Systematic and Model-Assisted Process Design for the Extraction and Purification of Artemisinin from Artemisia annua L.—Part II: Model-Based Design of Agitated and Packed Columns for Multistage Extraction and Scrubbing



Abstract
:1. Introduction
- Part 0:
- Sixt, M.; Strube, J. Systematic and model-assisted evaluation of solvent based- for pressurized hot water extraction for the extraction of Artemisinin from Artemisia annua L. Processes 2017, 5, 86, doi:10.3390/pr5040086 [5].
- Part I:
- Sixt, Schmidt et al. Systematic and model-assisted process design for the extraction and purification of Artemisinin from Artemisia annua L.—Part I: Conceptual process design and cost estimation. Processes 2018, 6, 161, doi:10.3390/pr6090161 [6].
- Part II:
- Schmidt, Sixt et al. Model-based design of agitated and packed columns for multistage extraction and scrubbing (this article);
- Part III:
- Mestmäcker, Schmidt et al. Systematic and model-assisted process design for the extraction and purification of Artemisinin from Artemisia annua L.—Part III: Chromatographic Purification. Processes 2018, 6, 180, doi:10.3390/pr6100180 [7];
- Part IV:
- Huter, Schmidt et al. Systematic and model-assisted process design for the extraction and purification of Artemisinin from Artemisia annua L.—Part IV: Crystallization. Processes 2018, 6, 181, doi:10.3390/pr6100181 [8].
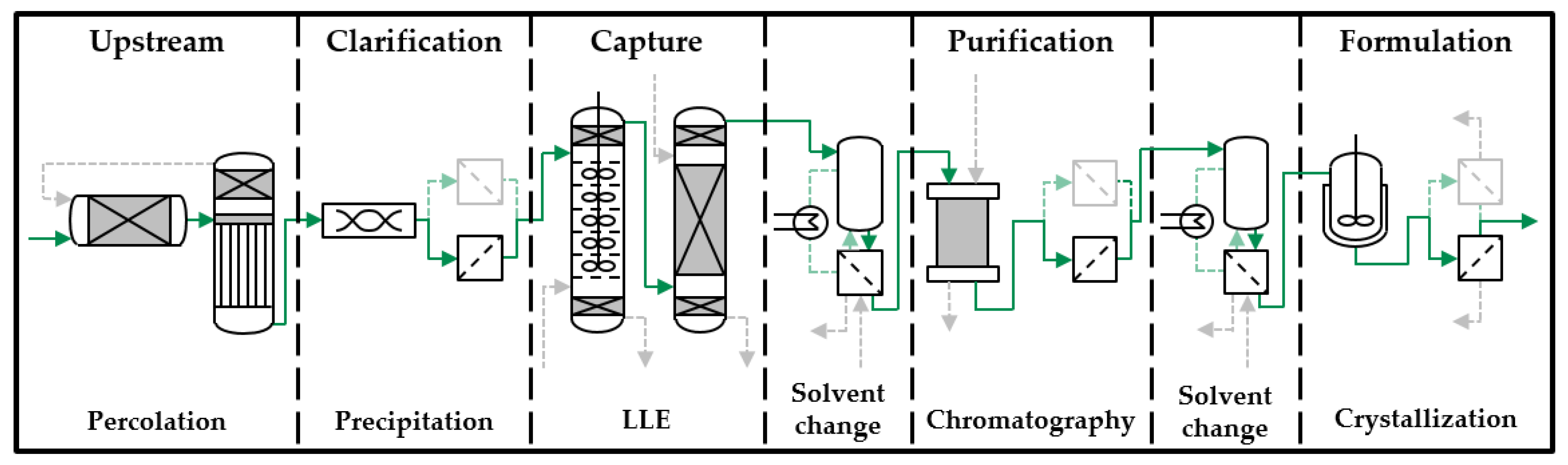
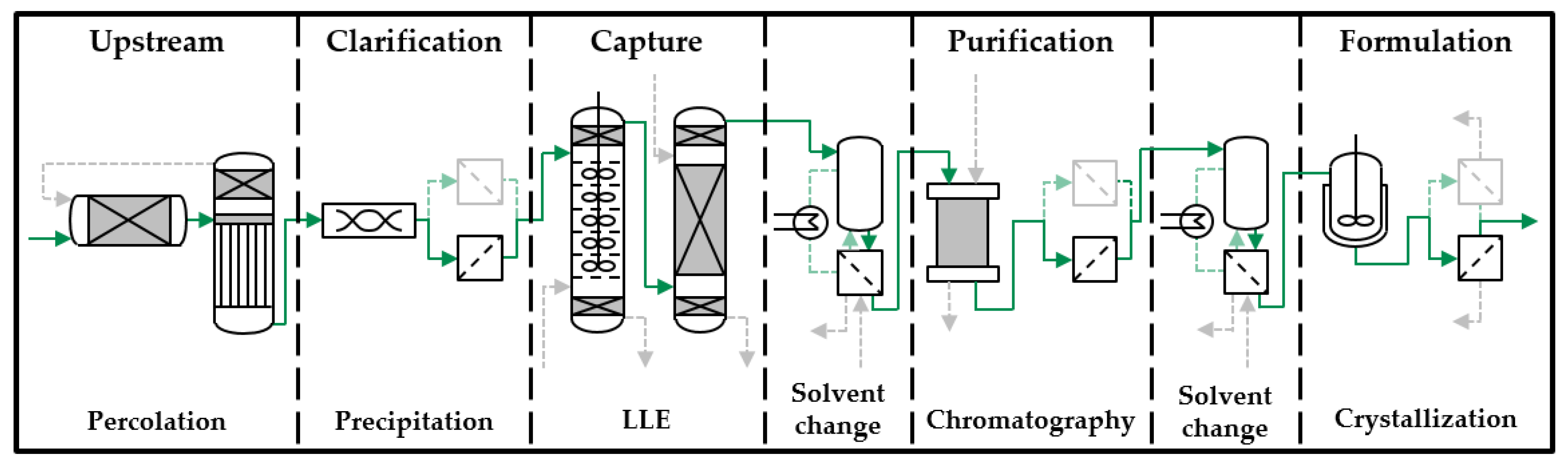
2. Model-Based Process Development Strategy
Distributed-Plug-Flow Model
3. Materials and Methods
3.1. Solid-Liquid Extraction and Precipitation
3.2. Shaking Experiments
3.3. Droplet Measurement
