3.1. CFD Simulation of the Turbulent Flame and Reaction Mechanism
In this study, the open-source code OpenFOAM v11 [
35] was used for the CFD simulations of the turbulent flame using the RANS equations. The information presented in this subsection can be found in more detail in [
36] and in the corresponding online user guide [
37]. For the simulation, the “multicomponentFluid” solver [
38] was used, which is suitable for reactive flows and combustion simulation. In the solution procedure, the PIMPLE algorithm was applied, which is a combination of the PISO (pressure implicit with split of operators) and SIMPLE (semi-implicit method for pressure-linked equations methods. The basic principle is based on the calculation of an approximated velocity from the momentum equation. An additional equation is used to calculate the pressure. To calculate the continuity equation, the pressure and velocity are then corrected. However, this correction step means that the momentum equation is no longer fulfilled. The process is continued iteratively until the deviations in both the continuity equation and the momentum equation are sufficiently low.
To solve the momentum equation (see Equation (8)), the class “momentumPredictor()” is used. This step is used to calculate an initial guess of the velocity, which is corrected in later steps of the PIMPLE algorithm to fulfill the continuity equation (see Equation (7)). In Equation (8), the variable
stands for the pressure, and
is the dynamic viscosity. Additionally, in the class “thermophysicalPredictor()” in OpenFOAM, the transport equations for the energy (see Equation (9)), based on the specific energy (see Equation (10)), and species (see Equation (1)) are solved. In the energy equation,
is the stress tensor,
is the thermal conductivity, and
is the enthalpy. The last term in the energy equation stands for the heat source from the chemical reactions and is related to the calculated species source terms from the chemistry calculation. Since the flames investigated in this study are of a turbulent nature, a turbulence model was used, which was the standard k-epsilon model proposed by Launder and Spalding [
39]. For the radiation model, the P1 model was activated [
40,
41]. The numerical grid, which was used for the simulations, is later shown in
Section 4.
As explained in
Section 2, the source term for each chemical species
has to be determined. Considering Equation (2), the volume fraction of the fine structure and the time scale will be determined based on the results from the turbulence models (turbulent kinetic energy, dissipation rate of the kinetic energy). However, the mass fractions in the fine structure after the fine structure time scale have to be calculated by the chemistry solver (integrating the chemistry). In the present study, reference simulations of all combustion cases (see
Section 4) will be carried out using the “seulex” solver [
42] in OpenFOAM, which is based on the linearly implicit Euler method with step size control [
43]. For comparison, in
Section 5, the simulations with this solver will be denoted as the “standard” chemistry (SC) solver.
For the prediction of the reaction kinetics within the fine structure of a numerical cell, a so-called reaction mechanism has to be chosen. In the reaction mechanism, the species involved in the chemical reactions, as well as the equations of the chemical reactions, are defined. So, for each reaction
, the reaction rate of a species
can be calculated by Equation (11). In this equation
and
are the stoichiometric coefficients of the species
at the reactant and the product side of the chemical equation,
and
are the reaction rate coefficients for the forward and backward direction of reaction
, and
is the molar concentration of the species
in the reaction
. Furthermore,
stands for the overall number of species in the reaction mechanism, and
as well as
are the exponents forming the reaction order. In case of elementary reactions, the exponents are equal to the stoichiometric coefficients. The reaction rate coefficients can be derived by the Arrhenius equation (see Equation (12) as an example for a forward reaction). The Arrhenius approach includes the pre-exponential factor
, the temperature exponent
, the activation energy
, and the universal gas constant
.
The values used in Equation (12) can be found in the reaction mechanism. In the present study, the authors used a modified mechanism from Jones and Lindstedt (JL), which was optimized in the work by Frassoldati et al. [
44]. All parameters of the reaction mechanism can be found in
Table 1.
Although more detailed reaction mechanisms are available in the literature with several hundreds or even thousands of species and reactions, a rather simple mechanism with 10 species and six chemical reactions was used. Other authors have already used more detailed mechanisms for the training of their ANNs (e.g., [
6]). However, the applicability or feature space (temperature and species concentrations) is often limited (e.g., from 700 to 3100 K in DeepFlame). So, using a reduced reaction mechanism allows the training of the ANNs in the present study for a much wider range of the feature space without extensively increasing the size of the training data or training time. The size of the data for a detailed reaction mechanism for conventional natural gas combustion is already quite large. Extending the training data sets to different turbulence levels (affecting the time scale in the EDC), different fuel mixtures, and oxy-fuel combustion would lead to a large data size. Furthermore, important for an accurate prediction of the chemistry by the ANNs is that the AI-based method can handle the complexity of the reaction kinetics based on the high variation on the time scales and level of occurrence of some minor species. For example, in
Table 1, it can be seen that reaction 6 is much faster than reactions 1, 2, or 3 by several orders of magnitude. Additionally, the reaction mechanism comprises major species, such as CH
4, with high concentrations (mass fractions) and minor species, such as OH, with low amounts. In between the major and minor species, several orders of magnitude also occur. These two aspects are the most critical parts for an accurate training of the ANNs and can be covered with the chosen reaction mechanism. The proposed training methodology described in the next sections should be applicable to larger reaction mechanisms, which will be worth investigating in future work.
