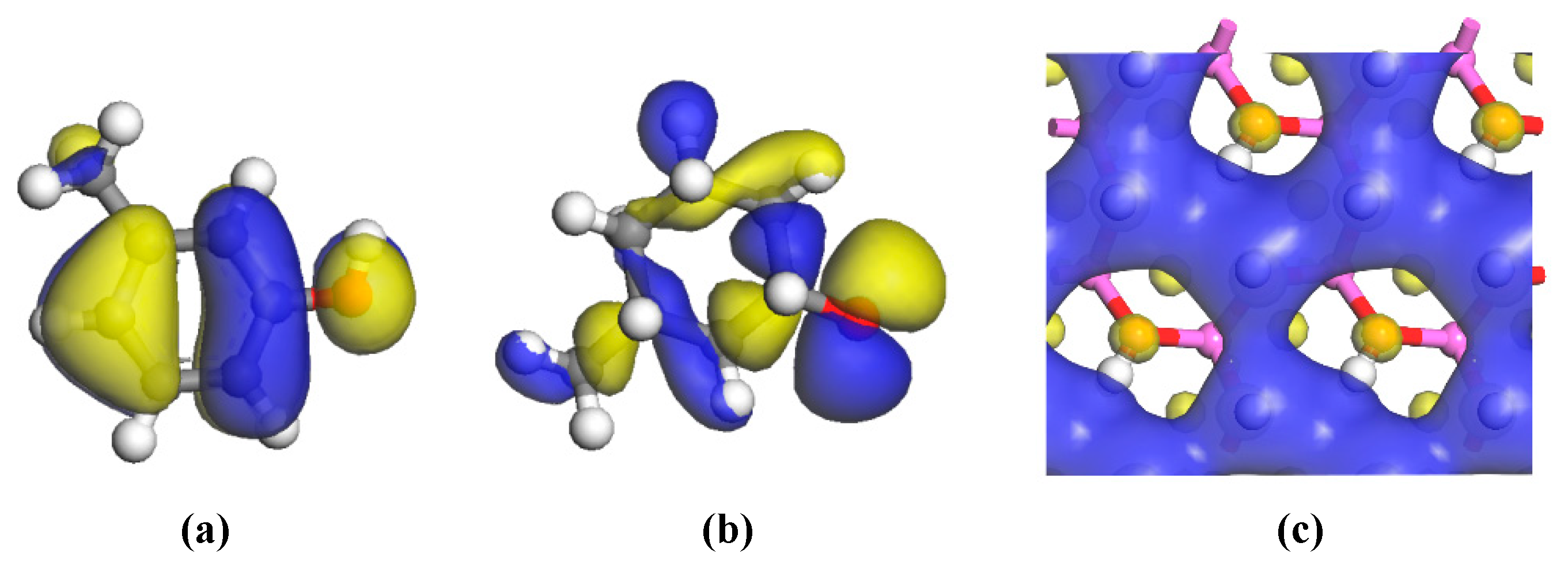
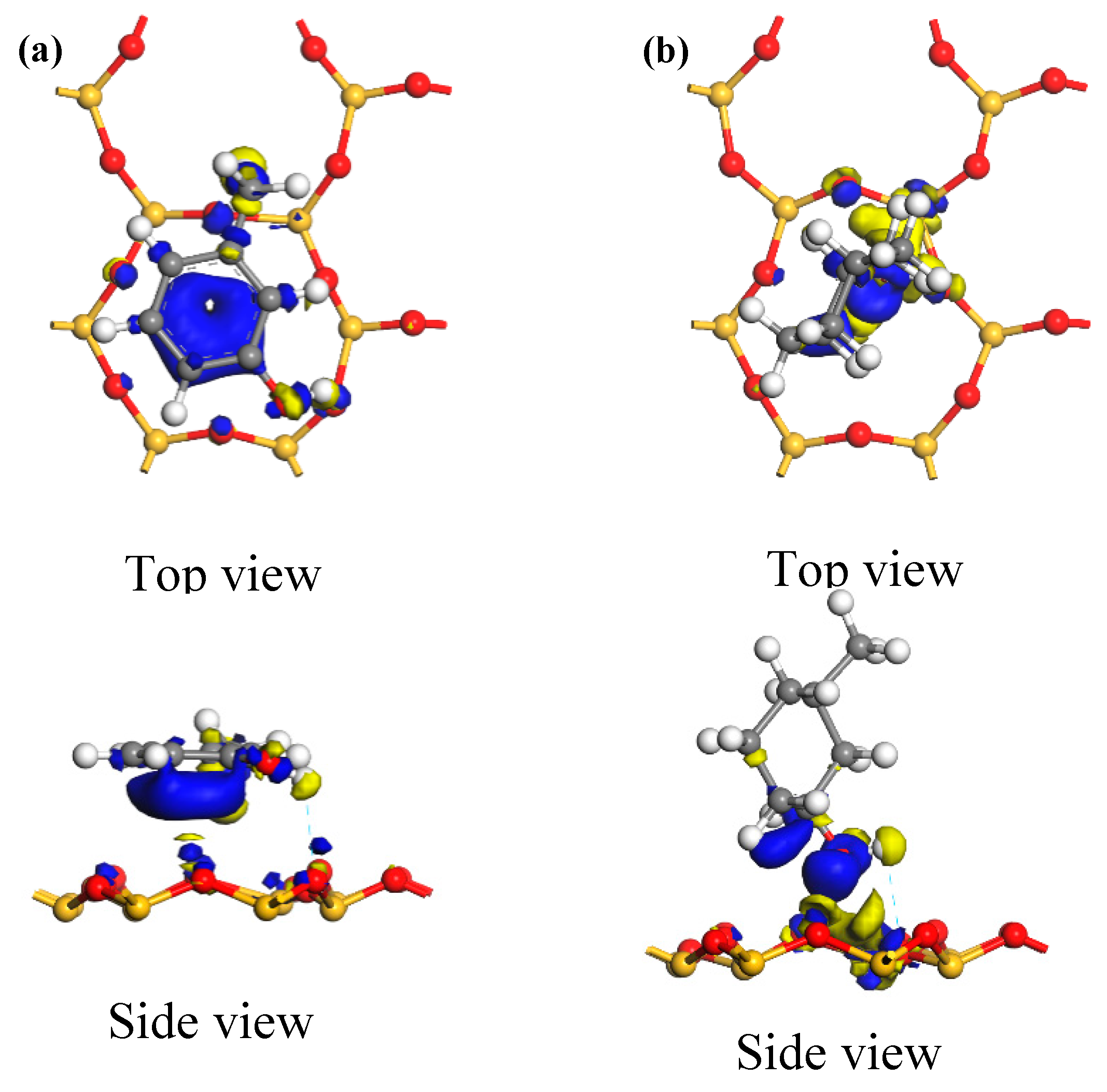
3.2.3. Electric Charge Analysis
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chen, J.; Min, F.; Liu, L.; Peng, C.; Lu, F. Hydrophobic aggregation of fine particles in high muddied coal slurry water. Water Sci. Technol. 2016, 73, 501–510. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Min, F.; Lu, F.J.; Zhang, M.; Song, S. A novel method for the determination of the point of zero net proton charge of colloidal kaolinite in aqueous solutions. Surf. Rev. Lett. 2016, 23, 1650023. [Google Scholar] [CrossRef]
- Israelachvili, J.N.; McGuiggan, P.M. Forces between surfaces in liquids. Science 1988, 241, 795–800. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Song, S.; Fort, T. Study on hydration layers near a hydrophilic surface in water through AFM imaging. Surf. Interface Anal. 2006, 38, 975–980. [Google Scholar] [CrossRef]
- Liu, L.; Shen, L.; Li, W.; Min, F.; Lu, F. Study on the aggregation behavior of kaolinite particles in the presence of cationic, anionic and non-ionic surfactants. PLoS ONE 2018, 13, e0204037. [Google Scholar] [CrossRef]
- Cui, J.R.; Fang, Q.X.; Huang, G.Z. Crystal structures and surface properties of diaspore and kaolinite. Nonferrous Met. 1999, 51, 25–30. [Google Scholar]
- Jiang, H.; Sun, Z.; Xu, L.; Hu, Y.; Huang, K.; Zhu, S. A comparison study of the flotation and adsorption behaviors of diaspore and kaolinite with quaternary ammonium collectors. Miner. Eng. 2014, 65, 124–129. [Google Scholar] [CrossRef]