3.4. Batch-Settling Experiments
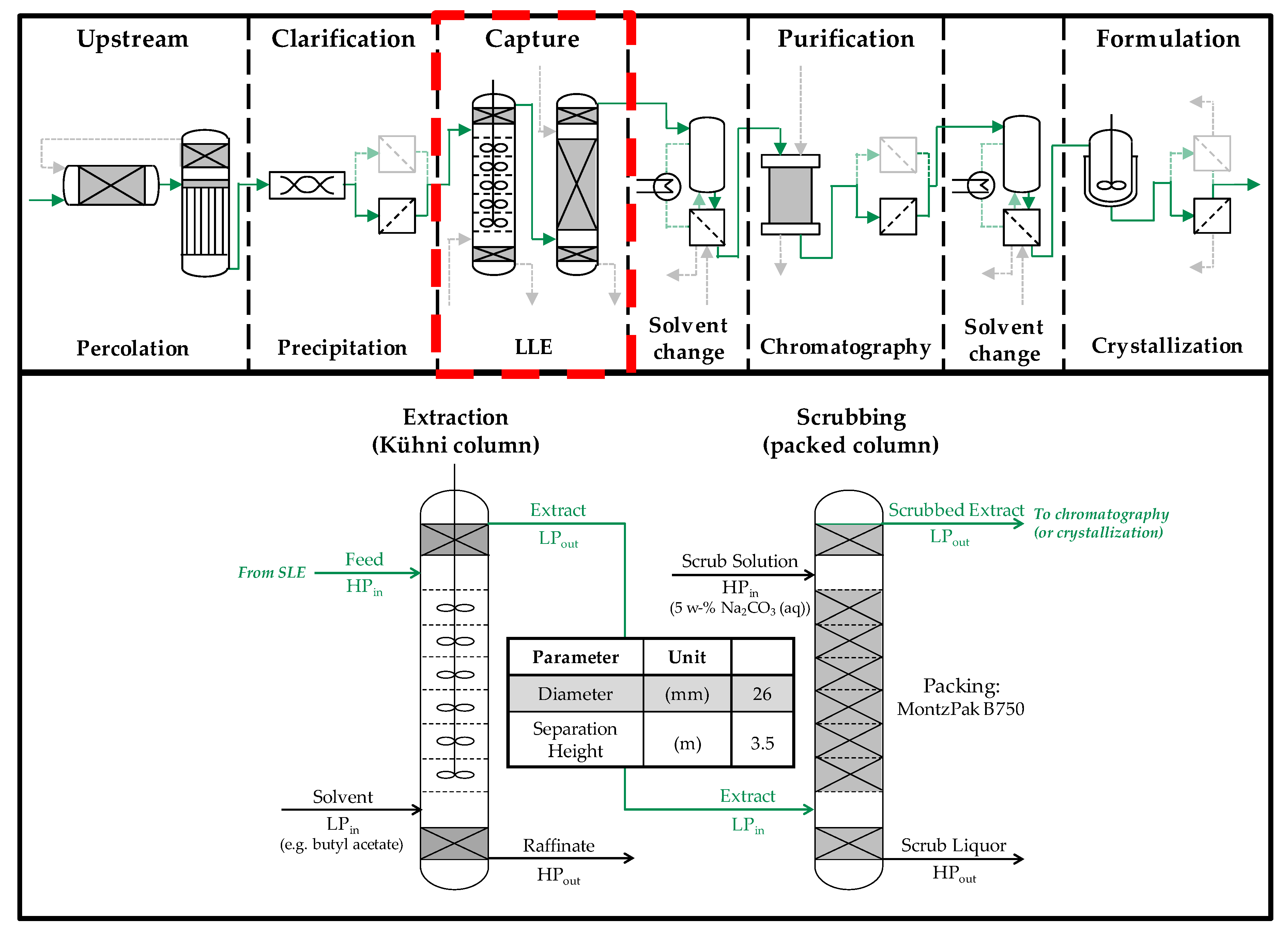
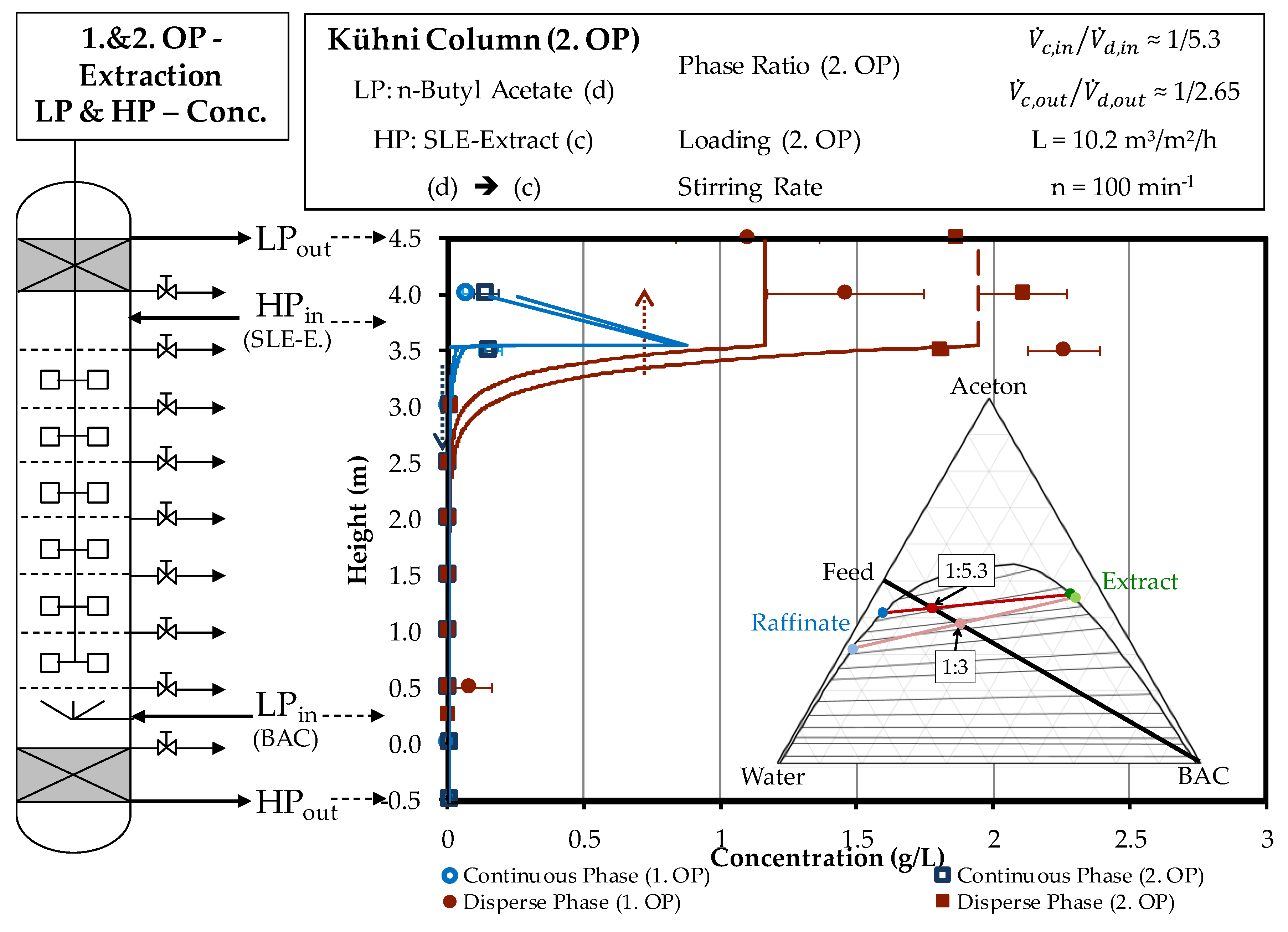
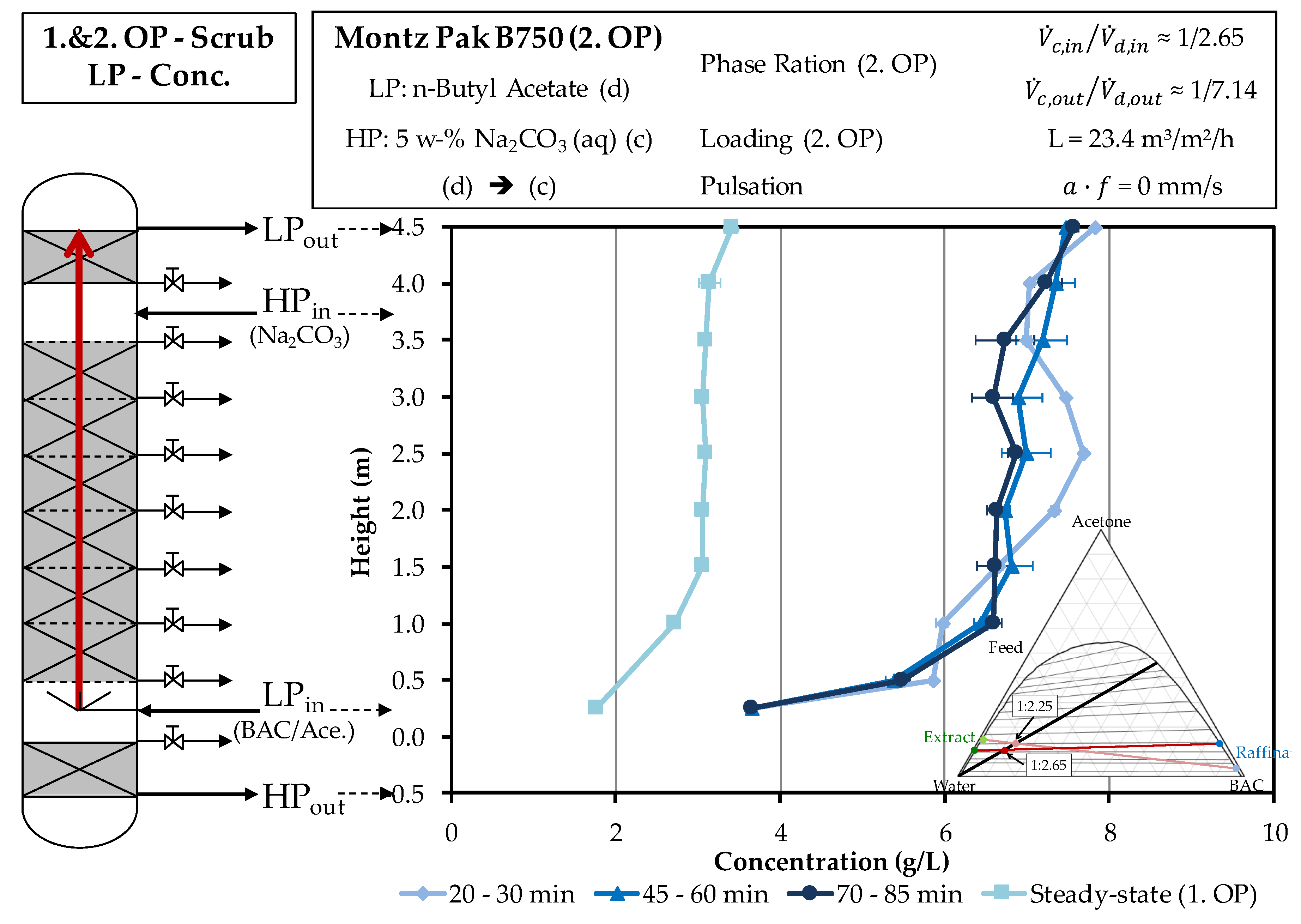
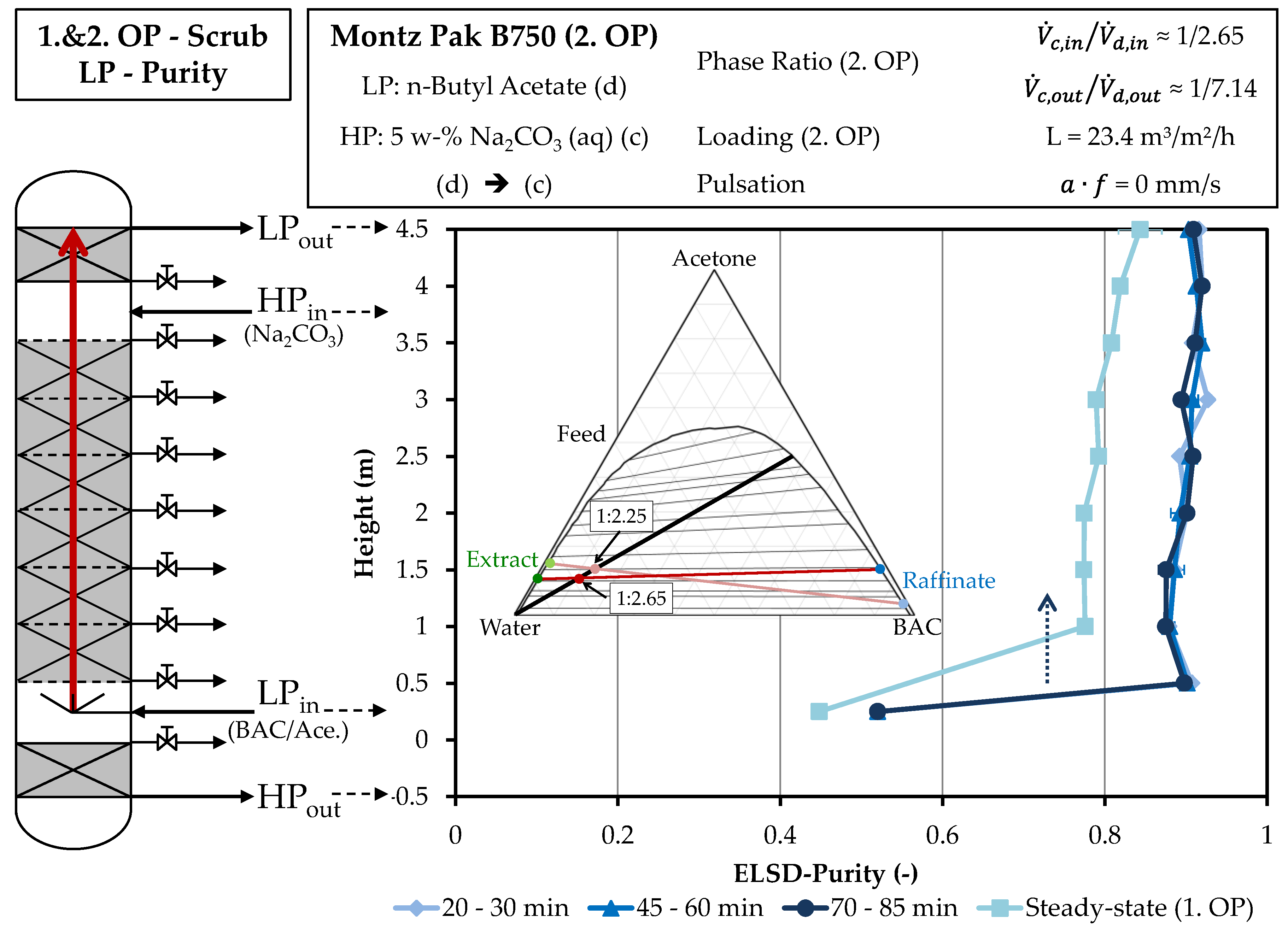
3.5. Column Experiments
3.6. Determination of the Concentration Profile
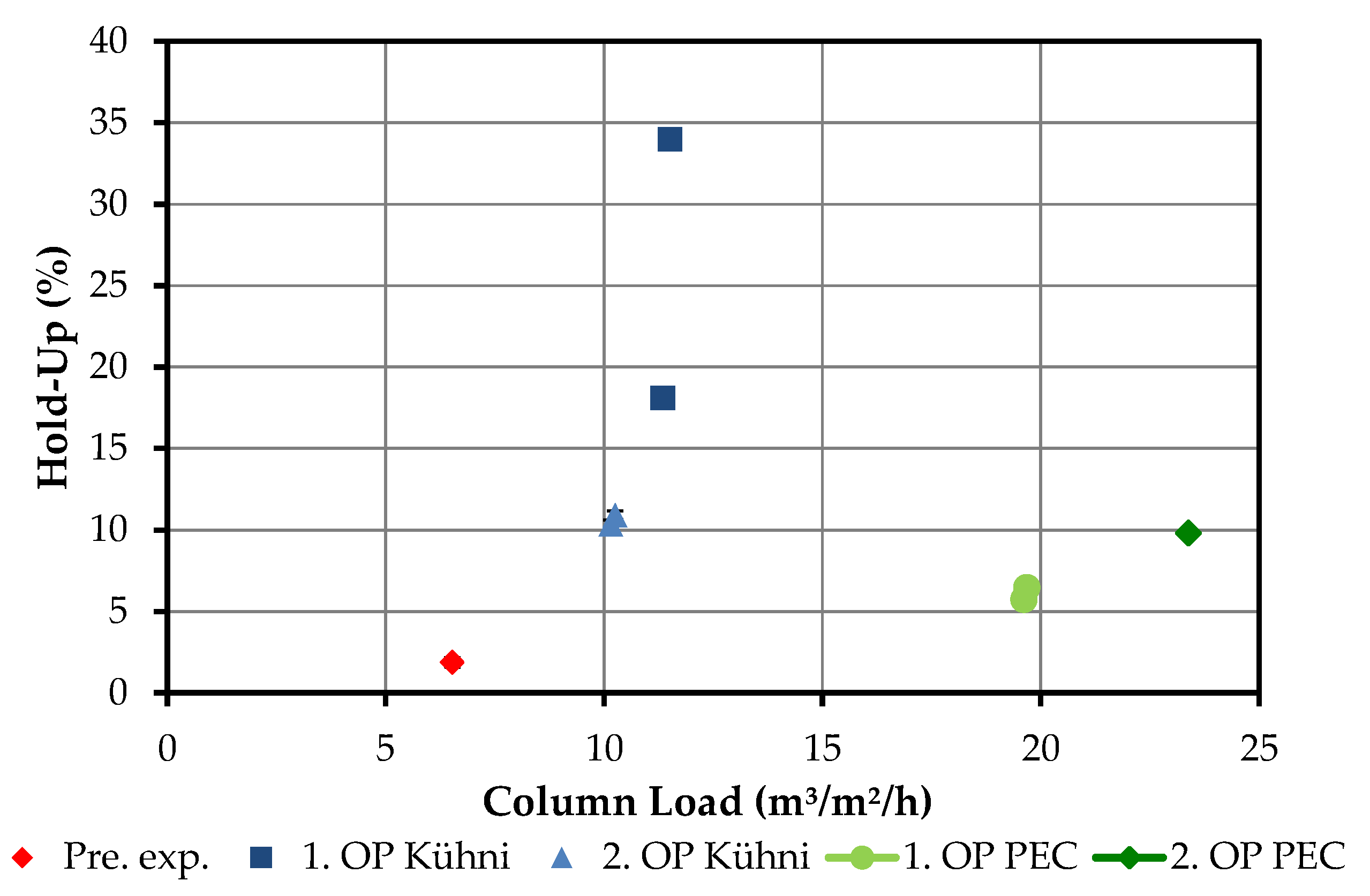
3.7. Determination of the Disperse Phase Hold-Up
3.8. Analytics
4. Results and Discussion
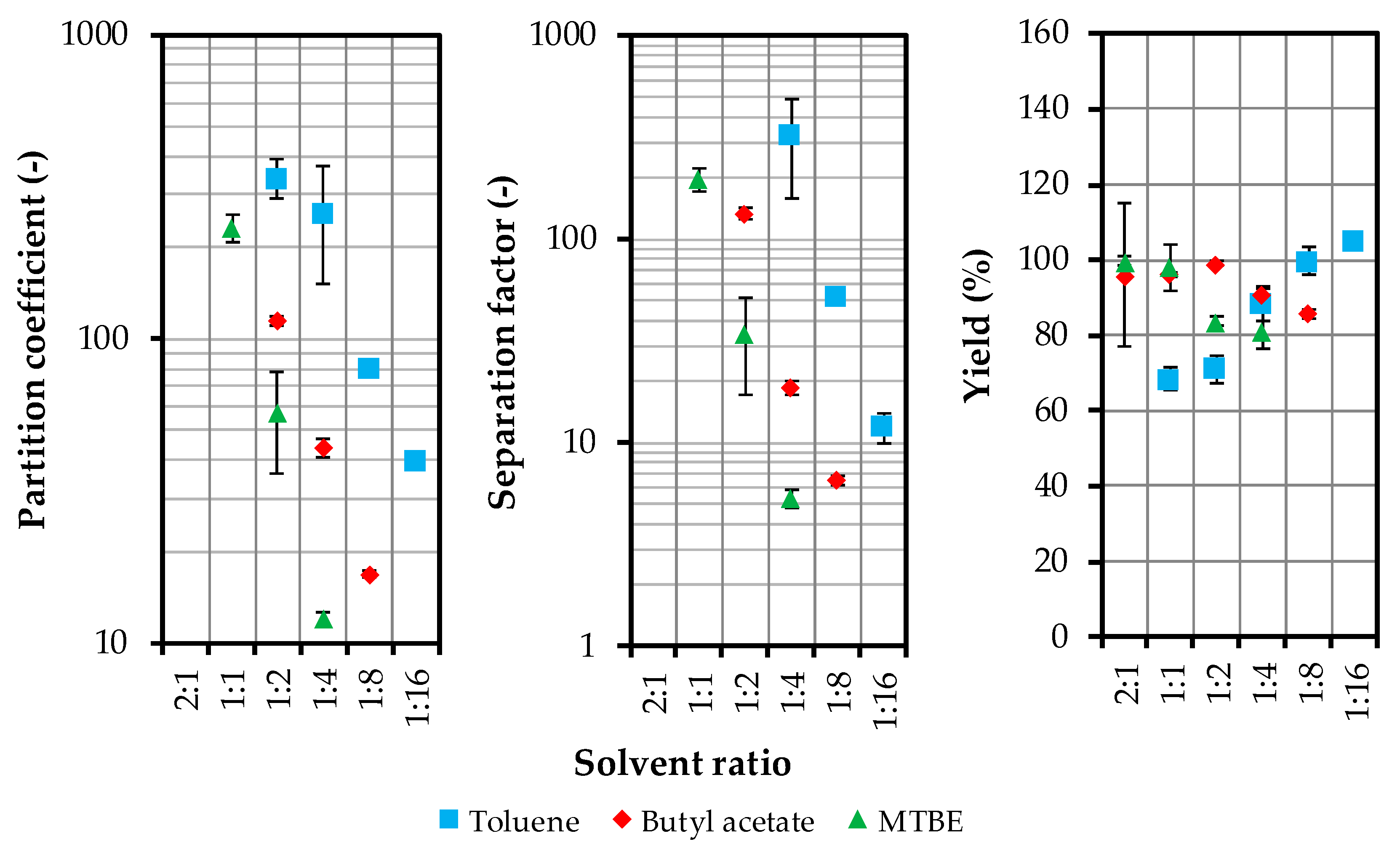
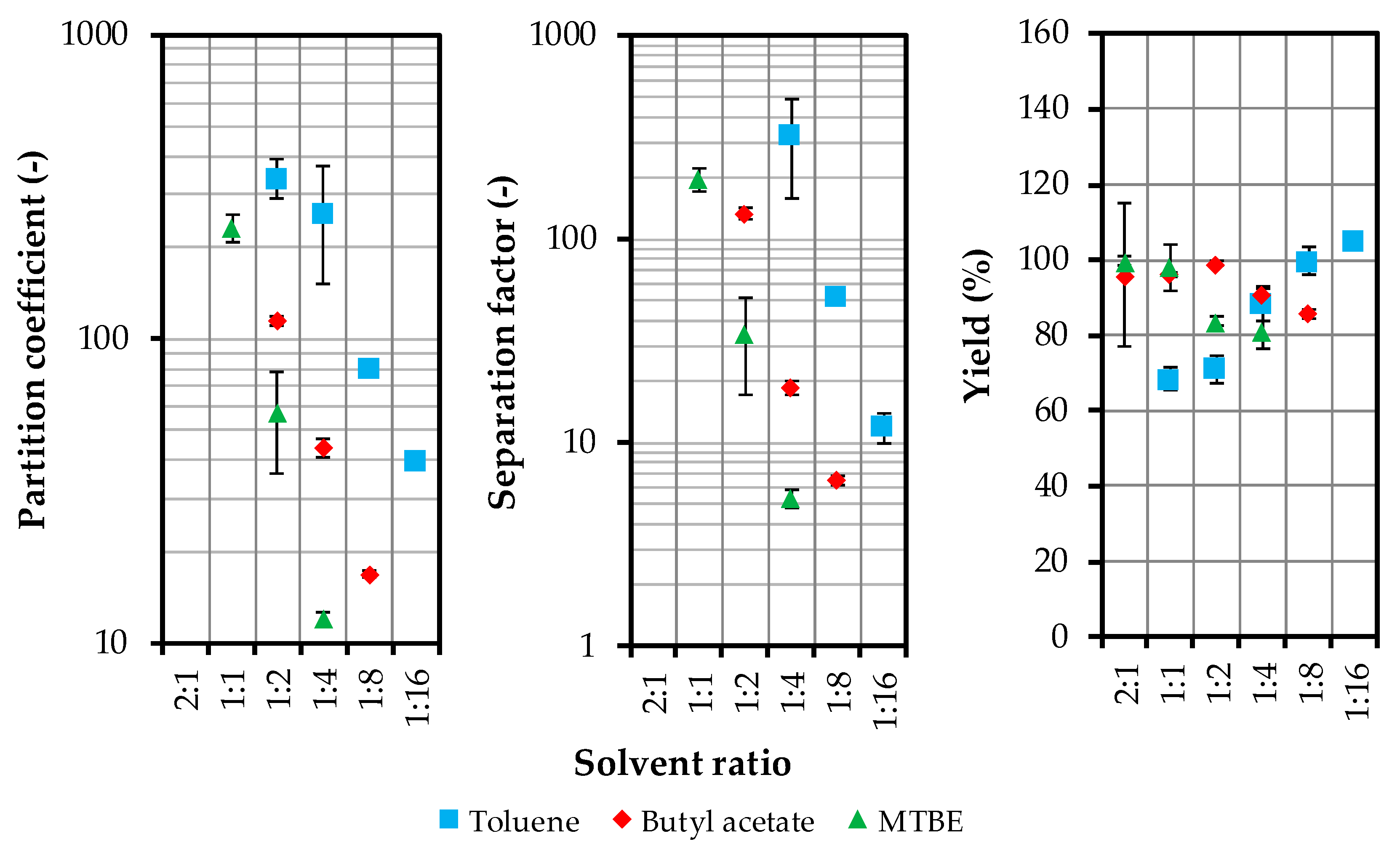
4.1. Solvent Selection and Model Parameter Determination
4.1.1. Distribution and Separation Factor