3.2. Data Generation and Pre-Processing
There are several factors influencing the ANN’s capability to predict the behavior of a certain system. In addition to the training process itself, the quality of the training data is also essential. Inadequate data can lead to ANNs with a poor prediction capability. Thus, the present section focuses on how the data for the training was created and the pre-processing before the training procedure since these steps are of high importance for the later accuracy of the ANNs. In the first step, the range of the input feature space will be determined, as well as the required distribution of the data in this space. In the second step, the pre-processing of the data will be described, finally leading to data that allows the training of ANNs with high prediction accuracy.
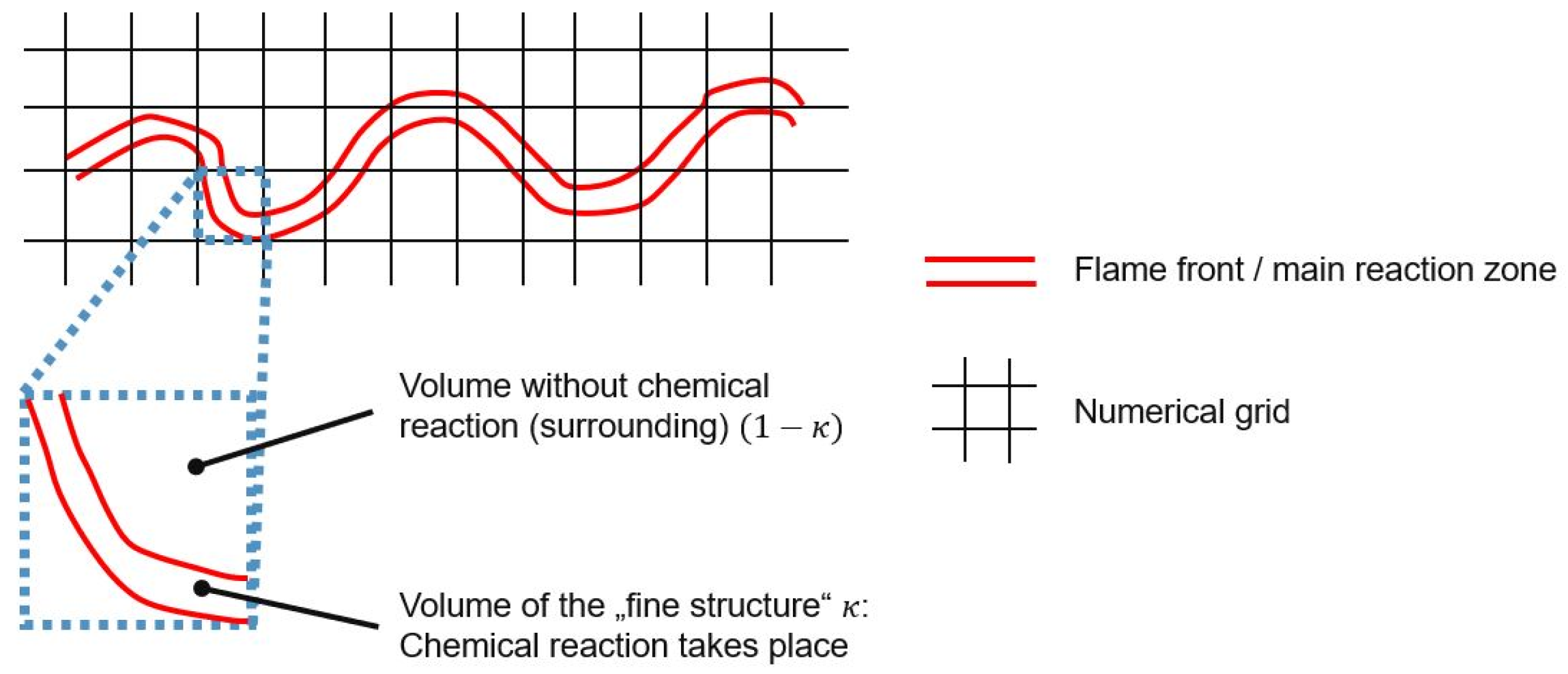
To avoid the usage of chemistry solvers for the predictions of the species source terms
, ANNs will be trained. It has to be mentioned that in the proposed framework, not the species source term
, will be predicted directly from the ANN. Looking at Equation (13), the ANNs in the present study will calculate the source term
. The multiplication with the fine structure volume fraction
is performed separately in OpenFOAM.
The resulting network function to approximate the species source term is denoted as in Equation (13), where represents the input feature space defined within the domain Since the reaction rate of a species within a reactor (or fine structure) depends on the temperature, species concentrations, and residence time (fine structure time scale), the input feature vector can be defined as . Subsequently, the vector for the species mass fractions is defined by all species involved in the reaction mechanism. During the CFD simulation, the input feature vector for the ANNs can be formed by the values from the previous iteration step , as well as the calculated time scale from the turbulence model, according to Equation (5) . It has to be mentioned that the pressure also affects the reaction rates and should be considered in the input feature vector. However, in the present study, the combustion takes place at ambient pressure without high gradients. Thus, the pressure is not part of the input feature space here.
