FluxVisualizer, a Software to Visualize Fluxes through Metabolic Networks


Abstract
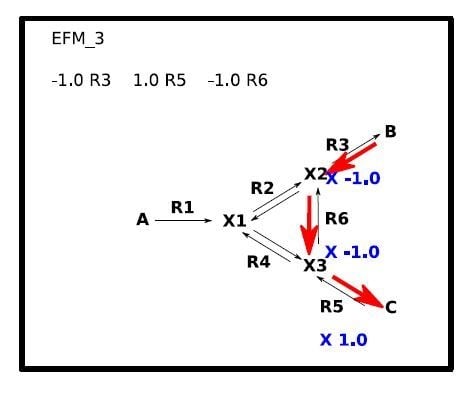
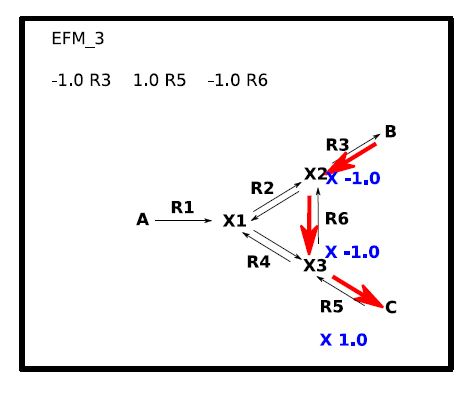
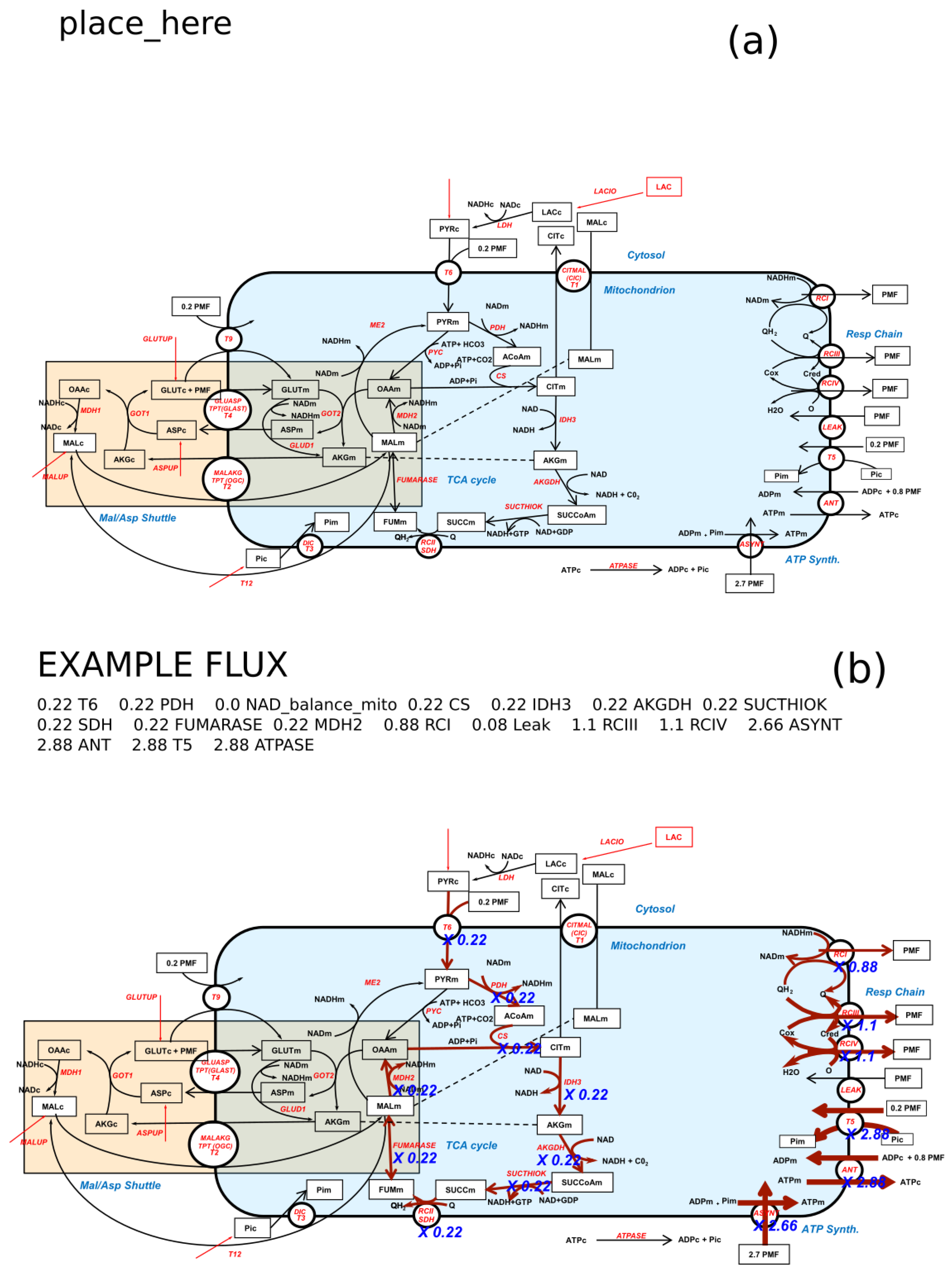
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
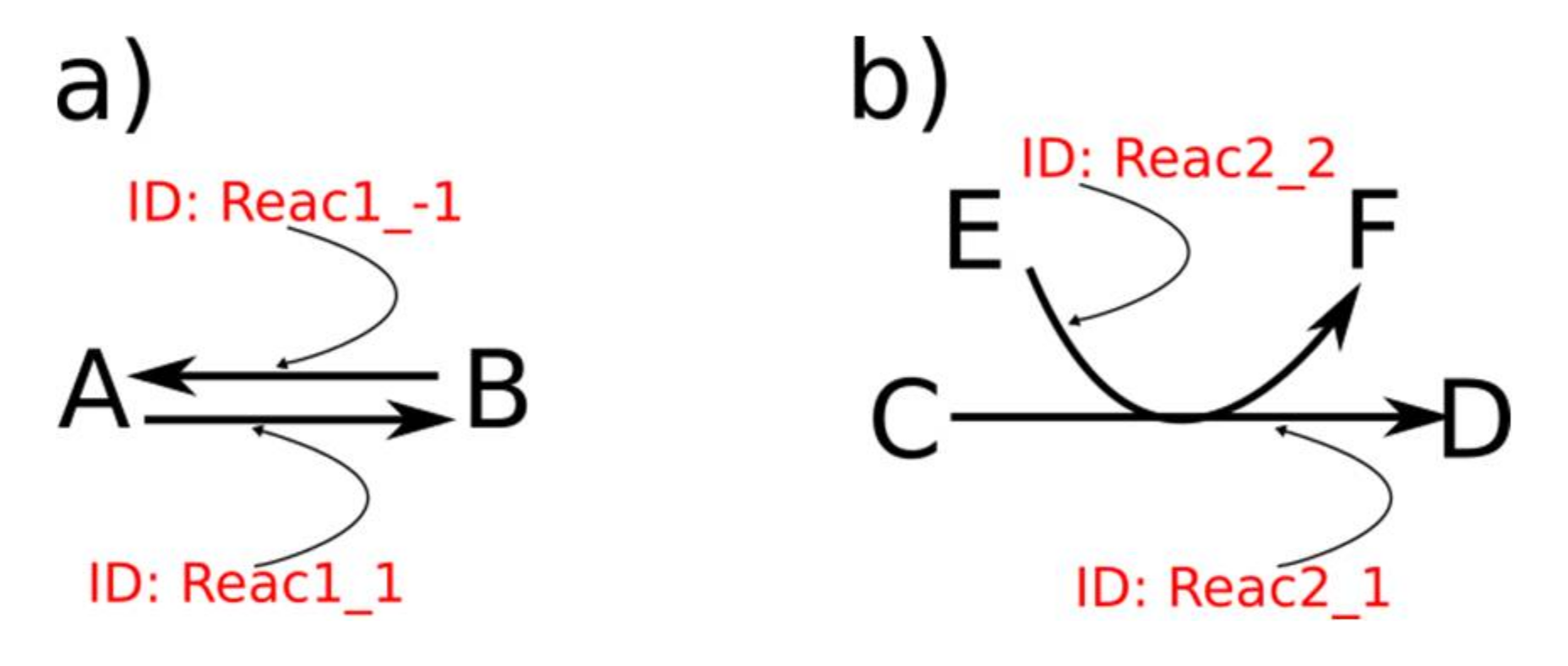{kind=link}
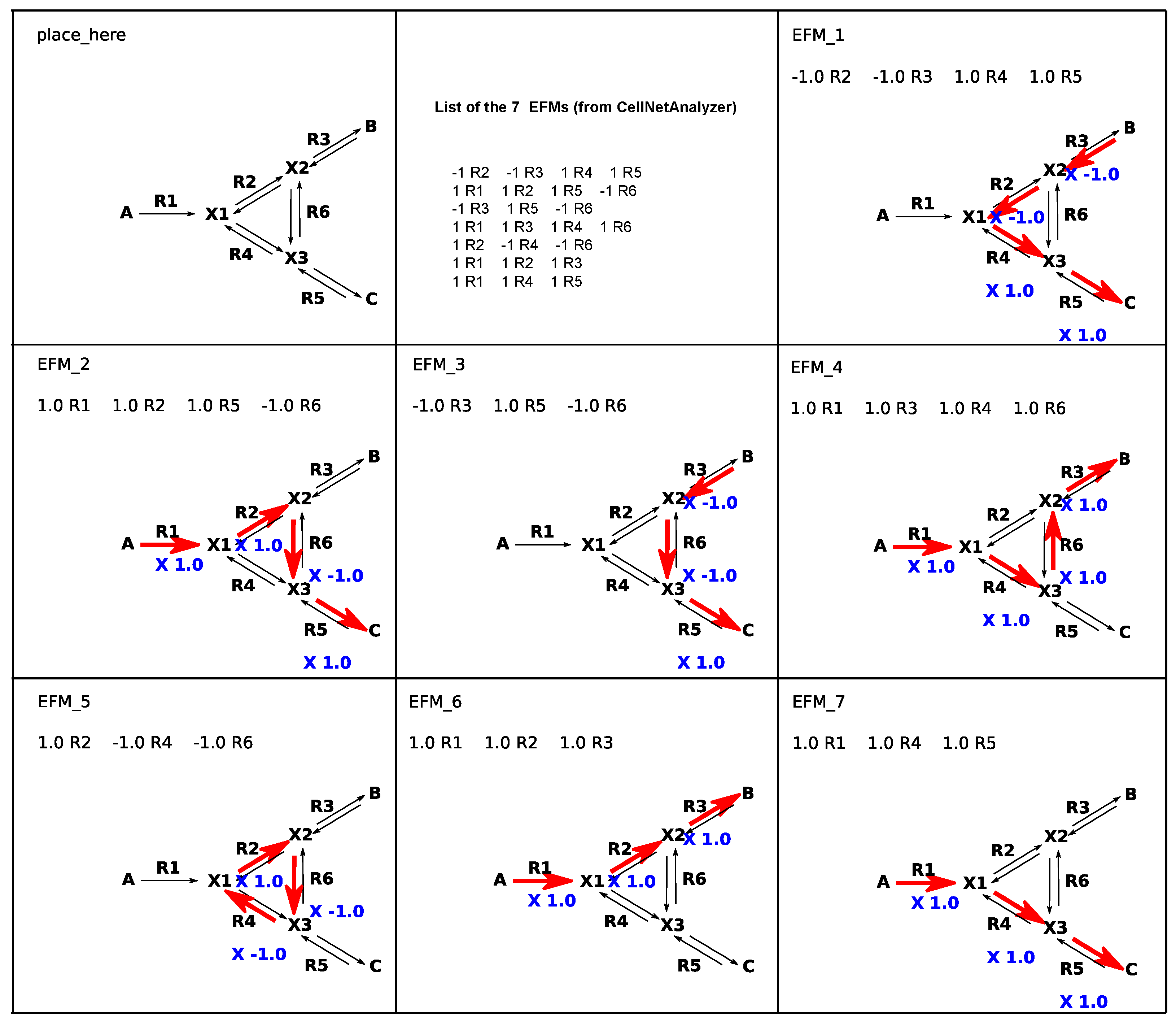{kind=link}
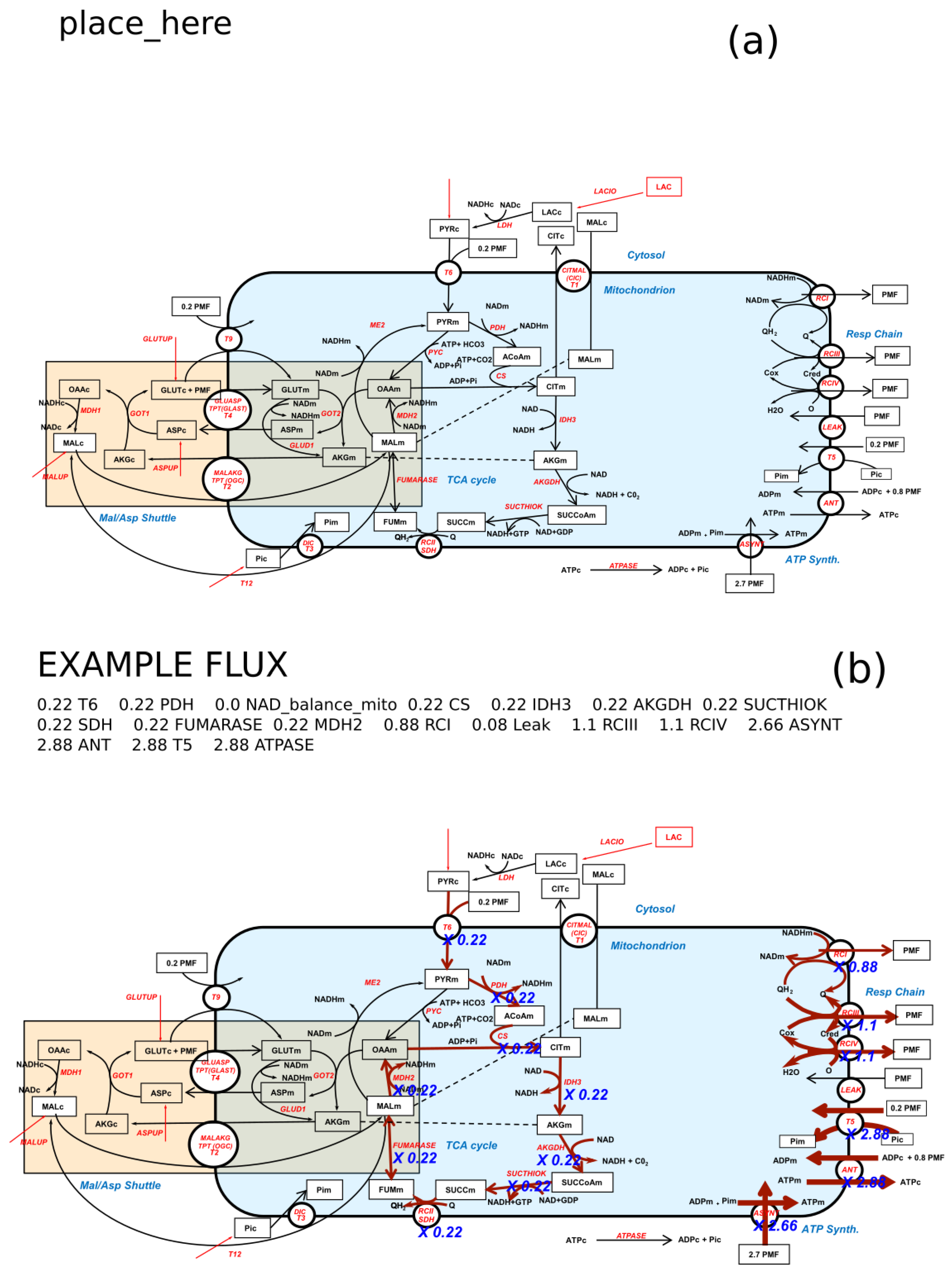
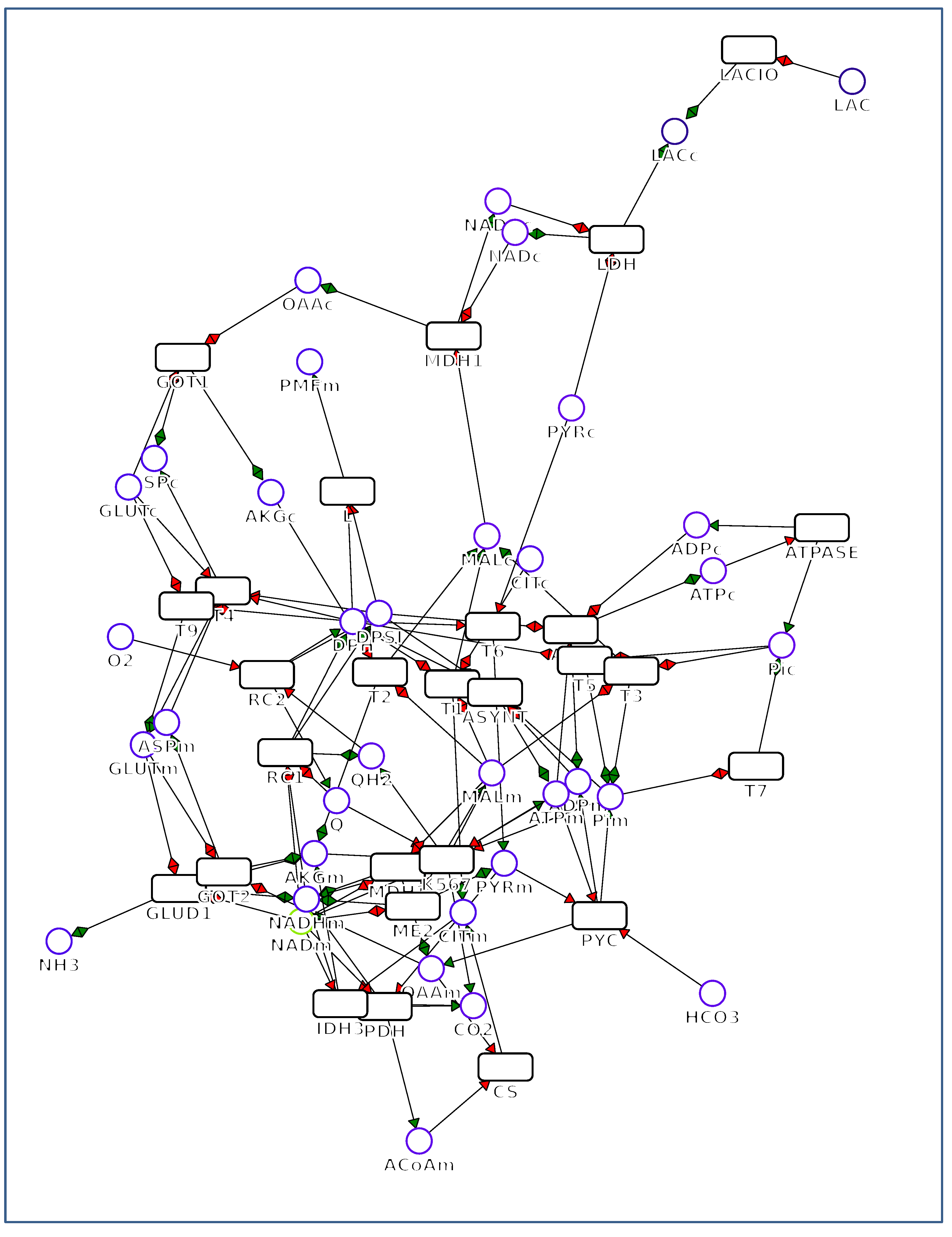{kind=link}
1. Introduction
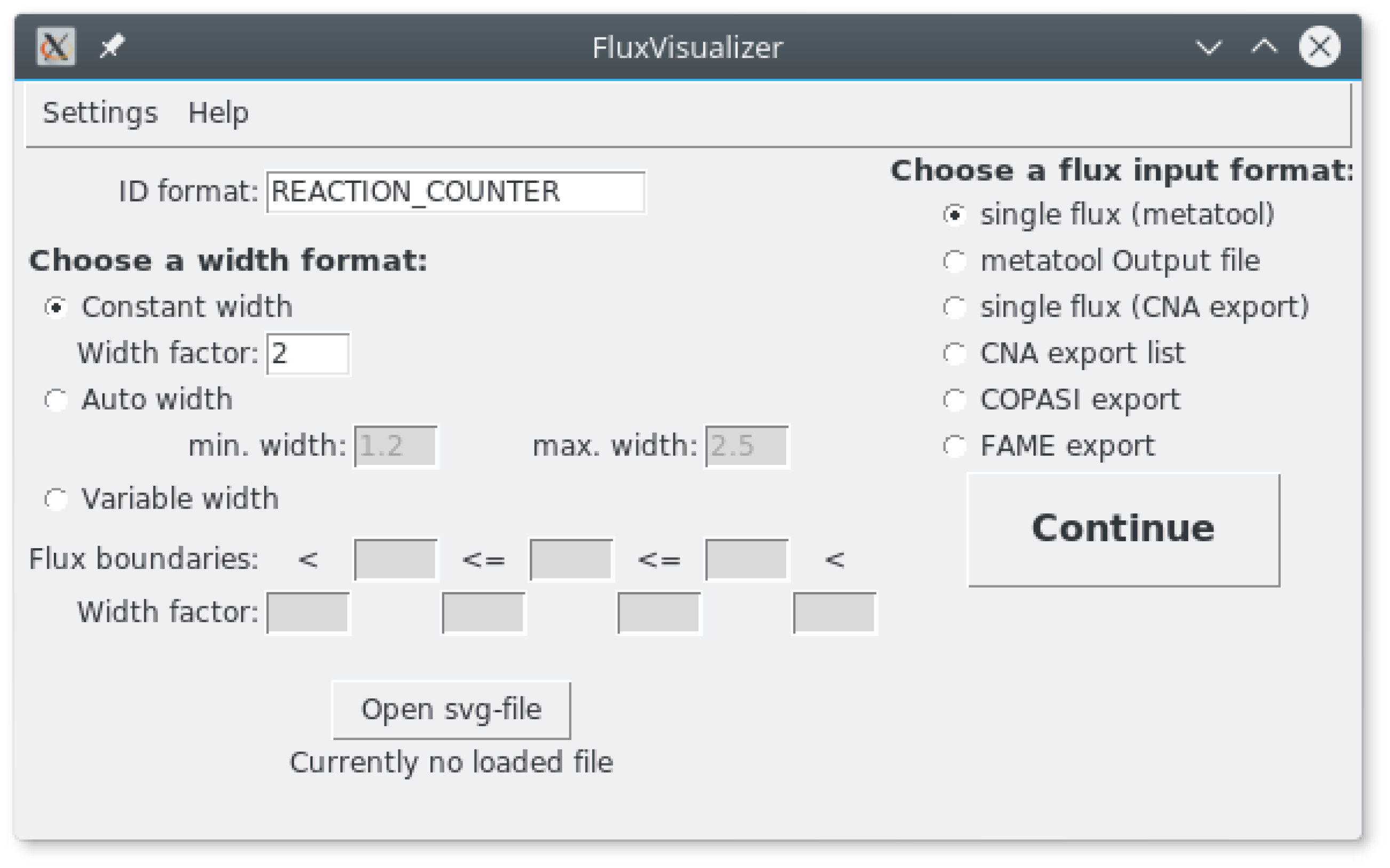
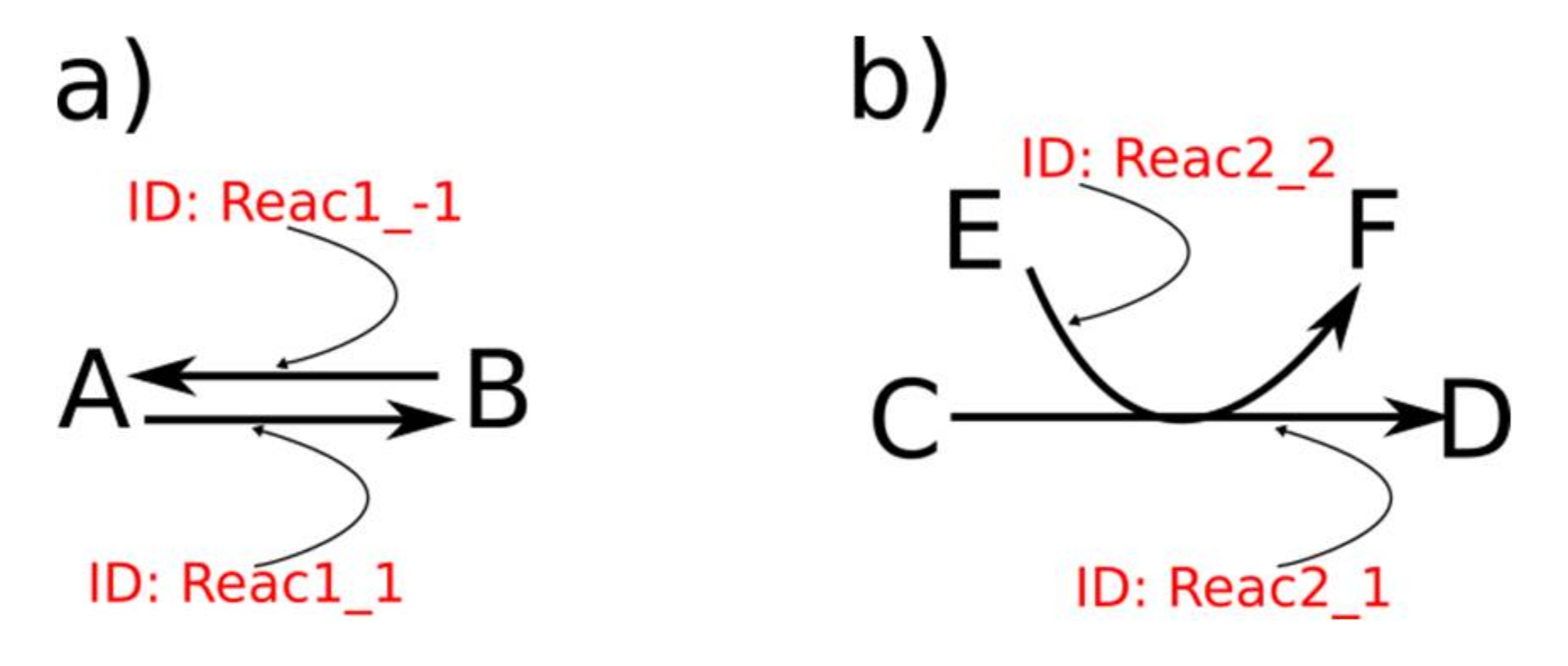
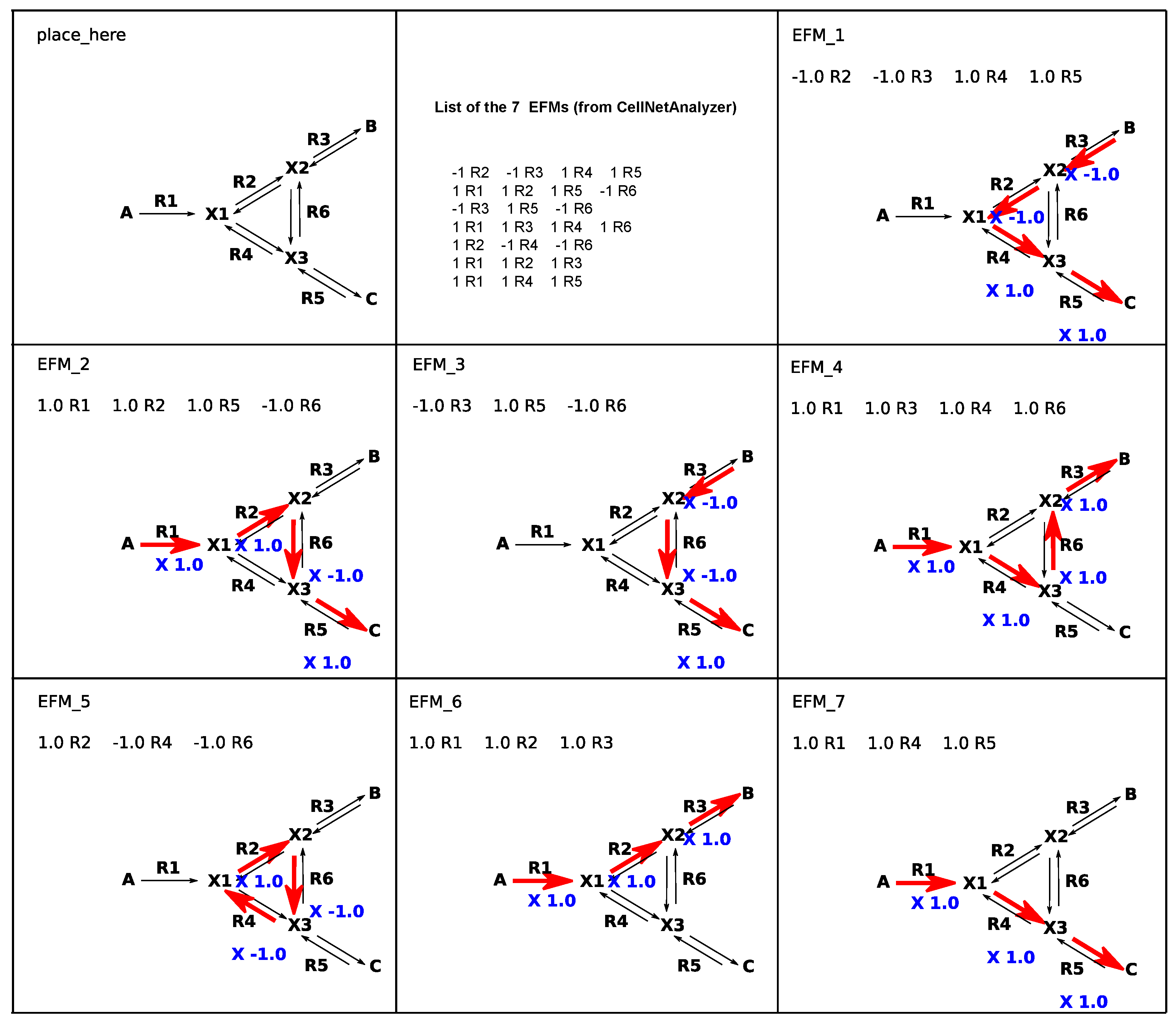
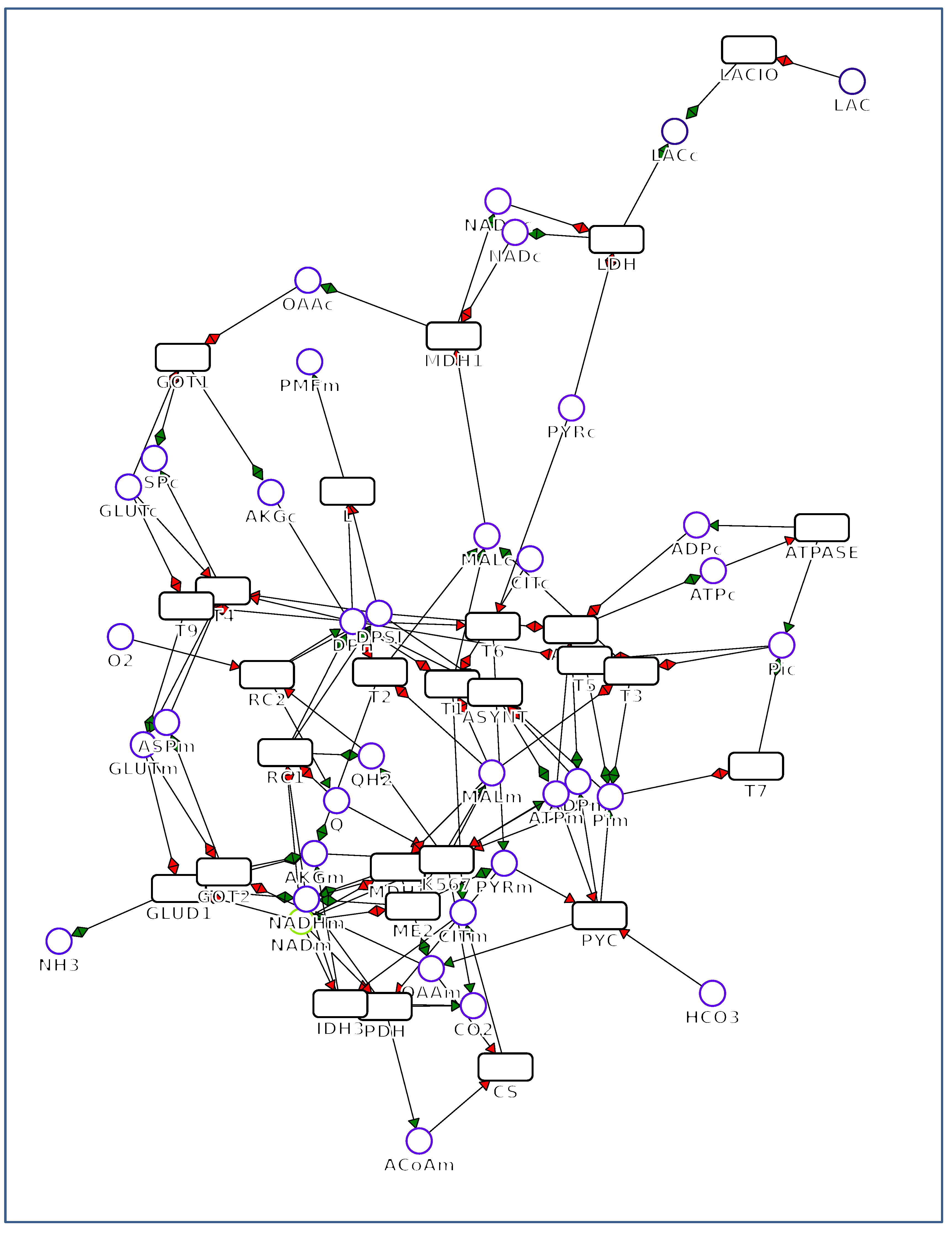
2. Overview of FluxVisualizer
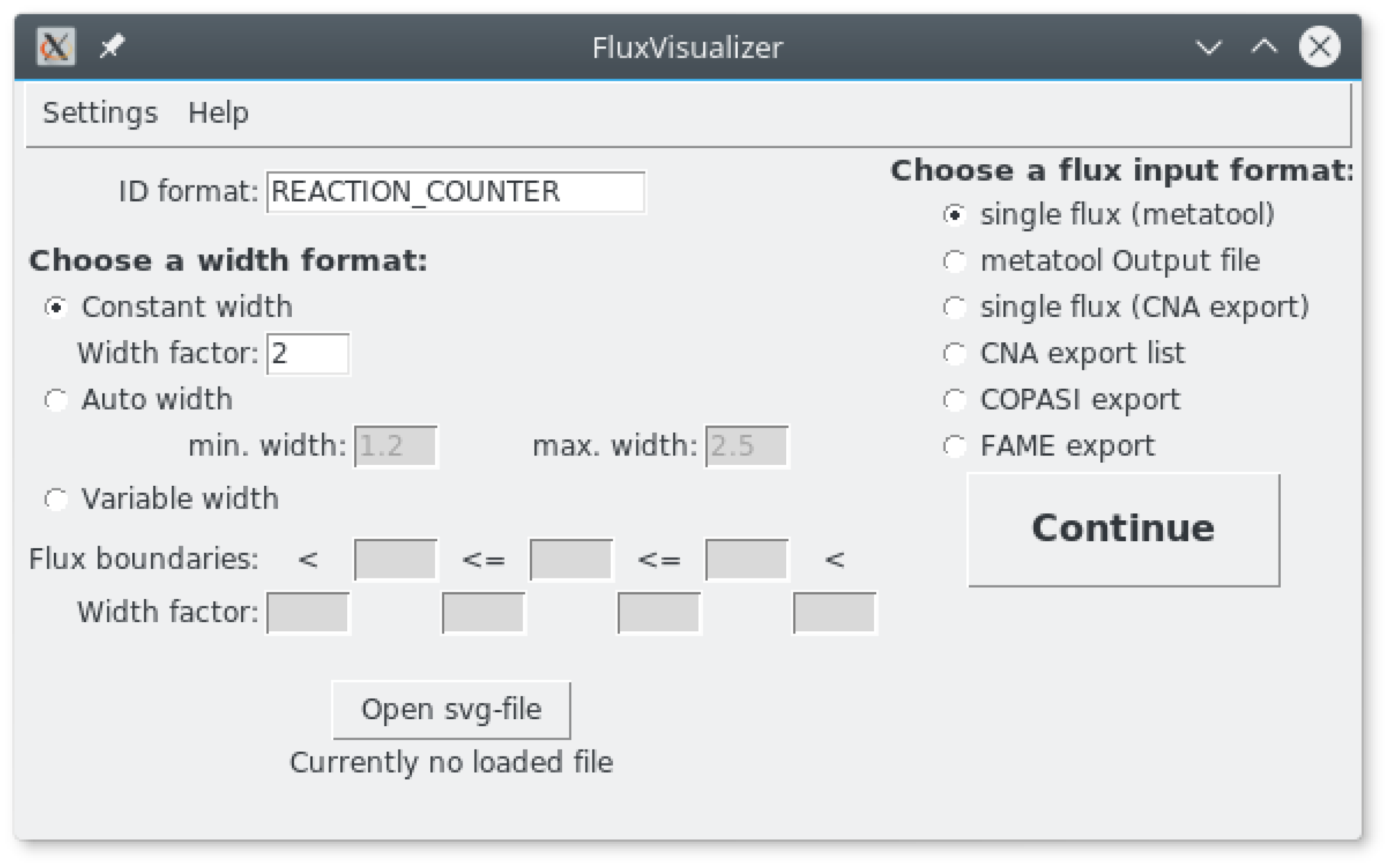
2.1. Main Window (Figure 2)
2.2. Secondary Windows (Figure 3)
2.3. Additional Options