- Perdew, J.P.; Kurth, S. Density functionals for non-relativistic coulomb systems in the New Century. Lect. Notes Phys. 2003, 620, 1–51. [Google Scholar]
- Wang, X.; Qian, P.; Song, K.; Zhang, C.; Dong, J. The DFT study of adsorption of 2, 4-dinitrotoluene on kaolinite surfaces. Comput. Theor. Chem. 2013, 1025, 16–23. [Google Scholar] [CrossRef]
- Liu, L.; Min, F.; Chen, J.; Lu, F.; Shen, L. The adsorption of dodecylamine and oleic acid on kaolinite surfaces: Insights from DFT calculation and experimental investigation. Appl. Surf. Sci. 2019, 470, 27–35. [Google Scholar] [CrossRef]
- Chen, J.; Min, F.; Liu, L.; Liu, C.; Lu, F. Experimental investigation and DFT calculation of different amine/ammonium salts adsorption on kaolinite. Appl. Surf. Sci. 2017, 419, 241–251. [Google Scholar] [CrossRef]
- Zhao, H.; Wang, B.; Li, Y.; Song, Q.; Zhao, Y.; Zhang, R.; Hu, Y.; Liu, S.; Wang, X.; Shu, X. Effect of chemical fractionation treatment on structure and characteristics of pyrolysis products of Xinjiang long flame coal. Fuel 2018, 234, 1193–1204. [Google Scholar] [CrossRef]
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.I.; Refson, K.; Payne, M.C. First principles methods using CASTEP. Z. Krist. 2005, 220, 567–570. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Vanderbilt, D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B 1990, 41, 7892–7895. [Google Scholar] [CrossRef]
- Han, Y.; Liu, W.; Chen, J.; Han, Y. Adsorption mechanism of hydroxyl calcium on two kaolinite (001) surface. J. China Coal Soc. 2016, 41, 743–750. [Google Scholar]