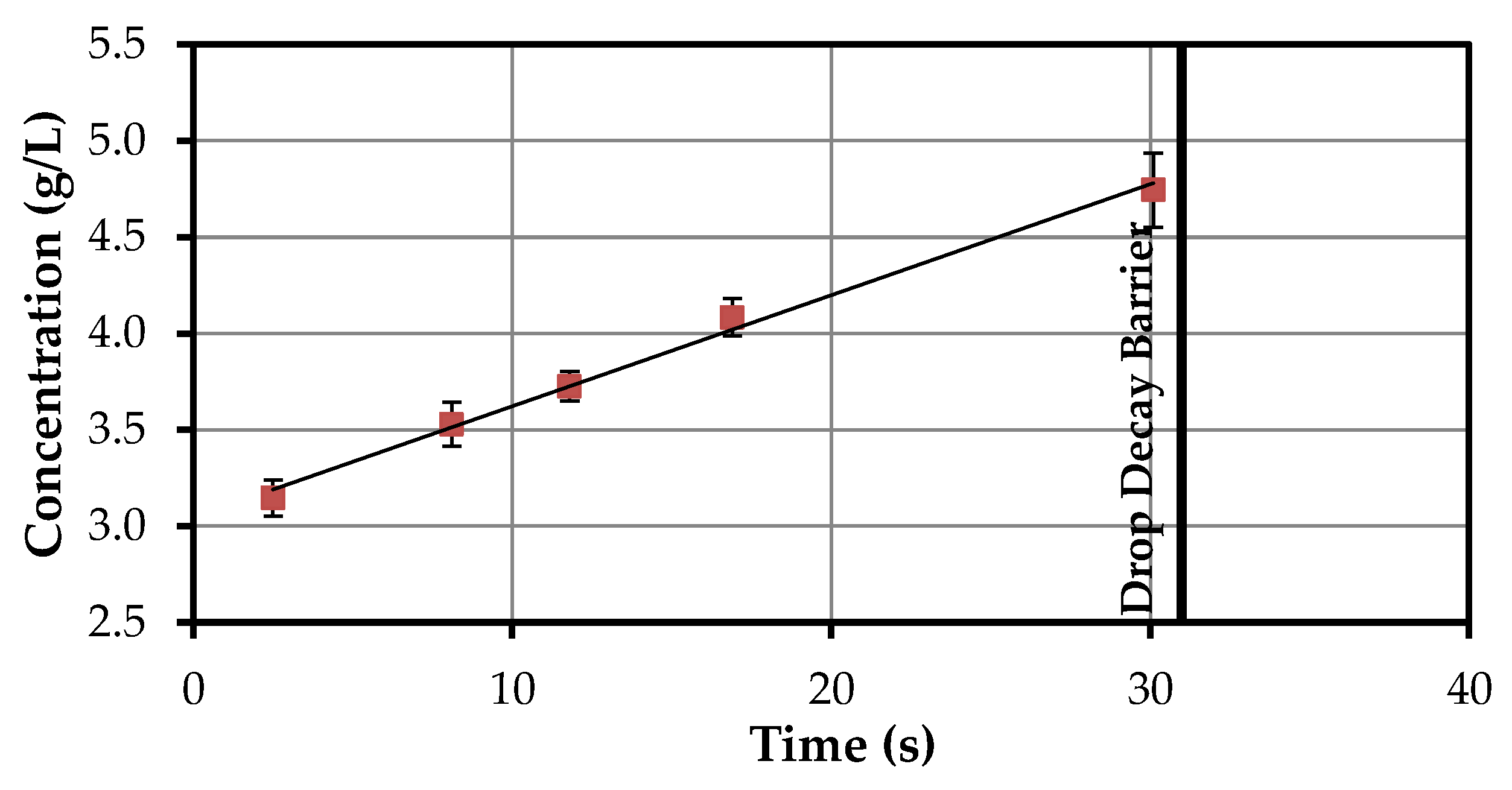
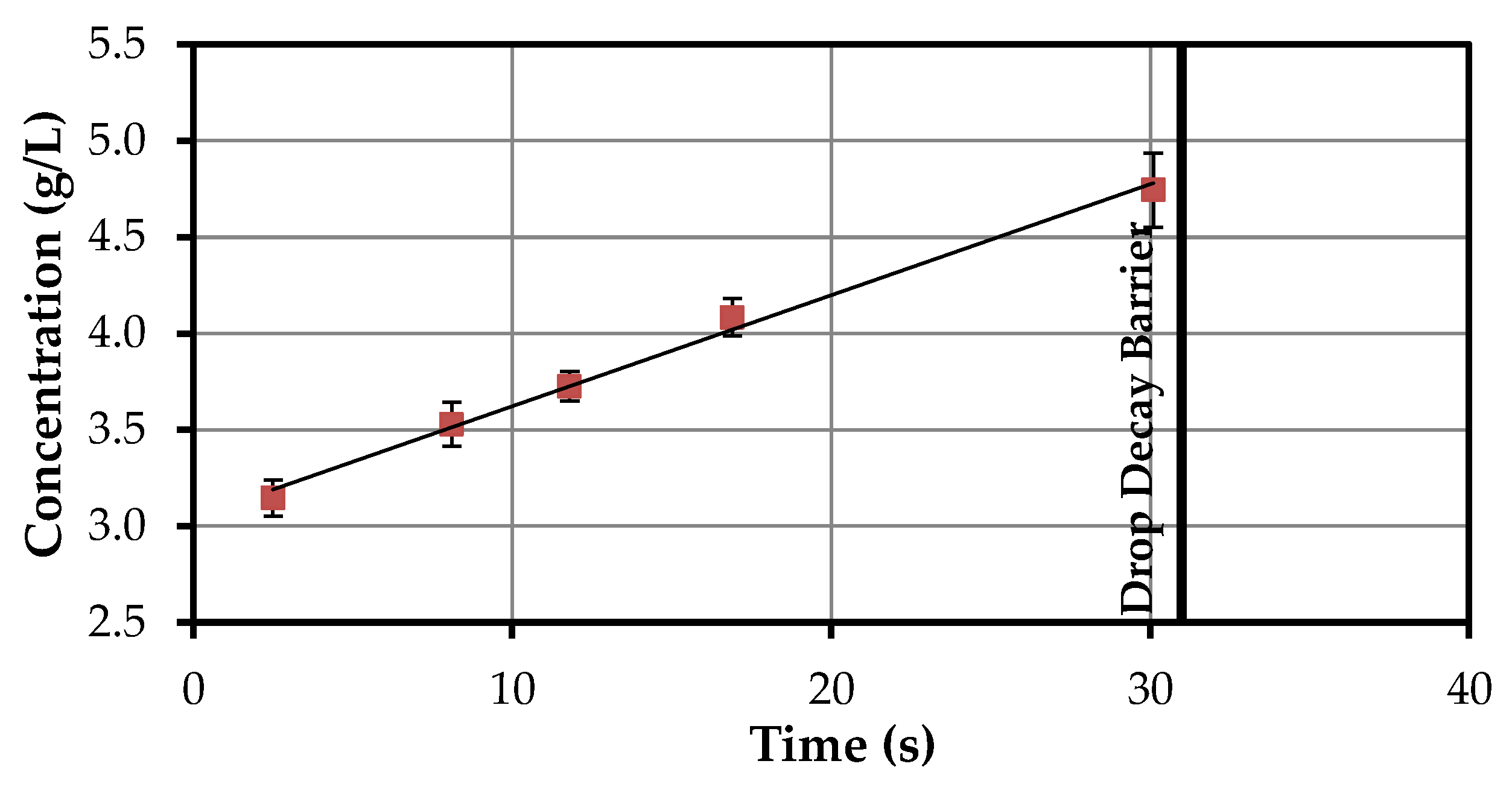
4.1.2. Volumetric Mass Transfer Coefficient
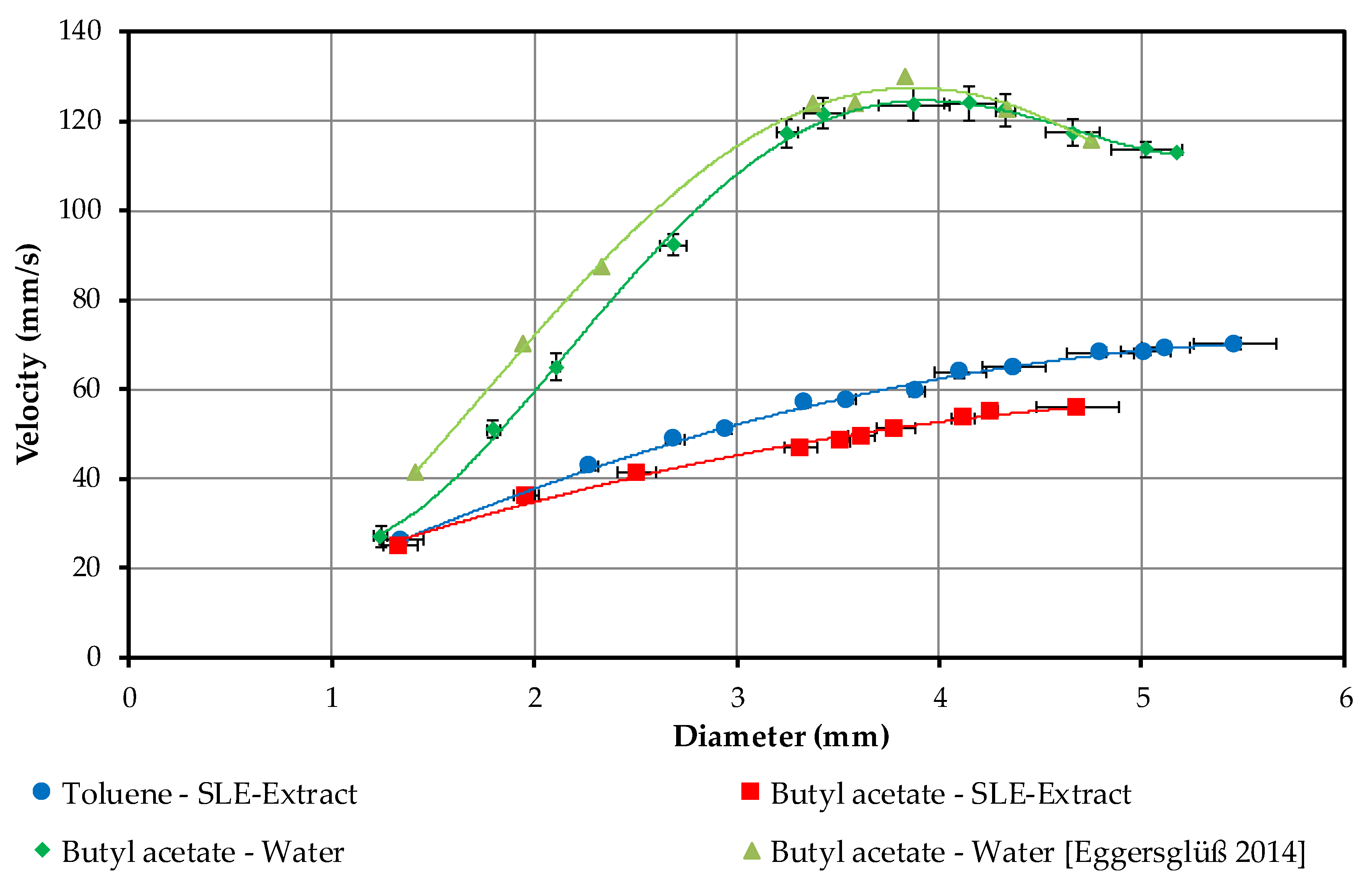
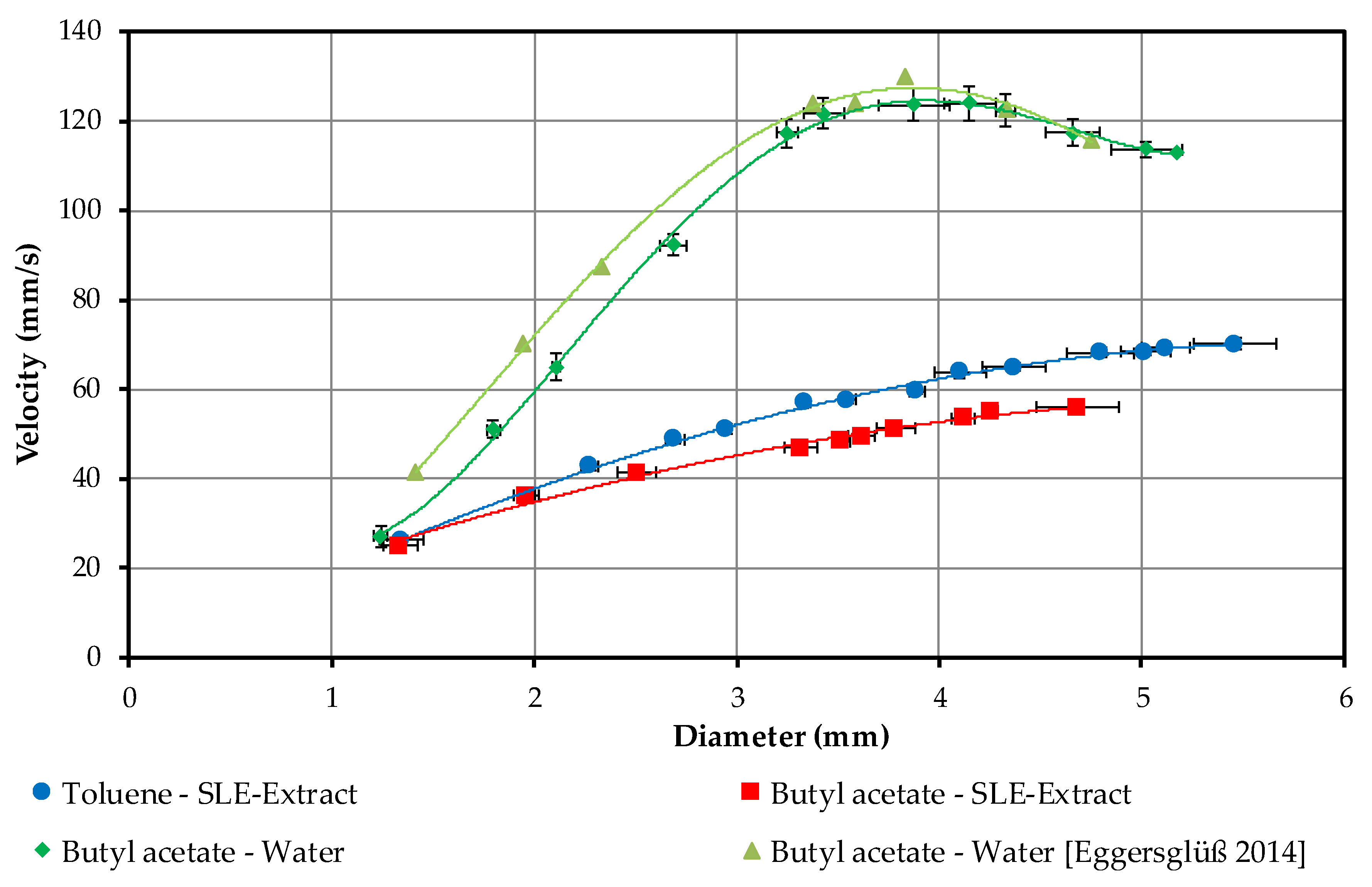
4.1.3. Droplet Sedimentation Velocity
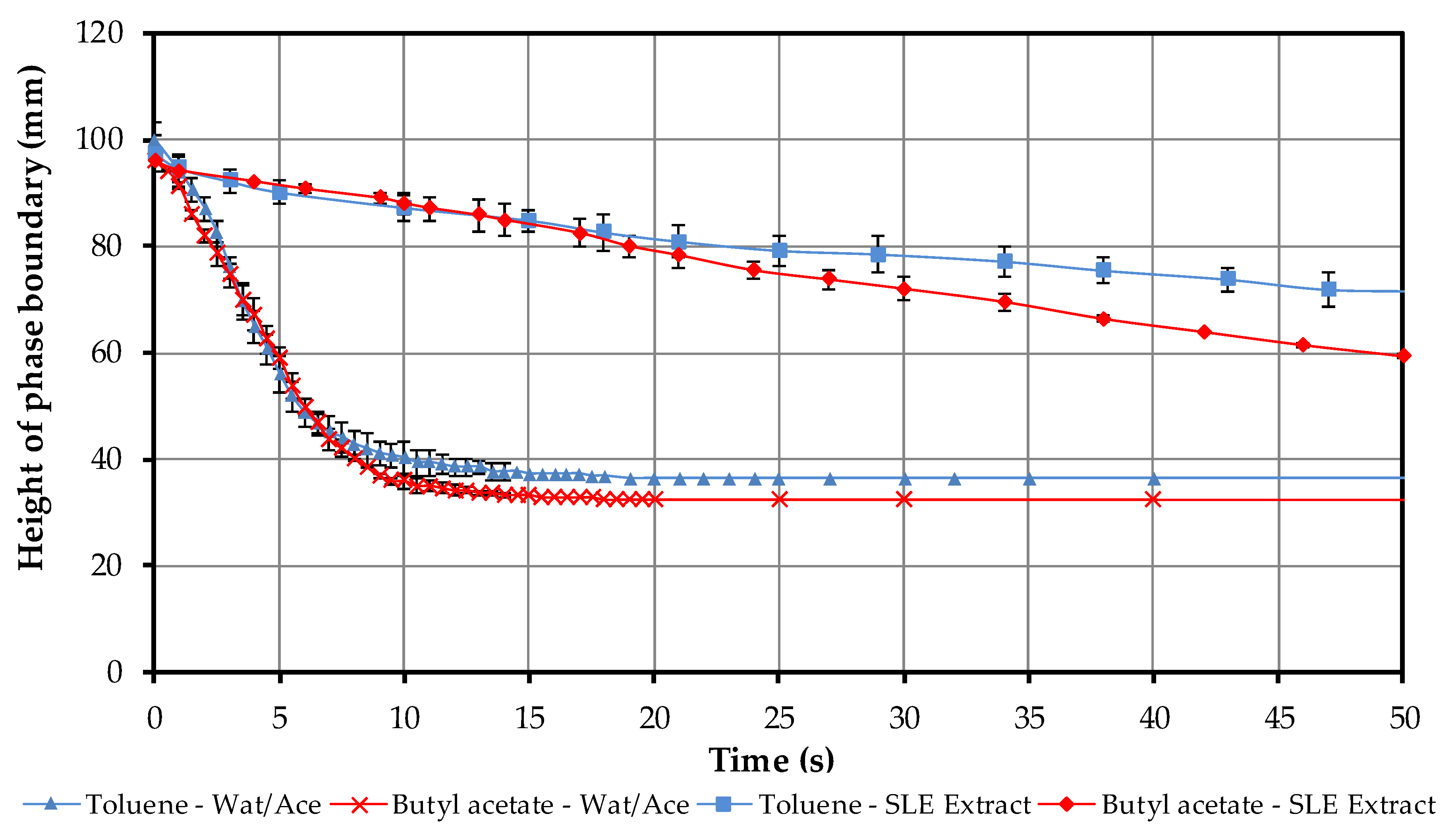
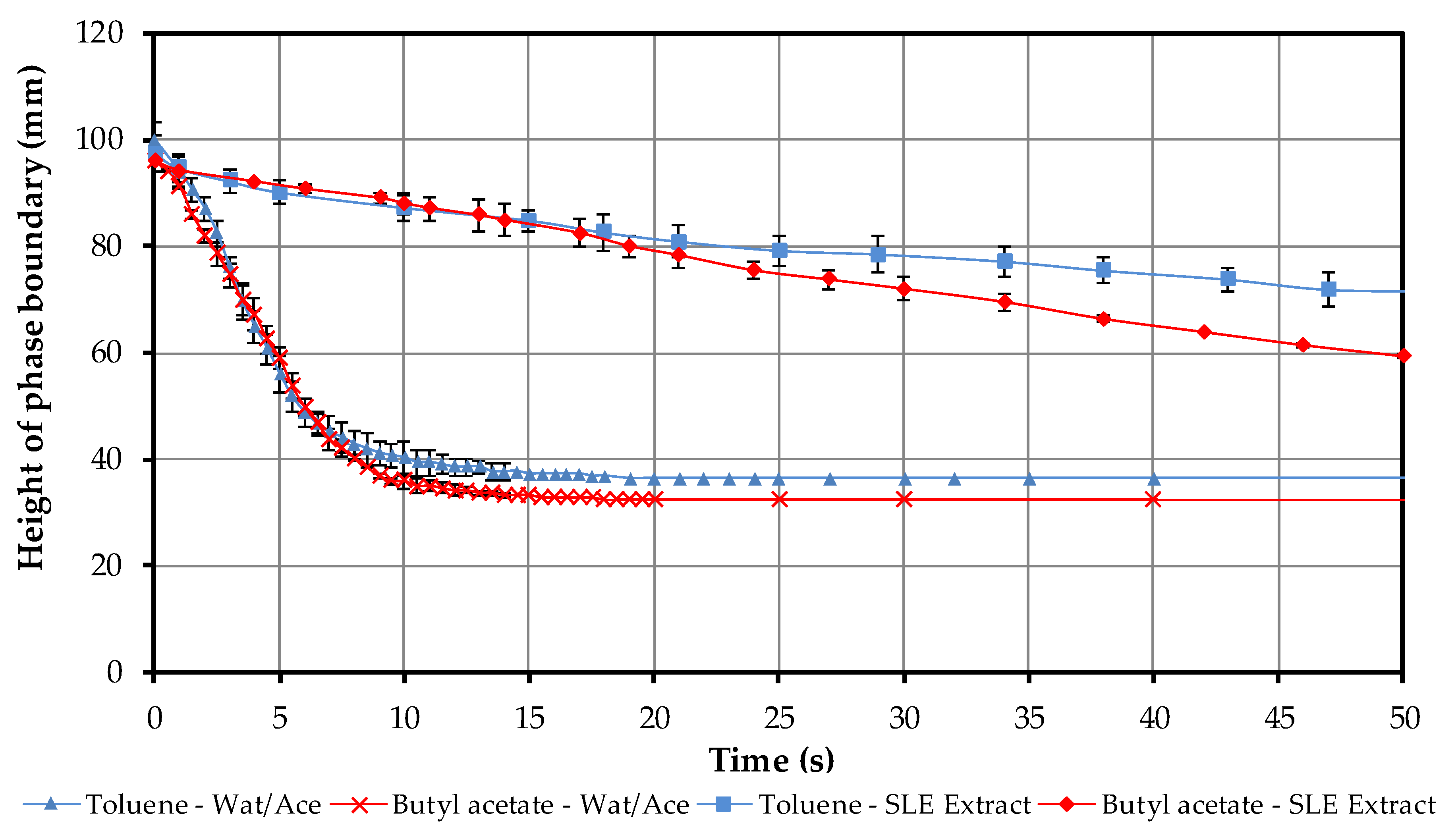
4.1.4. Batch-Settling Behavior
4.1.5. Model Calculations
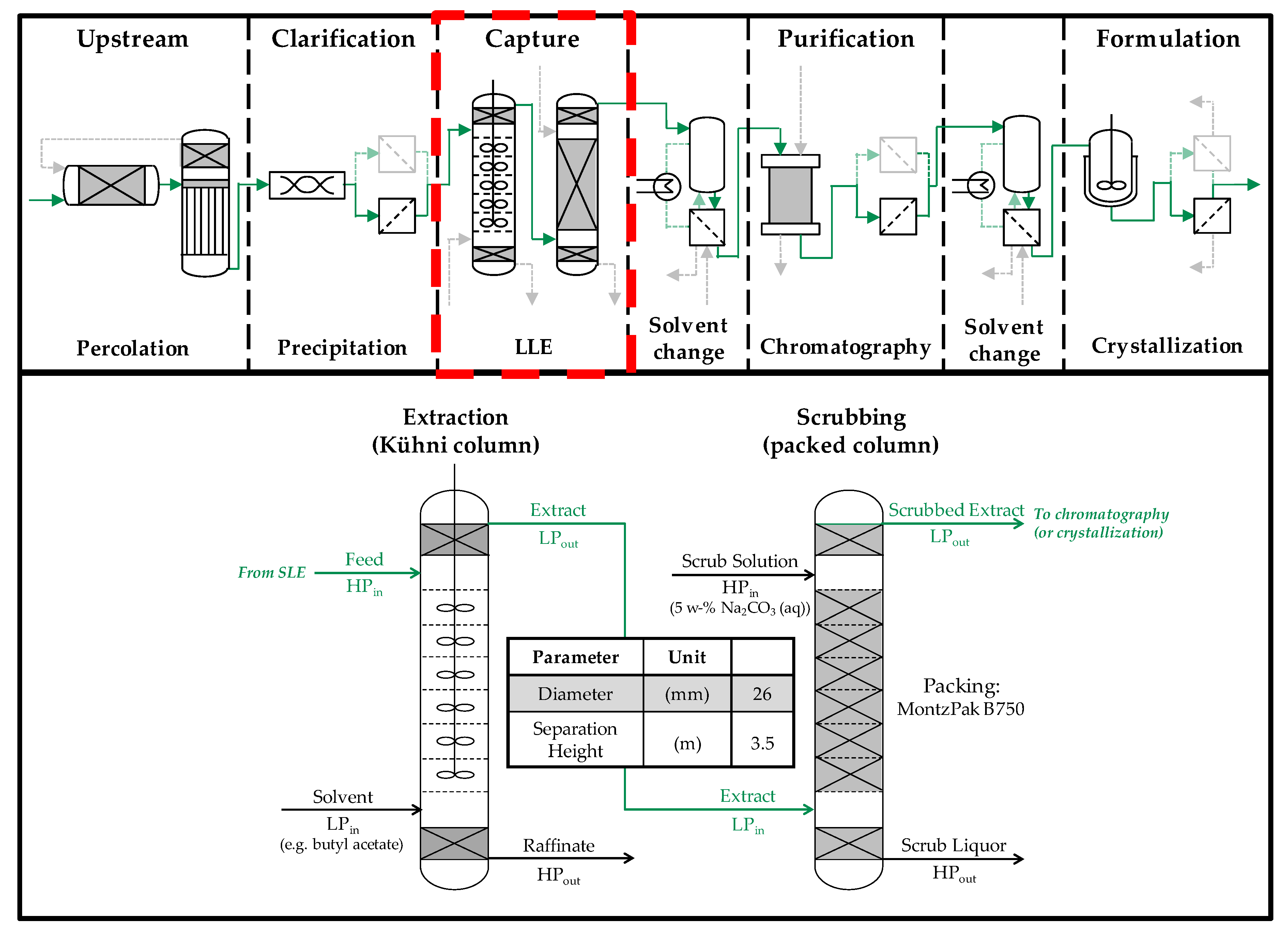
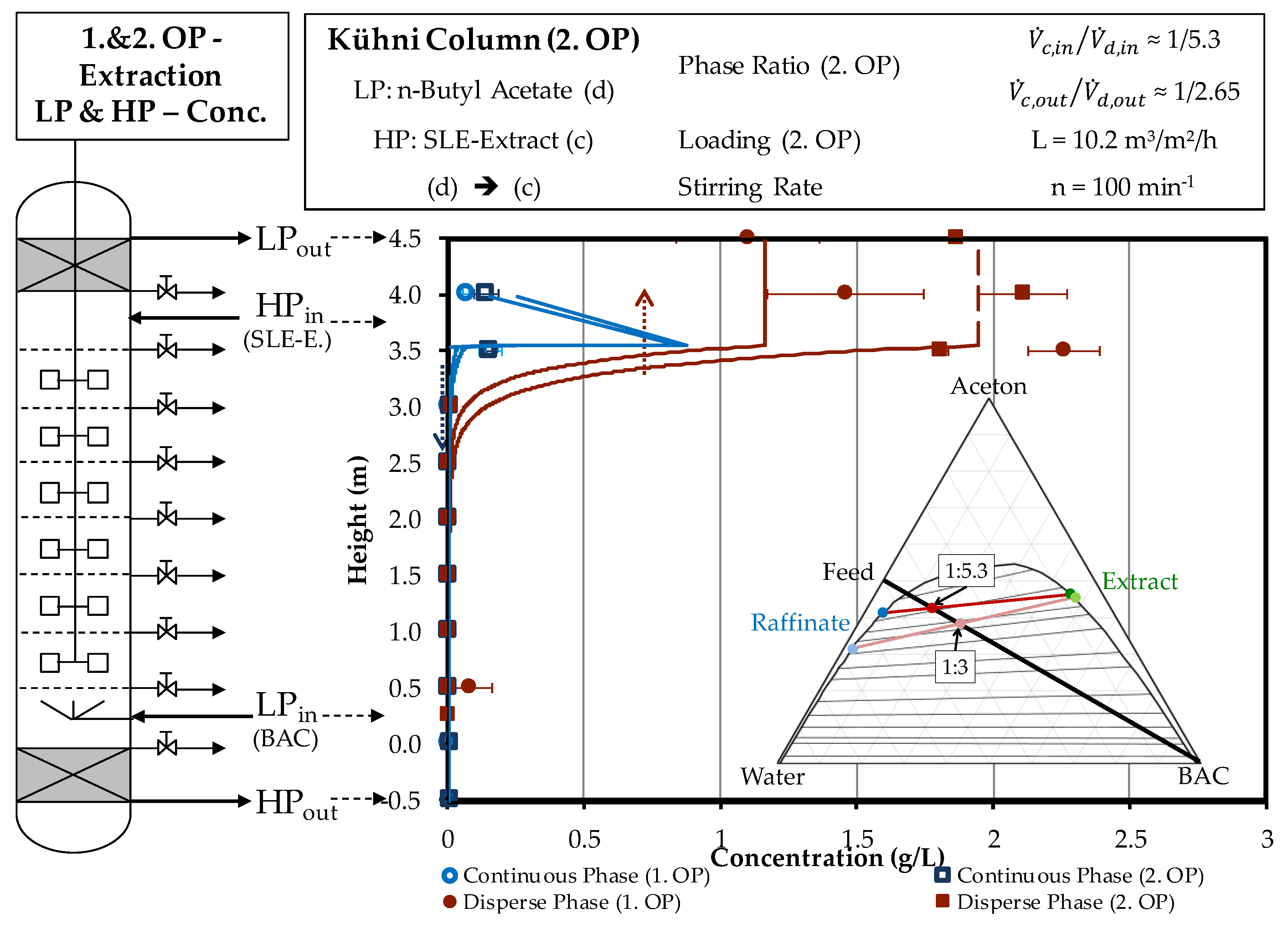
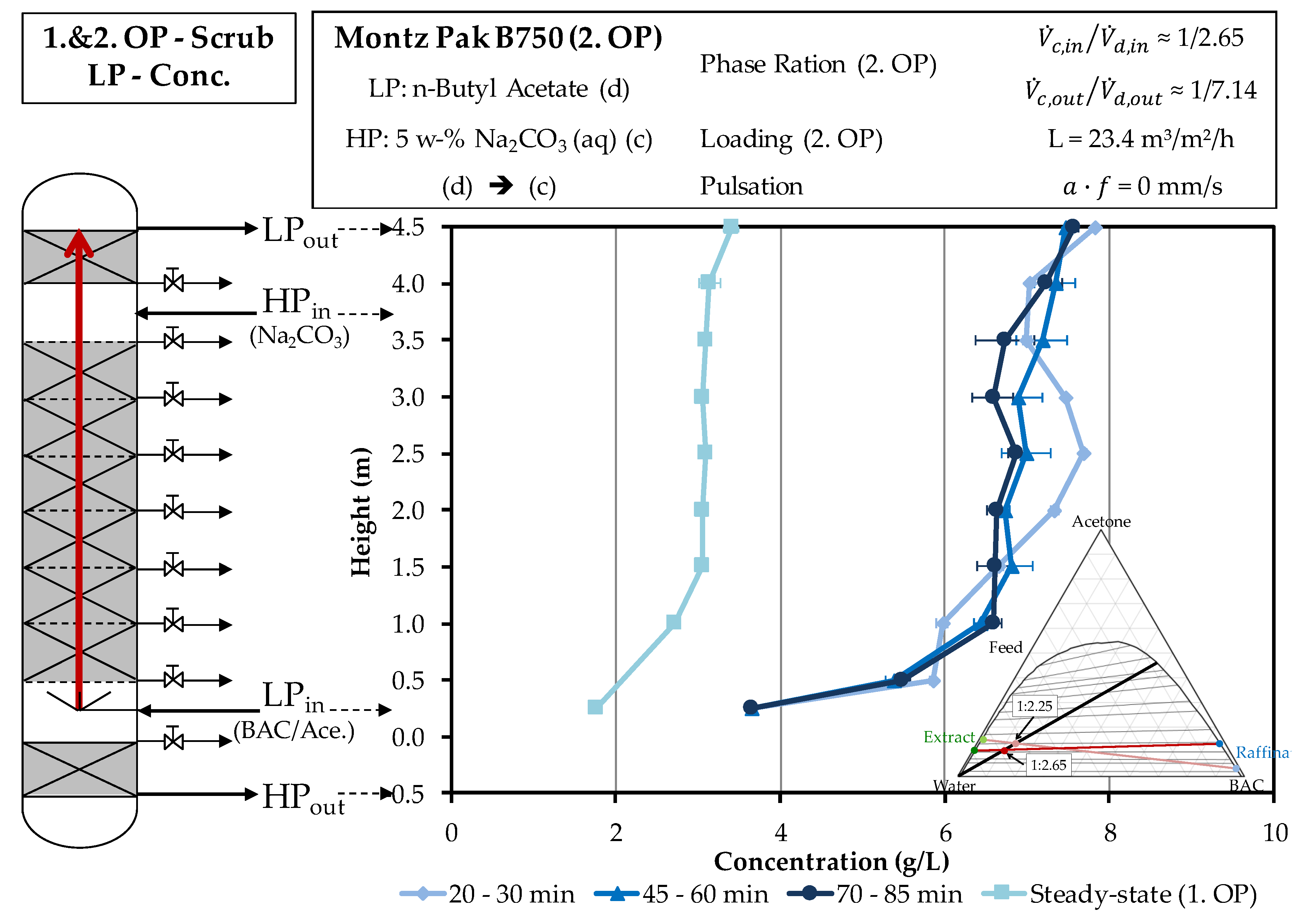
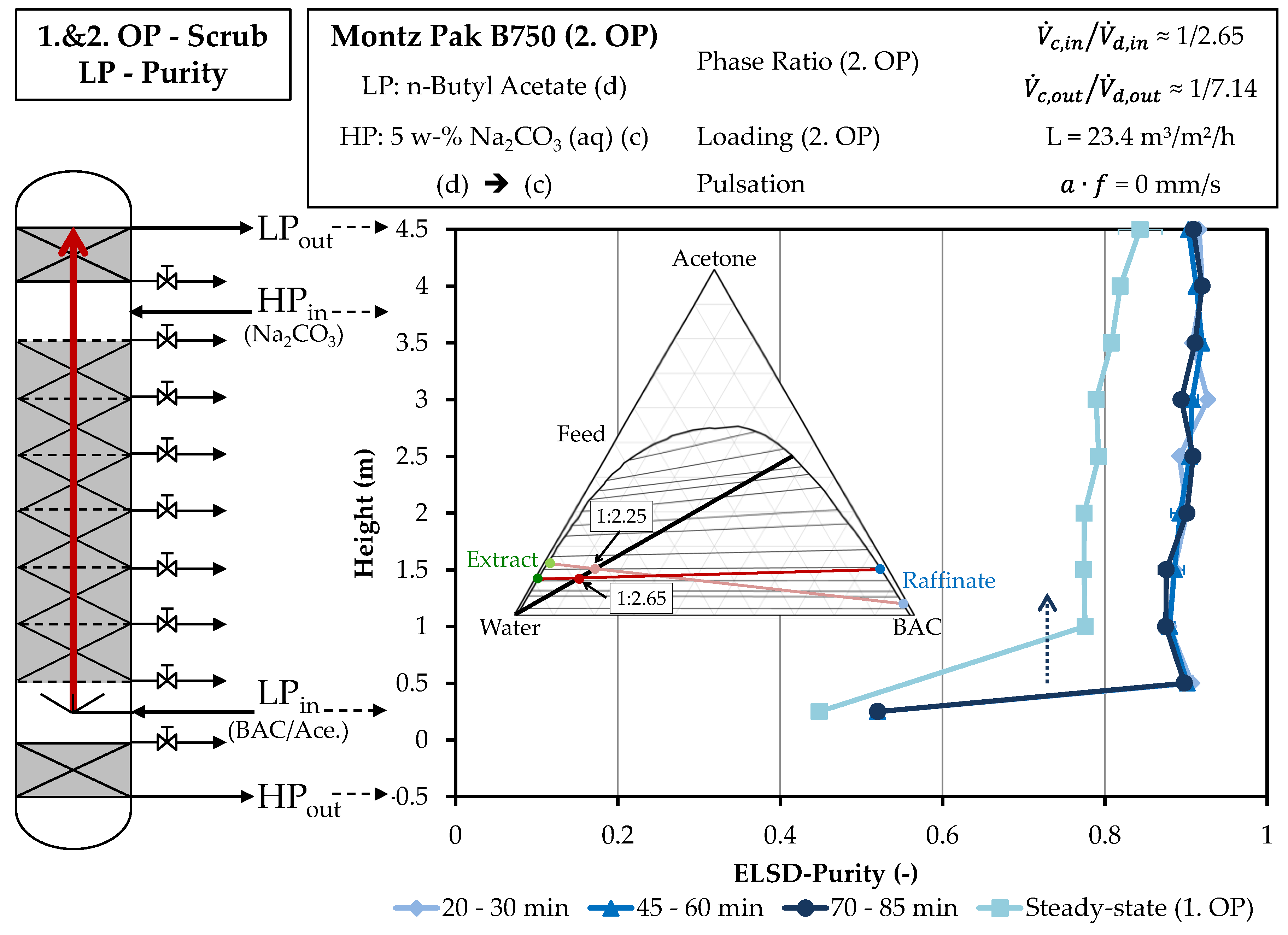