For the supervised learning of the ANNs, corresponding outputs are necessary. For this purpose, the open-source software Cantera [
7] was used. As mentioned in
Section 2, the chemical reactions in the fine structures of a numerical cell can be approximated by a PSR or PFR. Due to the different modeling of the chemistry in the fine structures, the results of the two concepts also deviate from each other under the same initial conditions. When considering the fine structures as PSR, small eddies that arise on the surface or diffusion effects enable mass transfer with the environment of the main reaction zone. In the PFR, there is no mass transfer with the environment, and the reactions can be considered as isolated from the environment. Accordingly, the consideration as PFR leads to a higher mean reaction rate compared to the PSR since there is no back-mixing of reactants in the PFR. Bösenhofer et al. [
45] indicate that modeling of the fine structures as a PFR provides good results under classical combustion conditions. If the focus is on a very detailed consideration of the reaction zone, the PSR approach should be chosen. Due to the lower numerical effort, the PFR approach was chosen for the data generation procedure with Cantera.
The PFR in Cantera is considered a 1D stationary tubular reactor with a constant cross-section and a constant flow rate. The fluid is considered to be homogeneous in the radial direction, and all diffusion processes in the radial and axial directions are neglected. Furthermore, the reactor is seen as operating under isobaric and adiabatic conditions. As an example, for the species concentrations over time (length) in the PFR,
Figure 2 is shown below. The inlet conditions for the PFR can be seen as the species concentrations and temperature from the previous iteration step (conditions of the surrounding of the fine structure in the cell—
). The horizontal axis in
Figure 2 represents the residence time of the reactants in the PFR, which is equal to the fine structure time scale
, which means that with one PFR simulation, several input features of the time scale can be derived. If a PSR was used, a single simulation for each
has to be carried out, which would have significantly increased the time for data generation. Based on the input features, temperature, and species concentrations, the species mass fractions in the fine structure
after the time scale
can be determined. As a consequence, the network prediction (output feature)
can be formed in accordance with Equation (13).
To define the input feature space, OpenFOAM simulations with the standard chemistry solver were carried out for all considered combustion cases (see
Section 4). During the simulations, the minimum and maximum values of all species concentrations, temperatures, and fine structure time scales were monitored. The observed maximum and minimum values were slightly extended to clearly avoid leaving the input feature space of the trained ANNs during the simulation. The range of each feature is shown in
Table 2. It can be seen that the temperature range is from below 0 °C to 2600 K, which considers the full range of possible temperatures in the considered combustion cases. Also, the oxygen mass fraction is significantly extended to 1. Thus, oxy-fuel combustion can also be considered by the ANNs. In combustion with pure oxygen, the possible nitrogen mass fraction is reduced to 0. Within the ranges defined in
Table 2, the input features were chosen by Monte Carlo sampling. Only nitrogen was determined by the fact that the sum of all mass fractions must be 1. In
Table 2, the distribution of the feature sampling is also shown. For the time scale
, logarithmic sampling was chosen. This means that for the 30 samples of
in one PFR simulation, more samples were defined for smaller time scales since the gradients of the species concentrations and temperature are higher. For larger time scales, the reaction moves towards equilibrium with low gradients. For the Monte Carlo sampling of the temperature and oxygen mass fraction, a homogeneous distribution of the random values was defined. The other distributions depend on whether the mass fractions of the species can reach larger values (l) or smaller values (s). For the species O and H (radicals), it can be seen in
Table 2 that the maximum mass fractions are below 0.005. So, these species are defined as small (s) and the other ones as large (l).
For data generation, the input feature vector is formed by Monte Carlo sampling, which is quite clear for the time scale, the temperature, and the oxygen mass fraction. At first glance, it might be the case that the species mass fractions in all numerical cells during the OpenFOAM simulations are homogeneously distributed. For example, this can be seen in
Figure 3 (left) by the purple area, highlighting the probability density of the mass fraction of CH
4. In the majority of the numerical cells, a very low mass fraction with an apparently Gaussian distribution is present. However, analyzing the results of the OpenFOAM simulations of all combustion cases showed that the mass fractions in all numerical cells of the simulation are not homogeneously distributed. After transformation of the species mass fractions using the Box–Cox transformation with
[
48] according to Equation (14), the distribution looks clearly different (see
Figure 3 (right)). Since the generated training data with Cantera should represent the “reality” (reference simulation with OpenFOAM) as well as possible, the distribution of the data sampling for the species mass fractions was defined as shown in
Table 3. It has to be mentioned that the data shown in
Figure 3 were based on classes of the mass fractions, leading to a histogram plot. However, it was decided to convert the histogram to a line plot. Thus, in the line plot, negative values for the mass fraction can occur, which are based on interpolation between the classes near a mass fraction of 0. The data sets contain no negative mass fractions.
In
Table 3, six ranges for the species mass fractions were defined with a certain probability that the Monte Carlo sampling uses the specific range for the sampling. Considering the small mass fractions, it can be seen that most input features for these mass fractions will be generated from 10
−4 to 10
−3. For large mass fractions, the majority of the input data will be generated in a range of 10
−3 to 10
−1, but very small mass fractions will also be considered during the sampling of the input features. With this sampling methodology for the input feature mass fraction, it is possible to generate training data, which is in close accordance with the “real” condition in the flame.