- Zhang, G.; Al-Saidi, W.A.; Myshakin, E.M.; Jordan, K.D. Dispersion-corrected density functional theory and classical force field calculations of water loading on a pyrophyllite(001) surface. J. Phys. Chem. C 2012, 116, 17134–17141. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Hu, X.L.; Michaelides, A. Water on the hydroxylated (001) surface of kaolinite: From monomer adsorption to a flat 2D wetting layer. Surf. Sci. 2008, 602, 960–974. [Google Scholar] [CrossRef]
- Bish, D.L. Rietveld refinement of the kaolinite structure at 1.5 K. Clays Clay Min. 1993, 41, 738–744. [Google Scholar] [CrossRef]
- Hu, Y.; Liu, L.; Min, F.; Zhang, M.; Song, S. Hydrophobic agglomeration of colloidal kaolinite in aqueous suspensions with dodecylamine. Colloids Surf. A Physicochem. Eng. Asp. 2013, 434, 281–286. [Google Scholar] [CrossRef]
- Liu, F.R.; Li, W.; Guo, H.Q.; Li, B.Q.; Bai, Z.Q.; Hu, R.S. XPS study on the change of carbon containing groups and sulfur transformation on coal surface. J. Fuel Chem. Technol. 2011, 39, 81–84. [Google Scholar]
- Shen, L.; Min, F.; Liu, L.; Zhu, J.; Xue, C.; Cai, C.; Zhou, W.; Wang, C. Application of gaseous pyrolysis products of the waste cooking oil as coal flotation collector. Fuel 2019, 239, 446–451. [Google Scholar] [CrossRef]
- Shi, Q.; Qin, B.; Liang, H.; Gao, Y.; Qu, B. Effects of igneous intrusions on the structure and spontaneous combustion propensity of coal: A case study of bituminous coal in Daxing Mine, China. Fuel 2018, 216, 181–189. [Google Scholar] [CrossRef]