3. Implementation and Requirements
4. Discussion and Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gehlenborg, N.; O’Donoghue, S.; Baliga, N.S.; Goesmann, A.; Hibbs, M.A.; Kitano, H.; Kohlbacher, O.; Neuweger, H.; Schneider, R.; Tenenbaum, D.; et al. Visualization of omics data for systems biology. Nat. Methods 2010, 7, S56–S68. [Google Scholar] [CrossRef] [PubMed]
- O’Donoghue, S.I.; Baldi, B.F.; Maier-Hein, L.; Stenau, E.; Hogan, J.M.; Humphrey, S.; Kaur, S.; McCarthy, D.J.; Moore, W.J.; Procter, J.B.; et al. Visualization of Biomedical Data. Annu. Rev. Biomed. Data Sci. 2018, in press. [Google Scholar]
- Schellenberger, J.; Park, J.O.; Conrad, T.M.; Palsson, B.Ø. BiGG: A Biochemical Genetic and Genomic knowledgebase of large scale metabolic reconstructions. BMC Bioinform. 2010, 11, 213. [Google Scholar] [CrossRef] [PubMed]
- Maarleveld, T.R.; Khandelwal, R.A.; Olivier, B.G.; Teusink, B.; Bruggeman, F.J. Basic concepts and principles of stoichiometric modeling of metabolic networks. Biotechnol. J. 2013, 8, 997–1008. [Google Scholar] [CrossRef] [PubMed]
- Antoniewicz, M.R. Methods and advances in metabolic flux analysis: A mini-review. J. Ind. Microb. Biotechnol. 2015, 42, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Kacser, H.; Burns, J.A. The control of flux. Symp. Soc. Exp. Biol. 1973, 32, 65–104. [Google Scholar] [CrossRef]
- Heinrich, R.; Rapoport, T.A. A linear steady-state treatment of enzymatic chains; its application for the analysis of the crossover theorem and of the glycolysis of human erythrocytes. Acta Biol. Med. Ger. 1973, 31, 479–494. [Google Scholar] [PubMed]