4.2. Column Experiments
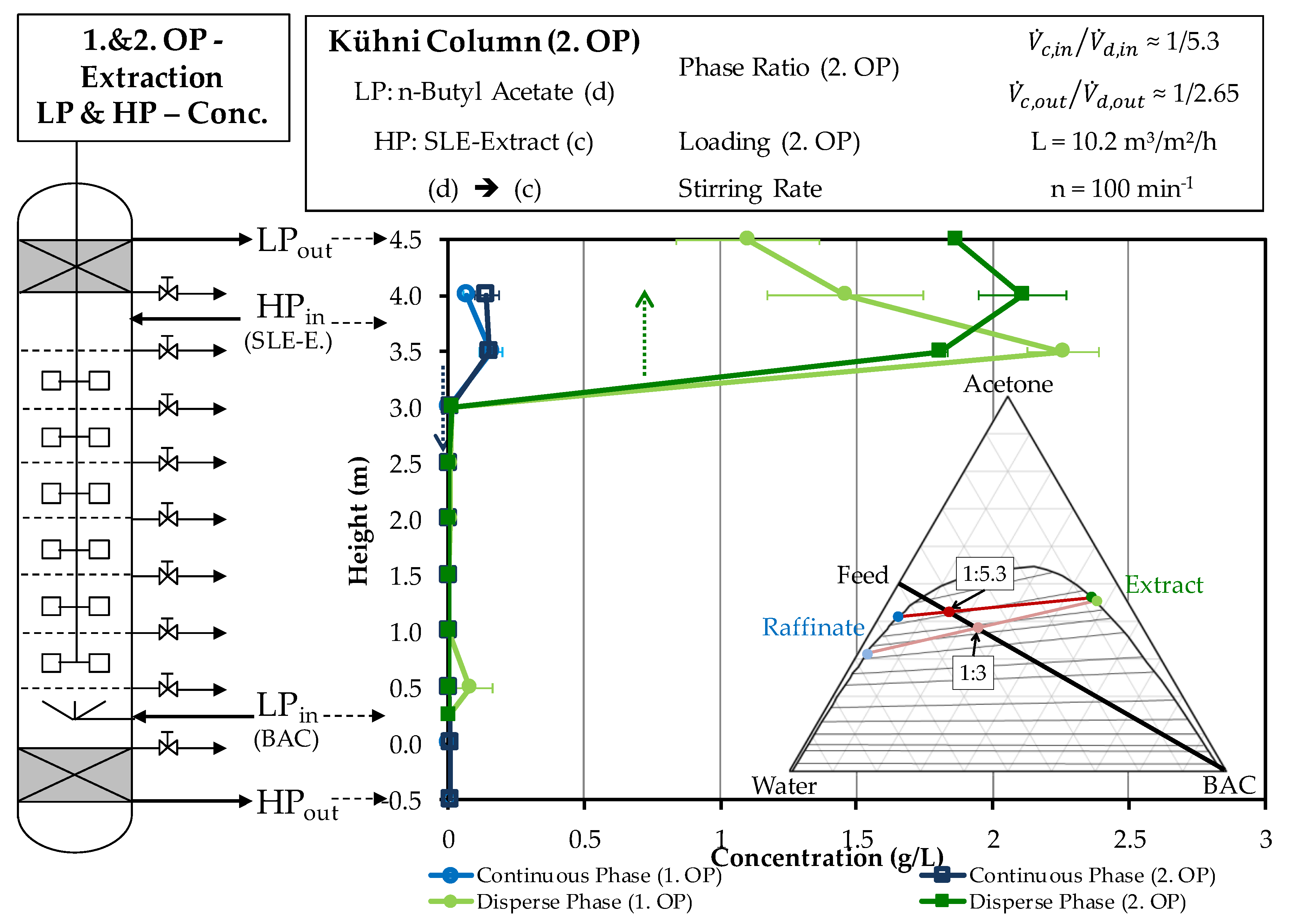
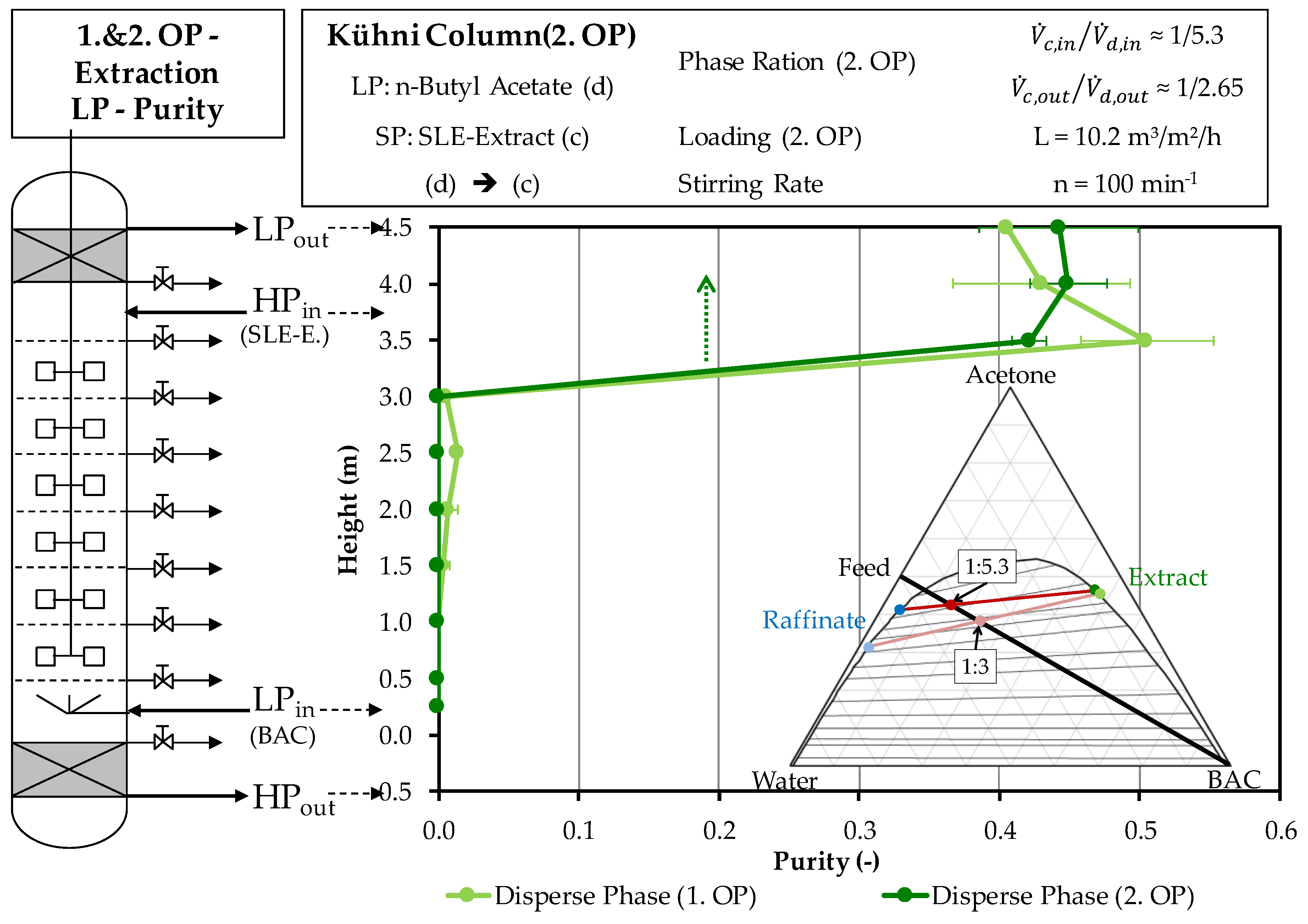
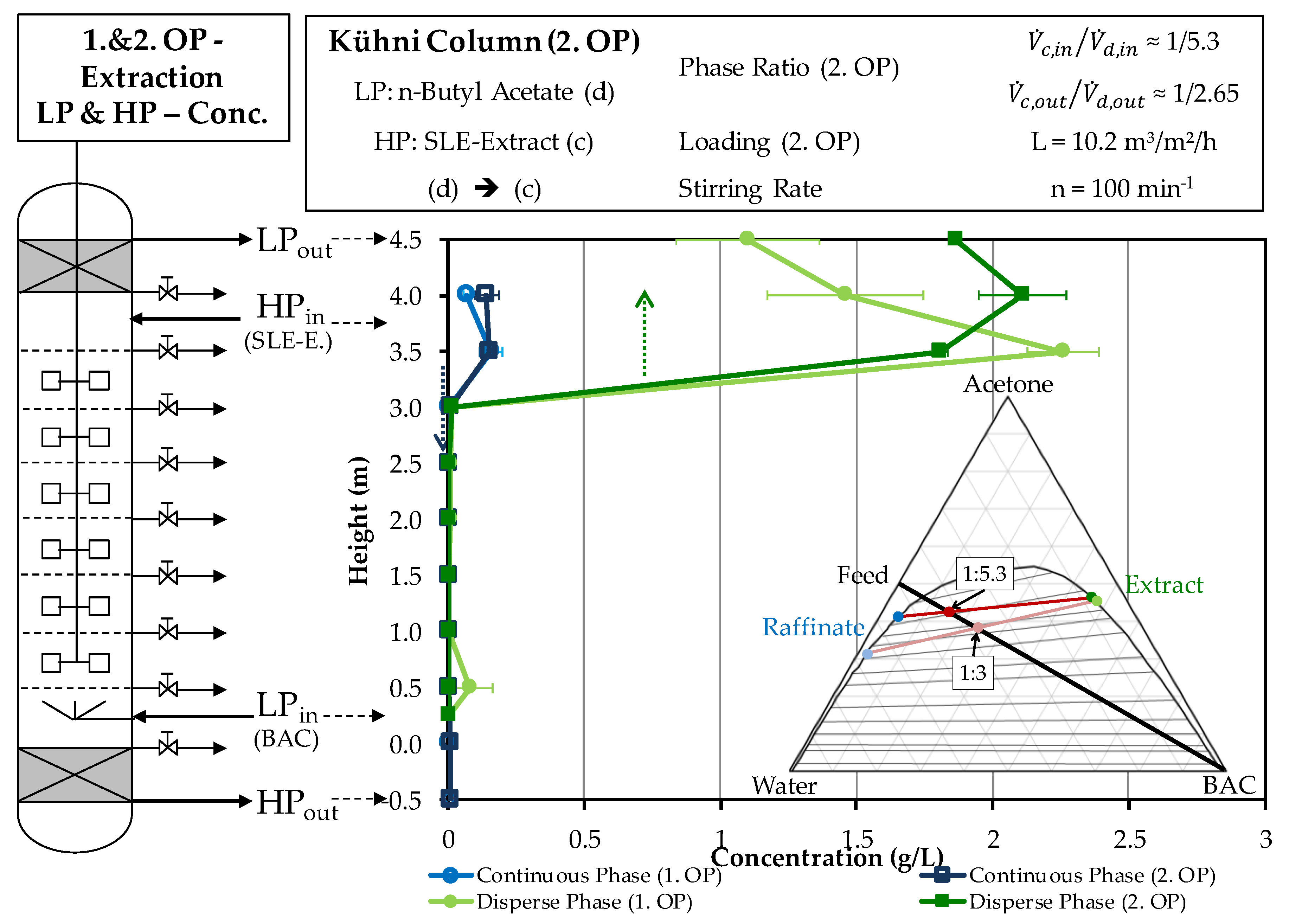
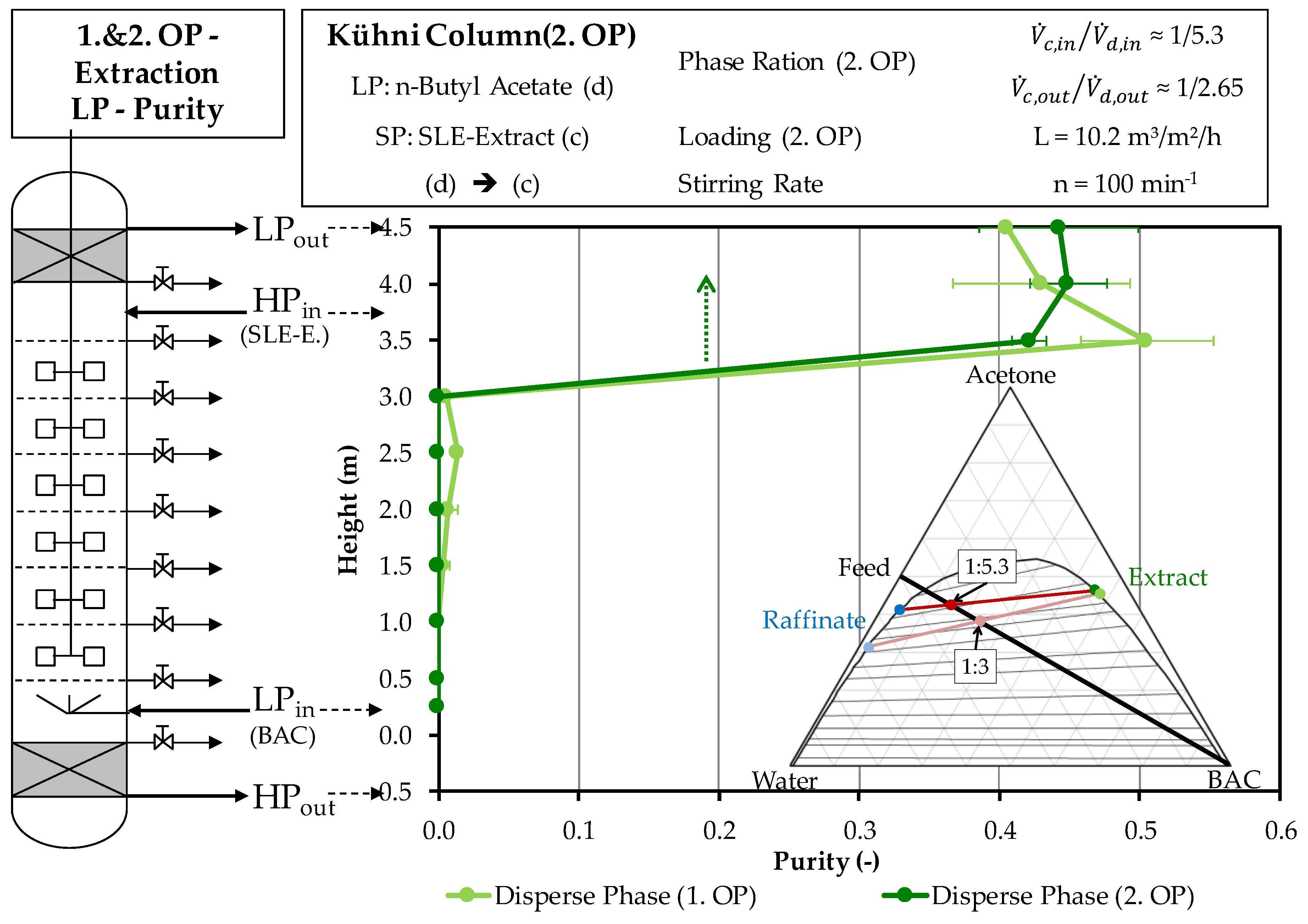
4.2.1. Extraction Column
4.2.2. Scrubbing Column
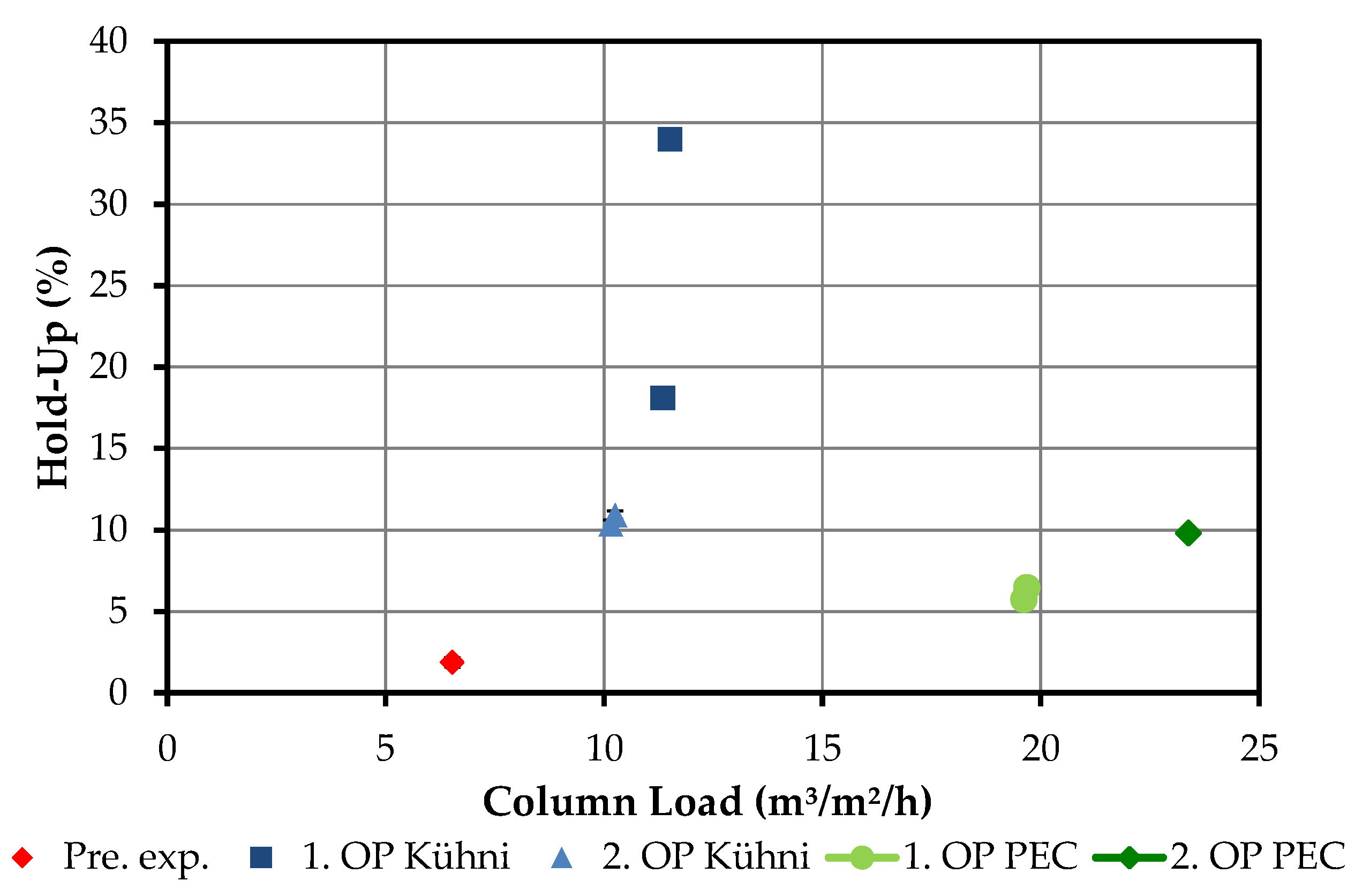
4.2.3. Hold-Up
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Litscher, D. Artemisinin. Traditionellen C hinesischen Medizin. Akupunktur Aurikulomedizin 2016, 28–31. [Google Scholar] [CrossRef]
- Dhingra, V.; Vishweshwar Rao, K.; Lakshmi Narasu, M. Artemisinin: Present status and perspectives. Biochem. Educ. 1999, 27, 105–109. [Google Scholar] [CrossRef]
- Bart, H.J.; Bäcker, W.; Bischoff, F.; Godecke, R.; Johannisbau, W.; Jordan, V.; Stockfleth, R.; Strube, J.; Wiesmet, V. Phytoextrakte–Produkte und Prozesse: Vorschlag für einen neuen, fachübergreifenden Forschungsschwerpunkt. Available online: https://dechema.de/dechema_media/Downloads/Positionspapiere/PP+Phytoextrakte+Okt_+2012-called_by-dechema-original_page-124930-original_site-dechema_eV-view_image-1-p-4254.pdf (accessed on 1 July 2018).