- Zhao, H.; Song, Q.; Liu, S.; Li, Y.; Wang, X.; Shu, X. Study on catalytic co-pyrolysis of physical mixture/staged pyrolysis characteristics of lignite and straw over an catalytic beds of char and its mechanism. Energy Convers. Manag. 2018, 161, 13–26. [Google Scholar] [CrossRef]
- Kelemen, S.R.; Afeworki, M.; Gorbaty, M.L.; Cohen, A.D. Charact erization of organically bound oxygen forms in lignites, peats, and pyrolyzed peats by X-ray photoelectron spectroscopy (XPS) and solid-state 13C NMR methods. Energy Fuels 2002, 16, 1450–1462. [Google Scholar] [CrossRef]
- Yang, Z.; Zhou, A.; Zhang, H.; Zhang, Q. XPS analysis of Shenfu coal with different density fraction under UV catalytic photo oxidation. J. China Univ. Min. Technol. 2010, 39, 98–103. [Google Scholar]
- Chen, J.H. The Solide Physics of Sulphide Minerals Flotation; Central South University Press: Changsha, China, 2015; pp. 61–62. [Google Scholar]
- Chen, J.; Min, F.; Liu, L. The interactions between fine particles of coal and kaolinite in aqueous, insights from experiments and molecular simulations. Appl. Surf. Sci. 2019, 467–468, 12–21. [Google Scholar] [CrossRef]
- Li, B.; Liu, S.; Guo, J.; Zhang, L. Interaction between low rank coal and kaolinite particles: A DFT simulation. Appl. Surf. Sci. 2018, 456, 215–220. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Computational Parameter | Lattice Parameters/Å | Cell Angles/° | Current Cell Volume/Å3 | ||||