- Reder, C. Metabolic control theory: A structural approach. J. Theor. Biol. 1988, 135, 175–201. [Google Scholar] [CrossRef]
- Sauro, H. Systems Biology: Introduction to Pathway Modeling; Ambrosius Publishing: Bangalore, India, 2014; ISBN 978-0-9824773-7-3. [Google Scholar]
- Klipp, E.; Liebermeister, W.; Wierling, C.; Kowald, A. Systems Biology: A Textbook, 2nd ed.; Wiley-Blackwell: Berlin, Germany, 2016; ISBN 978-3-527-33636-4. [Google Scholar]
- Ataman, M.; Hernandez Gardiol, D.F.; Fengos, G.; Hatzimanikatis, V. redGEM: Systematic reduction and analysis of genome-scale metabolic reconstructions for development of consistent core metabolic models. PLoS Comput. Biol. 2017, 13, e1005444. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.C.; Eyassu, F.; Mazat, J.-P.; Robinson, A.J. MitoCore: A curated constraint-based model for simulating human central metabolism. BMC Syst. Biol. 2017, 11, 114. [Google Scholar] [CrossRef] [PubMed]
- Orth, J.; Fleming, R.; Palsson, B. Reconstruction and Use of Microbial Metabolic Networks: The Core Escherichia coli Metabolic Model as an Educational Guide. EcoSal Plus 2010. [Google Scholar] [CrossRef] [PubMed]
- Omix Visualization | Welcome. Available online: https://www.omix-visualization.com/ (accessed on 21 April 2018).
- Jensen, P.A.; Papin, J.A. MetDraw: Automated visualization of genome-scale metabolic network reconstructions and high-throughput data. Bioinformatics 2014, 30, 1327–1328. [Google Scholar] [CrossRef] [PubMed]