Figure 3 (right) shows that the generated input feature for Cantera matches well with the OpenFOAM simulation data. Now, the input features cover the entire range for all combustion cases, and also the distribution of the features is in good agreement with “reality”. With these input features, Cantera simulations of the PFR will be performed.
Before the input and corresponding output data
can be used for the training of the ANNs, pre-processing steps have to be carried out. Data pre-processing is an important tool for improving the convergence of the training algorithm. For example, the data should be normalized to an order of magnitude of
to prevent difficulties during the network training, which would lead to a lower learning rate for reasons of stability, and thus slow down the learning process [
50]. It was proposed in [
51] that training usually converges faster when the data is close to a standard normal distribution. If the underlying data is not normally distributed, the data should be brought into an order of
. For the normalization of the input and output data, Equation (15) (e.g., for species mass fraction
) and (16) (for the reaction rates
) were used. Both were normalized within a range of 0…1. Additionally, for the output data, a root function was used. With the root function, the output data fits the normal distribution much better.
Finally, the pre-processed data set has a size of approx. 32 GB and consists of approx. 38 million data points
3.3. Subdivision of the Feature Space
The literature in
Section 1 highlighted that using one ANN might not be feasible for the prediction of chemistry in the full range of input features. Several authors dramatically increased the number of ANNs in conjunction with a classification methodology (e.g., SOM in [