- WHO. World Malaria Report 2017; WHO: Geneva, Switzerland, 2017. [Google Scholar]
- Sixt, M.; Strube, J. Systematic and model-assisted evaluation of solvent based- or pressurized hot water extraction for the extraction of Artemisinin from Artemisia annua L. Processes 2017, 5, 86. [Google Scholar] [CrossRef]
- Sixt, M.; Schmidt, A.; Mestmäcker, F.; Huter, M.J.; Uhlenbrock, L.; Strube, J. Systematic and model-assisted process design for the extraction and purification of Artemisinin from Artemisia annua L.—Part I: Conceptual process design and cost estimation. Processes 2018, 6, 161. [Google Scholar] [CrossRef]
- Huter, M.J.; Schmidi, A.; Mestmäcker, F.; Sixt, M.; Strube, J. Systematic and model-assisted process design for the extraction and purification of Artemisinin from Artemisia annua L.—Part III: Chromatographic Purification. Processes 2018, 6, 180. [Google Scholar] [CrossRef]
- Mestmäcker, F; Schmidi, A.; Huter, M.J.; Sixt, M.; Strube, J. Systematic and model-assisted process design for the extraction and purification of Artemisinin from Artemisia annua L.—Part IV: Crystallization. Processes 2018, 6, 181. [Google Scholar] [CrossRef]
- Bart, H.-J.; Garthe, D.; Grömping, T.; Pfennig, A.; Schmidt, S.; Stichlmair, J. Vom Einzeltropfen zur Extraktionskolonne. Chem. Ing. Tech. 2006, 78, 543–547. [Google Scholar] [CrossRef]
- Buchbender, F.; Schmidt, M.; Steinmetz, T.; Pfennig, A. Simulation von Extraktionskolonnen in der industriellen Praxis. Chem. Ing. Tech. 2012, 84, 540–546. [Google Scholar] [CrossRef]
- Schmidt, A.; Richter, M.; Rudolph, F.; Strube, J. Integration of aqueous two-phase extraction as cell harvest and capture operation in the manufacturing process of monoclonal antibodies. Antibodies 2017, 6, 21. [Google Scholar] [CrossRef]
- Eggersgluess, J.; Wellsandt, T.; Strube, J. Integration of aqueous two-phase extraction into downstream processing. Chem. Eng. Technol. 2014, 37, 1686–1696. [Google Scholar] [CrossRef]
- Eggersgluess, J.K.; Both, S.; Strube, J. Process development for the extraction of biomolecules application for downstream processing of proteins in aqueous two-phase systems. Chim. Oggi-Chem. Today 2012, 30, 32–36. [Google Scholar]
- Eggersgluess, J.K.; Richter, M.; Dieterle, M.; Strube, J. Multi-stage aqueous two-phase extraction for the purification of monoclonal antibodies. Chem. Eng. Technol. 2014, 37, 675–682. [Google Scholar] [CrossRef]
- Wellsandt, T.; Stanisch, B.; Strube, J. Efficient liquid/liquid extraction by a milli device based on hydrophobic membranes. Chem. Ing. Tech. 2015, 87, 1053. [Google Scholar] [CrossRef]
- Wellsandt, T.; Stanisch, B.; Strube, J. Characterization Method for Separation Devices Based on Micro Technology. Chem. Ing. Tech. 2015, 87, 150–158. [Google Scholar] [CrossRef]
- Helling, C.; Strube, J. Future processing and recycling strategies for rare earths. Chem. Ing. Tech. 2013, 85, 1272–1281. [Google Scholar] [CrossRef]
- Chartes, R.H.; Korchinsky, W.J. Modelling of liquid-liquid extraction columns predicting the influence of drop size distribution. Trans. Inst. Chem. Eng. 1975, 53, 247–254. [Google Scholar]
- Casamatta, G.; Vogelpohl, A. Modellierung der Fluiddynamik und des Stoffübergangs in Extraktionskolonnen. Chem. Ing. Tech. 1984, 56, 230–231. [Google Scholar] [CrossRef]
- Steiner, L. Rechnerische Erfassung der Arbeitsweise von FlüSsig-FlüSsig Extraktionskolonnen; Fortschritt-Berichte VDI: Düsseldorf, Germany, 1988. [Google Scholar]
- Houghton, P.A.; Madurawe, R.U.; Hatton, T.A. Convective and dispersion models for dispersed phase axial mixing—The significance of polydispersity effects in liquid-liquid contactors. Chem. Eng. Sci. 1988, 43, 617–639. [Google Scholar] [CrossRef]
- Leistner, J. Modellierung und Simulation Physikalischer, Dissoziativer und Reaktiver Extraktionsprozesse; Zugl.: Dortmund, Univ., Diss., 2004; Shaker: Aachen, Germany, 2005. [Google Scholar]
- Leistner, J.; Górak, A.; Bäcker, W.; Strube, J. Extraktion als mittel zur abwasseraufarbeitung. Chem. Ing. Tech. 2002, 74, 606. [Google Scholar] [CrossRef]
- Leistner, J.; Goerge, A.; Baecker, W.; Górak, A.; Strube, J. Modelling methodology for the simulation of solvent extraction processes–cobalt/nickel. In Proceedings of the International Solvent Extraction Conference, South Africa, 17–21 March 2002; pp. 958–963. [Google Scholar]
- Kolb, P. Hydrodynamik und Stoffaustausch in Einem Gerührten Miniplantextraktor der Bauart Kühni; Technische Universität Kaiserslautern: Kaiserslautern, Germany, 2004. [Google Scholar]
- Steinmetz, T. Tropfenpopulationsbilanzgestütztes Auslegungsverfahren zur Skalierung einer gerührten Miniplant-Extraktionskolonne; VDI-Verl.: Düsseldorf, Germany, 2007. [Google Scholar]
- Danckwerts, P.V. Continuous flow systems. Chem. Eng. Sci. 1953, 2, 1–13. [Google Scholar] [CrossRef]
- Levenspiel, O. Chemical Reaction Engineering; Wiley: New York, NY, USA, 1999. [Google Scholar]
- Lapkin, A.A.; Walker, A.; Sullivan, N.; Khambay, B.; Mlambo, B.; Chemat, S. Development of HPLC analytical protocol for artemisinin quantification in plant materials and extracts. J. Pharm. Biomed. Anal. 2009, 49, 908–915. [Google Scholar] [CrossRef] [PubMed]
- Qu, H.; Christensen, K.B.; Fretté, X.C.; Tian, F.; Rantanen, J.; Christensen, L.P. Chromatography-Crystallization Hybrid Process for Artemisinin Purification from Artemisia annua. Chem. Eng. Tech. 2010, 33, 791–796. [Google Scholar] [CrossRef]
- Eggersglüß, J. Entwicklung und Auslegung von Flüssig-Flüssig Bioextraktionsprozessen. Antikörperaufreinigung mit wässrigen Zweiphasensystemen. 2013 u.d.T.: Prozessentwicklung und Auslegung von Flüssig-Flüssig Bioextraktionsprozessen: Antikörperaufreinigung mit wässrigen Zweiphasensystemen; Shaker: Aachen, Germany, 2014. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time Interval | Time Differece | Concentration Difference | kL·a |
|---|---|---|---|
| (s) | (s) | (g/L) | (1/s) |
| 2.5–8.1 | 5.6 | 0.382 | 0.000285 |
| 8.1–11.8 | 3.7 | 0.198 | 0.000265 |
| 11.8–16.9 | 5.1 | 0.358 | 0.000233 |
| 16.9–30.1 | 13.2 | 0.660 | 0.000210 |
| Ø | 0.000248 |
| Parameter | Dimension | BAC (d) | SLE-Extract (c) |
|---|---|---|---|
| Inlet parameters first extraction operating point. | |||
| Mass Flow | (g/min) | 18.86 ± 0.91 | 59.65 ± 0.42 |
| Volume Flow | (mL/min) | 21.65 ± 1.06 | 64.14 ± 0.73 |
| Density | (kg/m³) | 871 ± 0.5 | 931 ± 0.5 |
| Concentration | (g/L) | --- | 0.84 ± 0.02 |
| ELSD-Purity | (%) | --- | 35.39 ± 1.41 |
| Outlet parameters first extraction operating point. | |||
| Mass Flow | (g/min) | 38.09 ± 3.93 | 40.99 ± 3.55 |
| Volume Flow | (mL/min) | 44.50 ± 4.74 | 42.30 ± 3.82 |
| Density | (kg/m³) | 856 ± 0.5 | 969 ± 0.5 |
| Concentration | (g/L) | 1.26 ± 0.07 | 0.01 ± 0.01 |
| ELSD-Purity | (%) | 43.40 ± 2.52 | 0.045 ± 0.045 |
| Inlet parameters second extraction operating point. | |||
| Mass Flow | (g/min) | 10.64 ± 0.28 | 60.01 ± 0.01 |
| Volume Flow | (mL/min) | 12.21 ± 0.32 | 64.53 ± 0.02 |
| Density | (kg/m³) | 871 ± 0.5 | 931 ± 0.5 |
| Concentration | (g/L) | --- | 0.87 ± 0.01 |
| ELSD-Purity | (%) | --- | 41.84 ± 0.51 |
| Outlet parameters second extraction operating point. | |||
| Mass Flow | (g/min) | 18.60 ± 2.30 | 52.01 ± 2.63 |
| Volume Flow | (mL/min) | 21.35 ± 2.72 | 55.93 ± 3.03 |
| Density | (kg/m³) | 856 ± 0.5 | 969 ± 0.5 |
| Concentration | (g/L) | 1.67 ± 0.17 | 0.01 ± 0.01 |
| ELSD-Purity | (%) | 49.88 ± 0.40 | 0.08 ± 0.08 |
| Extraction | |||||
| Component | Dimension | BAC | SLE-Extract | LP Extract | HP Raffinate |
| Acetone | (wt.-%) | 0 | 50 | 46 ± 2 | 42 ± 2 |
| Water | (wt.-%) | 0 | 50 | 11 ± 3 | 56 ± 3 |
| Butylacetat | (wt.-%) | 100 | 0 | 43 ± 1 | 2 ± 1 |
| Scrubbing | |||||
| Component | Dimension | LP Extract | HP Scrub Solution | LP Scrubbed | HP Scrub Liqour |
| Acetone | (wt.-%) | 46 ± 2 | 0 | 4 ± 2 | 12 ± 2 |
| Water | (wt.-%) | 11 ± 3 | 95 | 2 ± 2 | 88 ± 2 |
| Butyl Acetate | (wt.-%) | 43 ± 1 | 0 | 94 ± 1 | 1 ± 1 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schmidt, A.; Sixt, M.; Huter, M.J.; Mestmäcker, F.; Strube, J. Systematic and Model-Assisted Process Design for the Extraction and Purification of Artemisinin from Artemisia annua L.—Part II: Model-Based Design of Agitated and Packed Columns for Multistage Extraction and Scrubbing. Processes 2018, 6, 179. https://doi.org/10.3390/pr6100179
Schmidt A, Sixt M, Huter MJ, Mestmäcker F, Strube J. Systematic and Model-Assisted Process Design for the Extraction and Purification of Artemisinin from Artemisia annua L.—Part II: Model-Based Design of Agitated and Packed Columns for Multistage Extraction and Scrubbing. Processes. 2018; 6(10):179. https://doi.org/10.3390/pr6100179
Chicago/Turabian StyleSchmidt, Axel, Maximilian Sixt, Maximilian Johannes Huter, Fabian Mestmäcker, and Jochen Strube. 2018. "Systematic and Model-Assisted Process Design for the Extraction and Purification of Artemisinin from Artemisia annua L.—Part II: Model-Based Design of Agitated and Packed Columns for Multistage Extraction and Scrubbing" Processes 6, no. 10: 179. https://doi.org/10.3390/pr6100179
APA StyleSchmidt, A., Sixt, M., Huter, M. J., Mestmäcker, F., & Strube, J. (2018). Systematic and Model-Assisted Process Design for the Extraction and Purification of Artemisinin from Artemisia annua L.—Part II: Model-Based Design of Agitated and Packed Columns for Multistage Extraction and Scrubbing. Processes, 6(10), 179. https://doi.org/10.3390/pr6100179