- Chazalviel, M.; Frainay, C.; Poupin, N.; Vinson, F.; Merlet, B.; Gloaguen, Y.; Cottret, L.; Jourdan, F. MetExploreViz: Web component for interactive metabolic network visualization. Bioinformatics 2017, 34, 312–313. [Google Scholar] [CrossRef] [PubMed]
- König, M.; Holzhütter, H.G. Fluxviz—Cytoscape plug-in for visualization of flux distributions in networks. Genome Inform. 2010, 24, 96–103. [Google Scholar] [PubMed]
- Granger, B.R.; Chang, Y.C.; Wang, Y.; DeLisi, C.; Segrè, D.; Hu, Z. Visualization of metabolic interaction networks in microbial communities using VisANT 5.0. PLoS Comput. Boil. 2016, 12, e1004875. [Google Scholar] [CrossRef] [PubMed]
- Turkay, C.; Jeanquartier, F.; Holzinger, A.; Hauser, H. On computationally-enhanced visual analysis of heterogeneous data and its application in biomedical informatics. In Interactive Knowledge Discovery and Data Mining in Biomedical Informatics; Holzinger, A., Jurisica, I., Eds.; Springer: Berlin/Heidelberg, Germany, 2014; pp. 117–140. [Google Scholar]
- Pfeiffer, T.; Sánchez-Valdenebro, I.; Nuño, J.C.; Montero, F.; Schuster, S. METATOOL: For studying metabolic networks. Bioinformatics 1999, 15, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Klamt, S.; Saez-Rodriguez, J.; Gilles, E.D. Structural and functional analysis of cellular networks with CellNetAnalyzer. BMC Syst. Biol. 2007, 1, 2. [Google Scholar] [CrossRef] [PubMed]
- Hoops, S.; Sahle, S.; Gauges, R.; Lee, C.; Pahle, J.; Simus, N.; Singhal, M.; Xu, L.; Mendes, P.; Kummer, U. COPASI—A COmplex PAthway SImulator. Bioinformatics 2006, 22, 3067–3074. [Google Scholar] [CrossRef] [PubMed]
- Boele, J.; Olivier, B.G.; Teusink, B. FAME, the Flux Analysis and Modeling Environment. BMC Syst. Biol. 2012, 6, 8. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rose, T.D.; Mazat, J.-P. FluxVisualizer, a Software to Visualize Fluxes through Metabolic Networks. Processes 2018, 6, 39. https://doi.org/10.3390/pr6050039
Rose TD, Mazat J-P. FluxVisualizer, a Software to Visualize Fluxes through Metabolic Networks. Processes. 2018; 6(5):39. https://doi.org/10.3390/pr6050039
Chicago/Turabian StyleRose, Tim Daniel, and Jean-Pierre Mazat. 2018. "FluxVisualizer, a Software to Visualize Fluxes through Metabolic Networks" Processes 6, no. 5: 39. https://doi.org/10.3390/pr6050039
APA StyleRose, T. D., & Mazat, J.-P. (2018). FluxVisualizer, a Software to Visualize Fluxes through Metabolic Networks. Processes, 6(5), 39. https://doi.org/10.3390/pr6050039