27]). An alternative is given by the DeepFlame framework [
6], which reduced the range for the input features for the single ANN trained for chemistry prediction. Prieler et al. [
29] only trained two ANNs for the prediction of the chemical reactions in laminar counter-flow diffusion flames. The ANNs were trained for cases where ignition occurs and cases without ignition. Since this study revealed promising results with a limited number of ANNs, significantly reducing the training time, a similar approach was used in the present study. Volgger [
52] stated that the applicability range for the separate ANNs can be related to the input feature of the temperature or the fine structure time scale. The temperature highly affects the reaction rates in the fine structure caused by the definition of the Arrhenius approach (see Equation (12)). However, the time scale was used as a criterion to determine which ANN should be used in the numerical cell for the prediction of the reaction kinetics.
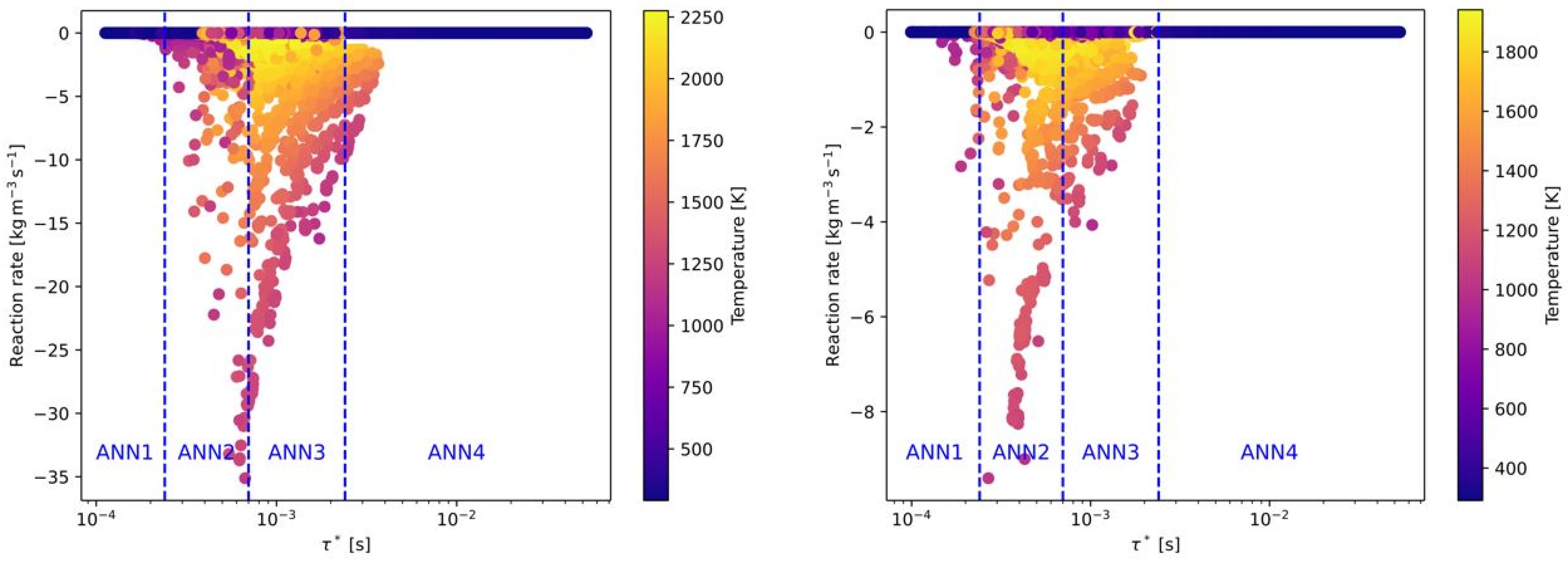
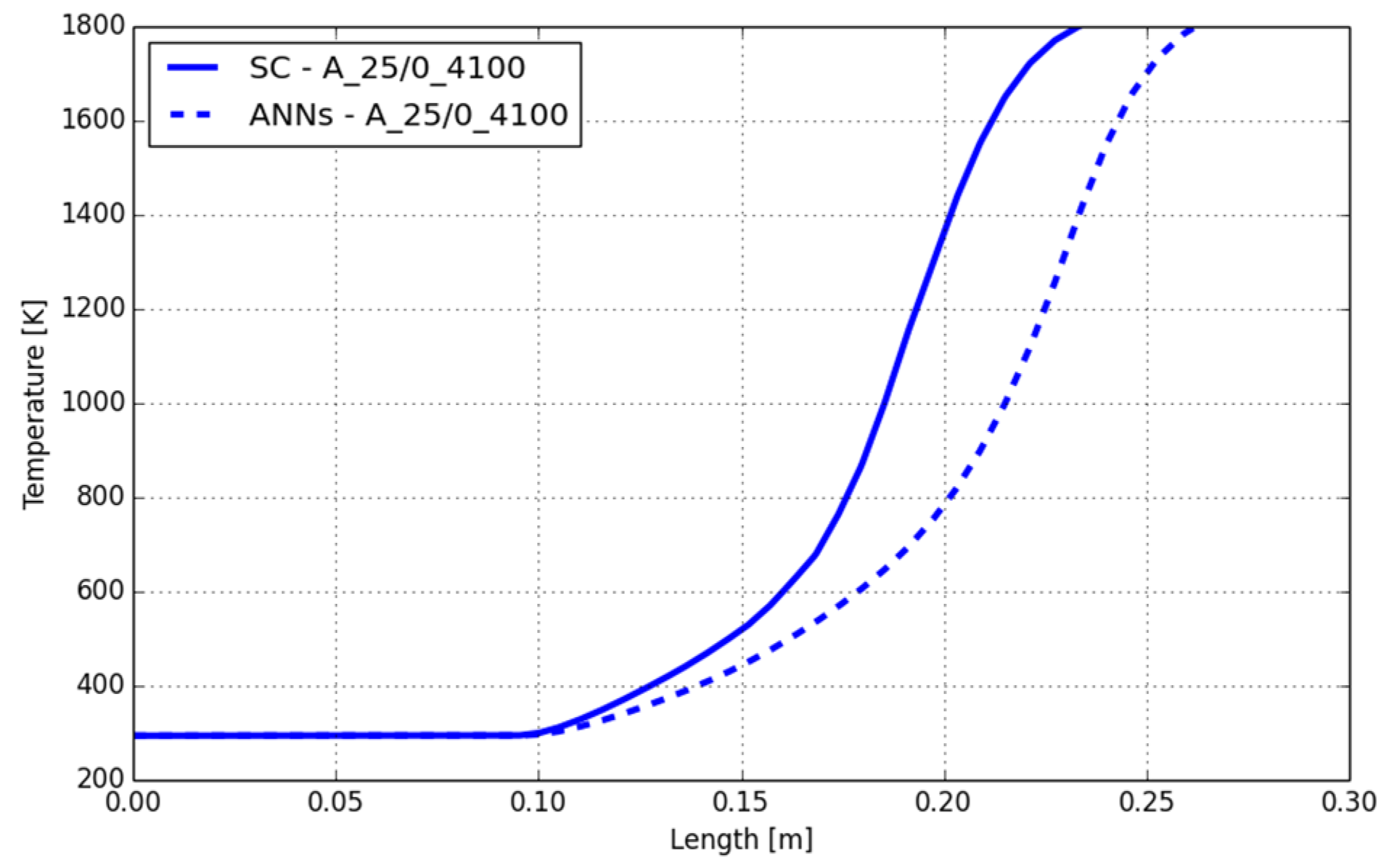
Four different ANNs were trained for pre-defined ranges of the fine structure time scale. In the following figures, the reaction rates of all OpenFOAM simulations were presented, depending on the fine structure time scale. The final regions of the time scale for each ANN (ANN1 to ANN4) are already marked in these figures and summarized in
Table 4. In
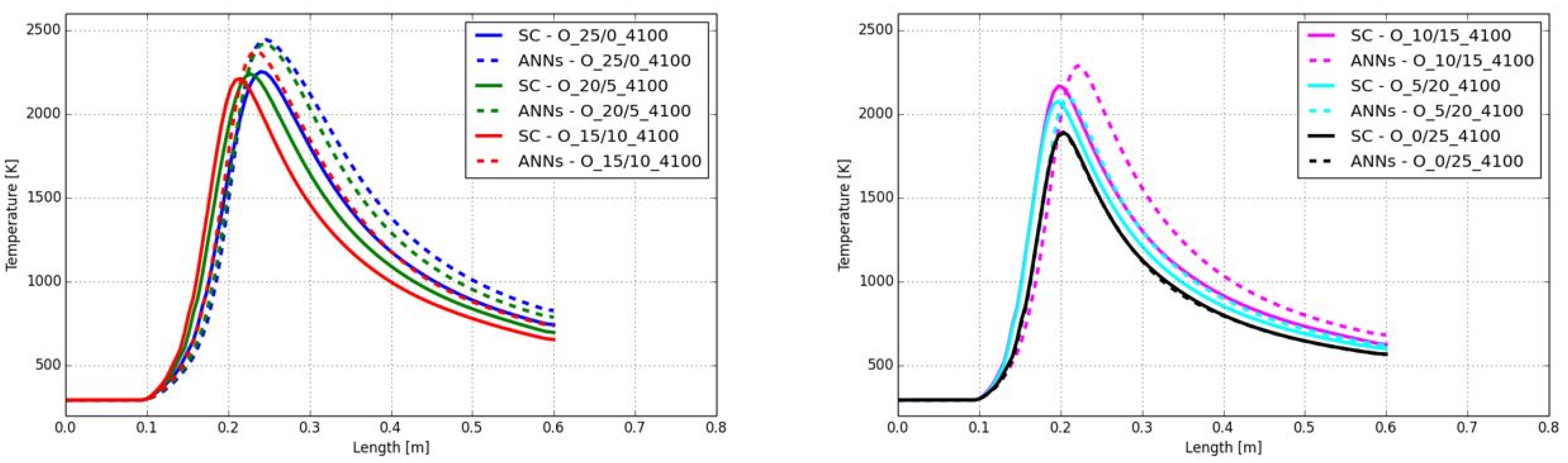
Figure 4, all reaction rates from the OpenFOAM simulations are presented for the combustion of CH
4 and H