|---|---|---|---|---|---|---|---|
| a | b | c | α | β | γ | ||
| GGA/400 eV | 5.196 | 9.007 | 7.372 | 93.029 | 105.983 | 89.866 | 331.221 |
| (GGA/700 eV) [19] | 5.196 | 9.021 | 7.485 | 91.700 | 104.720 | 89.780 | 339.170 |
| Experimental test value [20] | 5.153 | 8.942 | 7.391 | 91.926 | 105.046 | 89.797 | 329.910 |
| Analysis Method | C | O | Al | Si | ||||
|---|---|---|---|---|---|---|---|---|
| W% | A% | W% | A% | W% | A% | W% | A% | |
| Surface analysis | 80.45 | 84.61 | 19.41 | 15.33 | 0.14 | 0.06 | 0 | 0 |
| Point analysis | 50.95 | 58.2 | 48.35 | 41.45 | 0.35 | 0.18 | 0.35 | 0.17 |
| Average | 65.7 | 71.405 | 33.88 | 28.39 | 0.245 | 0.12 | 0.175 | 0.085 |
| Sample | Atomic Concentration/% | ||||
|---|---|---|---|---|---|
| C 1s | O 1s | Si 2p | Al 2p | Fe 2p | |
| Carbon impurity | 29.93 | 31.68 | 18.12 | 10.94 | 9.33 |
| Kaolinite sample | 11.15 | 50.16 | 19.75 | 14.21 | 4.73 |
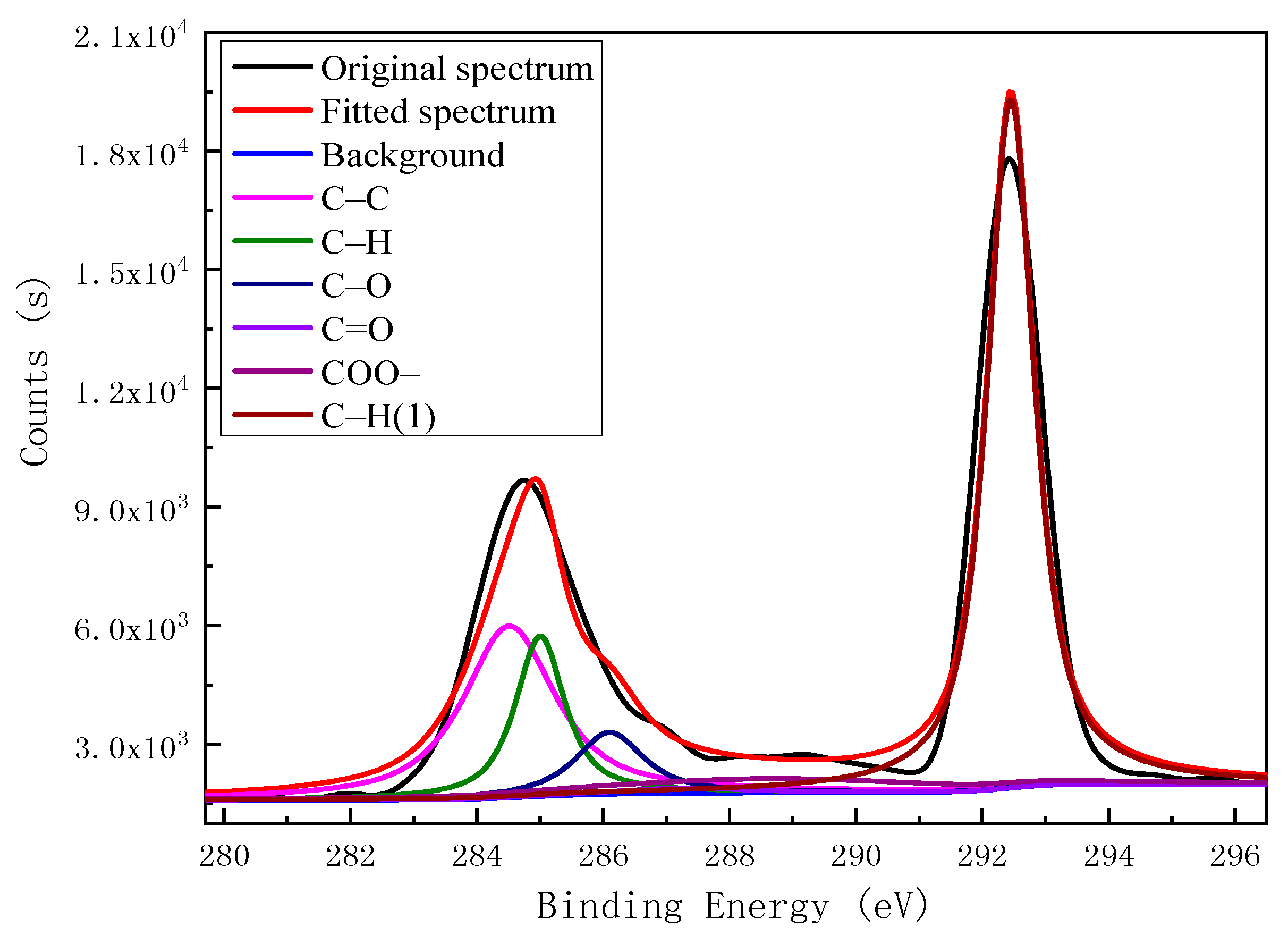
| Element | Binding Energy/eV | Functional Group | Peak Area | Relative Content % |
|---|---|---|---|---|
| C 1s | 284.50 | C–C | 10,971.27 | 23.96 |
| 284.99 | C–H | 5480.877 | 11.97 | |
| 286.10 | C–O | 2914.683 | 6.36 | |
| 287.30 | C=O | 442.6801 | 0.97 | |
| 288.60 | COO– | 3266.749 | 7.13 | |
| 292.50 | C–H | 22,716.64 | 49.61 | |
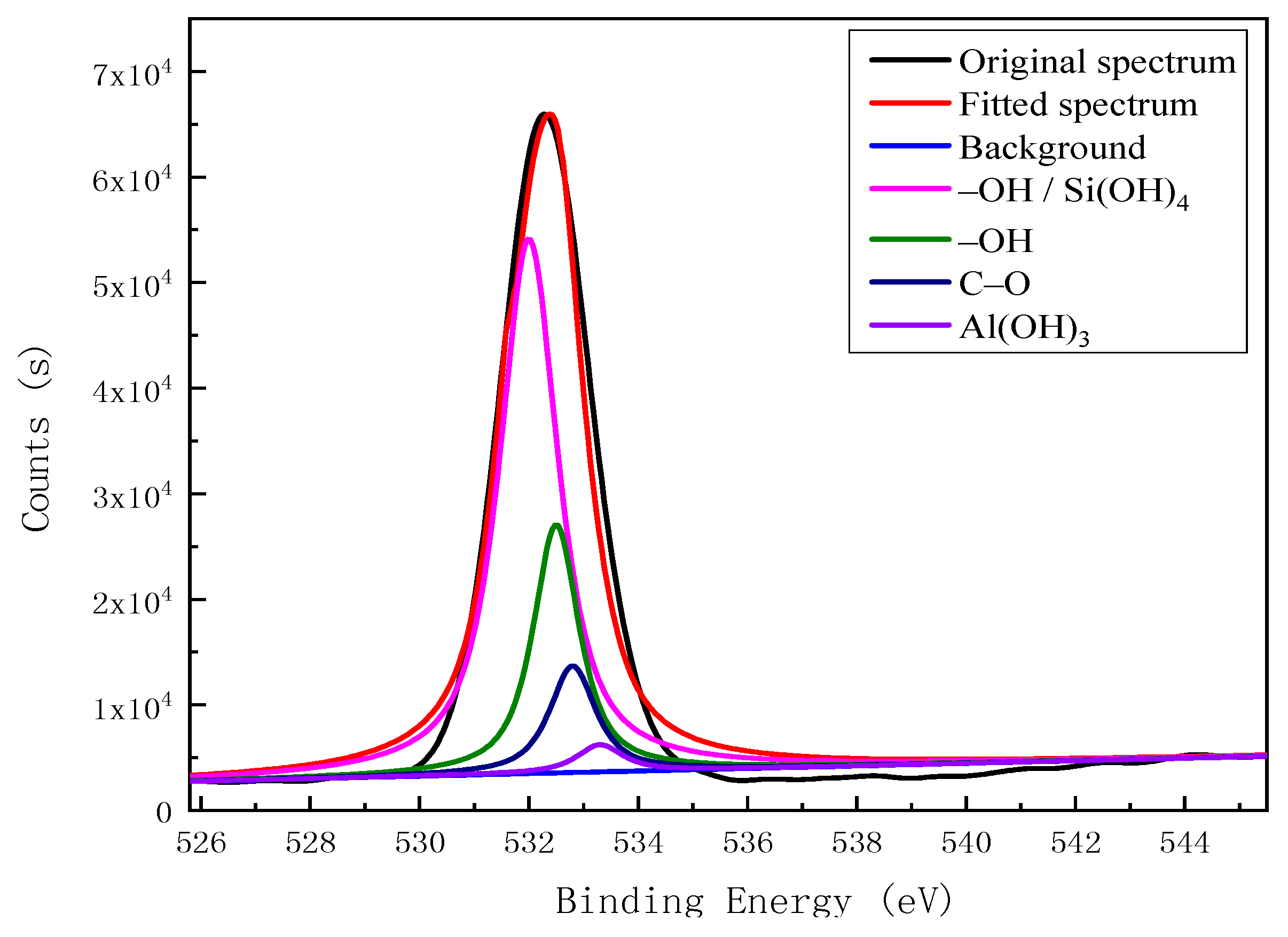
| O 1s | 531.90 | –OH, Si(OH)4 | 95,055.25 | 46.46 |
| 532.40 | –OH | 34,541.36 | 16.88 | |
| 532.80 | C–O | 50,132.20 | 24.50 | |
| 533.30 | Al(OH)3 | 24,872.81 | 12.16 |