2 with air. The reaction rates of these species were chosen because they are the main fuels in this study. The main target in defining the range of the time scale was that one or two ANNs should cover the range where it comes to combustion. The other ANNs should cover ranges without or with a minor number of combustion cases. This approach is similar to Prieler et al. [
29]. In
Figure 4, it can be seen that ANN2 and 3 cover the full range of the combustion cases with an equal distribution. ANN1 and ANN4 are considering time scales (nearly) without combustion. The highest reaction rates of CH
4 were found around a time scale of 0.0005 s
−1 and within a range of 0.0002 and 0.0011 s
−1. Thus, ANN2 and ANN3 were trained with data (input features) with these time scales. For the combustion of H
2, the maximum reaction rates occurred on a similar time scale (see
Figure 4 (right)).
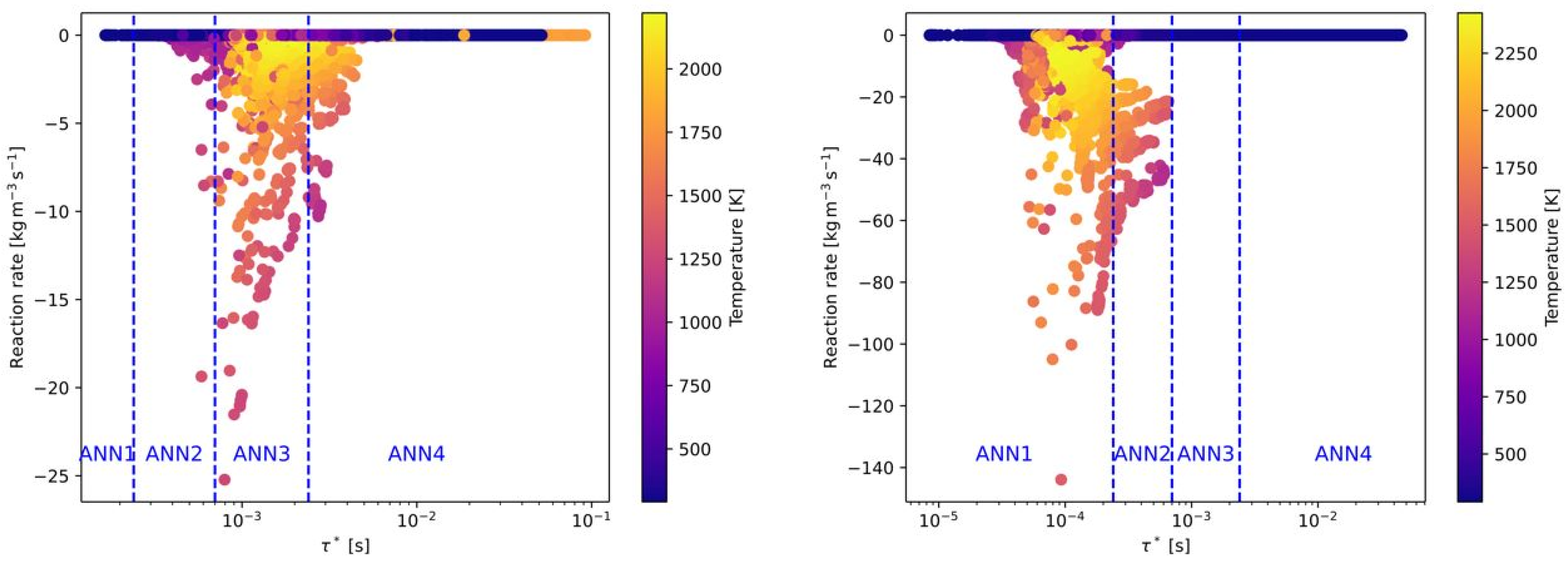
In
Figure 5, the same combustion cases (CH
4 and H
2) in
Figure 4 are presented, but with pure oxygen as an oxidizer instead of air (without nitrogen). Although the combustion with pure oxygen is in general related to a higher reactivity, the OpenFOAM data showed that the time scales are similar to the combustion with air as an oxidizer. Only a slight shift of the reaction rates to higher time scales can be observed. Therefore, the defined time scale ranges of the ANNs are also suitable for oxy-fuel combustion. It has to be mentioned that the values of the reaction rates between air-fuel and oxy-fuel combustion are clearly different for the combustion of CH
4 with air (see
Figure 4 (left) and
Figure 5 (left)). Whereas the maximum reaction rates with air are approximately 10 kg/(m
3s), the reaction rates with oxygen are more than 3 times higher. This difference between air-fuel and oxy-fuel combustion cannot be observed when H
2 is used as fuel.
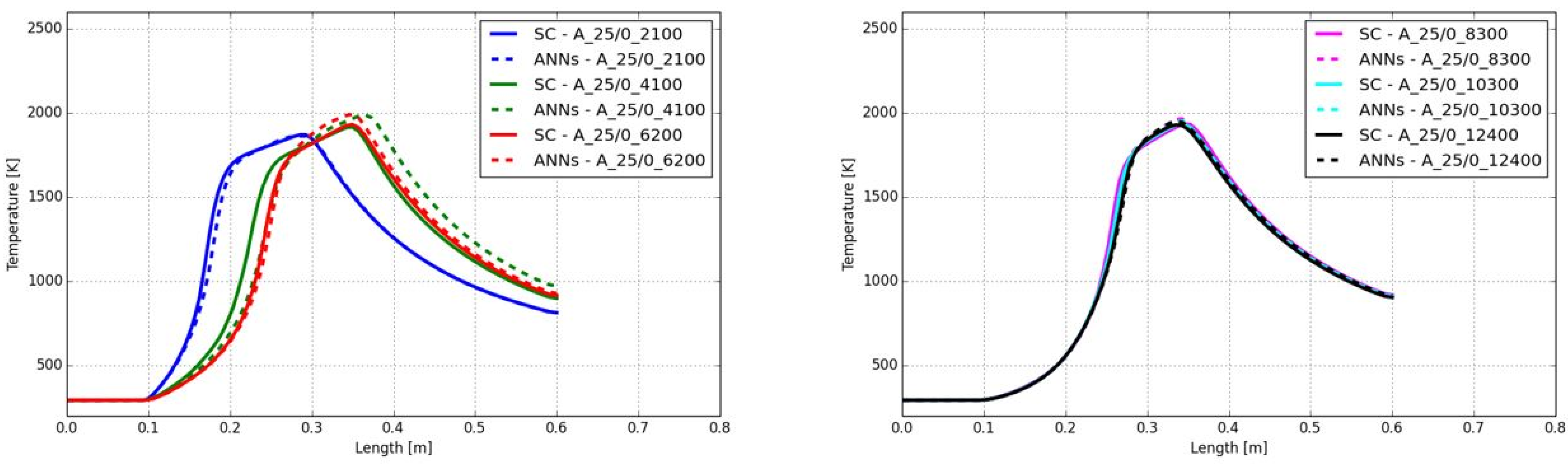
In addition to the composition of the fuel and oxidizer, the third effect to be covered by the ANNs is the level of turbulence. As described later in
Section 4, six Reynolds numbers, related to the flow conditions in the main jet of the burner, were investigated. In
Figure 6, the effect of the Reynolds numbers on the reaction rates and time scales for the combustion of CH
4 with air is presented. Compared to
Figure 4 (left), the time scale, where the highest reaction rates occurred, was shifted slightly to higher time scales (see
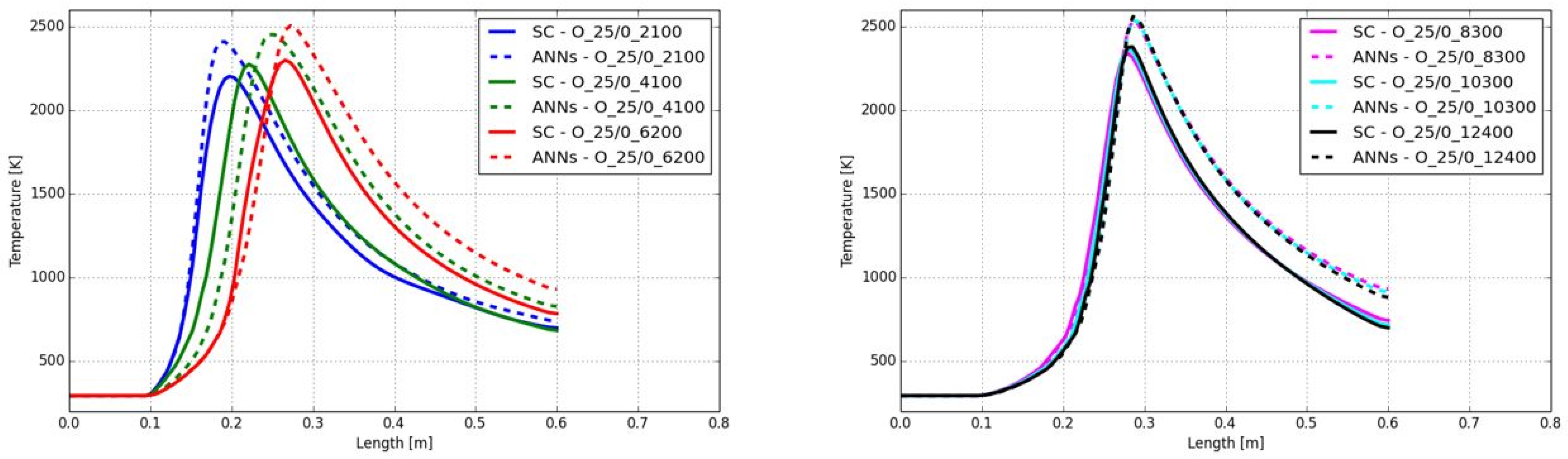
Figure 6 (left)). Thus, all cases with ignition are now located in ANN3. In contrast, higher turbulence in the main jet significantly decreases the time scales for high reaction rates. As a consequence, all burning cases are now in the range of ANN1. Similar to air-fuel and oxy-fuel combustion of different fuels (see
Figure 4 and
Figure 5), there is hardly any difference in the time scales when switching from air-fuel (see
Figure 6) to oxy-fuel combustion (see
Figure 7) under different turbulence levels. But, the maximum reaction rate increases again when oxy-fuel is used as an oxidizer instead of air.