| Adsorption Configuration | Adsorption Energy Eads/eV | |
|---|---|---|
| Kaolinite (001) Surface | Kaolinite (001) Surface | |
| Ph–OH | −1.19 | −1.02 |
| C–OH | −0.78 | −0.55 |
| Adsorption Configuration | Adsorption Status | Mulliken Charge/e | |
|---|---|---|---|
| (001) Surface | (001) Surface | ||
| Ph–OH | before | 0 | 0 |
| after | −0.52 | −0.31 | |
| Kaolinite surface | before | 0 | 0 |
| after | 0.52 | 0.31 | |
| C–OH | before | 0 | 0 |
| after | −0.29 | −0.17 | |
| Kaolinite surface | before | 0 | 0 |
| after | 0.29 | 0.17 | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, F.; Liu, L.; Min, F.; Chen, J.; Zhang, M. Density Functional Theory Analysis of the Adsorption Interactions of Carbon Impurities in Coal-associated Kaolinite. Processes 2019, 7, 782. https://doi.org/10.3390/pr7110782
Lu F, Liu L, Min F, Chen J, Zhang M. Density Functional Theory Analysis of the Adsorption Interactions of Carbon Impurities in Coal-associated Kaolinite. Processes. 2019; 7(11):782. https://doi.org/10.3390/pr7110782
Chicago/Turabian StyleLu, Fangqin, Lingyun Liu, Fanfei Min, Jun Chen, and Mingxu Zhang. 2019. "Density Functional Theory Analysis of the Adsorption Interactions of Carbon Impurities in Coal-associated Kaolinite" Processes 7, no. 11: 782. https://doi.org/10.3390/pr7110782
APA StyleLu, F., Liu, L., Min, F., Chen, J., & Zhang, M. (2019). Density Functional Theory Analysis of the Adsorption Interactions of Carbon Impurities in Coal-associated Kaolinite. Processes, 7(11), 782. https://doi.org/10.3390/pr7110782