Finally, the analysis using the OpenFOAM simulation data showed that the time scale is only affected by the turbulence levels. The effect of the fuel mixture and the oxidizer is minor. However, the oxidizer significantly affects the level of the maximum reaction rates. Thus, it can be concluded that the turbulence level had an effect on the ranges of the fine structure time scales for the different ANNs, but it had no effect on the other training parameters or data pre-processing steps.
For the air-fuel and oxy-fuel combustion cases with all fuel types, ANN1 was mainly used for higher Reynolds numbers when an ignition case was considered. With a decreasing turbulence level in the main jet, ANN1 is replaced by ANN2 and ANN3. For larger time scales, ANN4 is applied, which represents cases without ignition for all cases. In
Table 4, the ranges of the time scales for each ANN are summarized. Although OpenFOAM data were used to roughly determine the ranges of the residence times, the exact values shown in
Table 4 were determined by trial and error. The best performance was achieved with the ranges presented in
Table 4. But, it has to be mentioned that the differences between the trials were very small.
3.4. ANN Structure, Training, and Hyperparameter Tuning
In the present study, a feed-forward architecture without backward connections was used for the ANNs. This approach seems reasonable since all previous studies mentioned in
Section 1 also used the same architecture. The ADAM algorithm was used as the learning algorithm for all ANNs trained in this work. According to Kingma et al. [
53], this is a computationally efficient and robust stochastic optimization algorithm that is particularly suitable for problems with large data sets and many parameters. From the entire data set, 80% was used for training and 20% for validation. Also, other ratios between training and validation data sets were tested with a negligible effect on the performance of the ANNs. The learning rate is adjusted individually for different parameters, whereby the change in learning rates is also monitored using a weighted moving average. The batch size for the learning process has a significant effect on the generalization ability of the neural network. In general, the larger the batches, the worse the network generalizes. This means that the prediction quality of the network on a validation data set decreases (see, for example, [
54]). On the other hand, larger batches have the advantage that the calculation per iteration step can be better parallelized, which greatly reduces the training time. In the present study, the batch size was defined with a value of 256, and the training duration was 100 epochs. It was observed that after 100 epochs, the error was at a minimum level for all ANNs without a hint of over-fitting. Using smaller batch sizes had a minor effect on the training errors shown in
Table 5 but led to a higher training duration. Thus, the value of 256 was a good compromise.
To determine the structure of the ANNs with regard to the number of hidden layers and the number of neurons per layer, a hyperparameter tuning was carried out. The Keras tuner [
55] in TensorFlow [
56] was used to select suitable hyperparameters. Bayesian optimization was used to search for a set of hyperparameters that best approximates the data set. In the course of this work, an ANN structure of four hidden layers with 256 neurons per layer proved to be a good compromise between training effort and network performance for the tested ANNs. In addition to the four hidden layers, an input layer with 12 neurons (10 species, fine structure time scale, and temperature) and an output layer with 10 neurons (reaction rate for each species
) was used. The rectified linear unit (ReLU) was used as the activation function for the neurons of the input layer and the hidden layers. This function is suitable for training very deep neural networks with a large number of hidden layers [
57]. An alternative activation function would be the Gaussian error linear unit (GELU), as it was used in the DeepFlame framework. A comparison between the ReLU and GELU was not performed in the course of the present study. A linear activation was applied to the output layer. Although for the training of the final ANNs the size of the data set was 32 GB, smaller data sets were tested, and the errors were observed (see
Table 5).
The coupling of the ANNs, which are based on Python v3.11.10, with the C++ framework of OpenFOAM v11, was performed via the pybind11 library [
58].




















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}