
A Survey on the Chemical Recycling of Polyolefins into Monomers

 , ,
, , 

Abstract
1. Introduction
2. Microwave-Assisted Pyrolysis
2.1. Fundamentals of Microwave-Assisted Pyrolysis
2.2. Previous Works of Microwave-Assisted Pyrolysis
2.3. Perspectives on Microwave-Assisted Pyrolysis
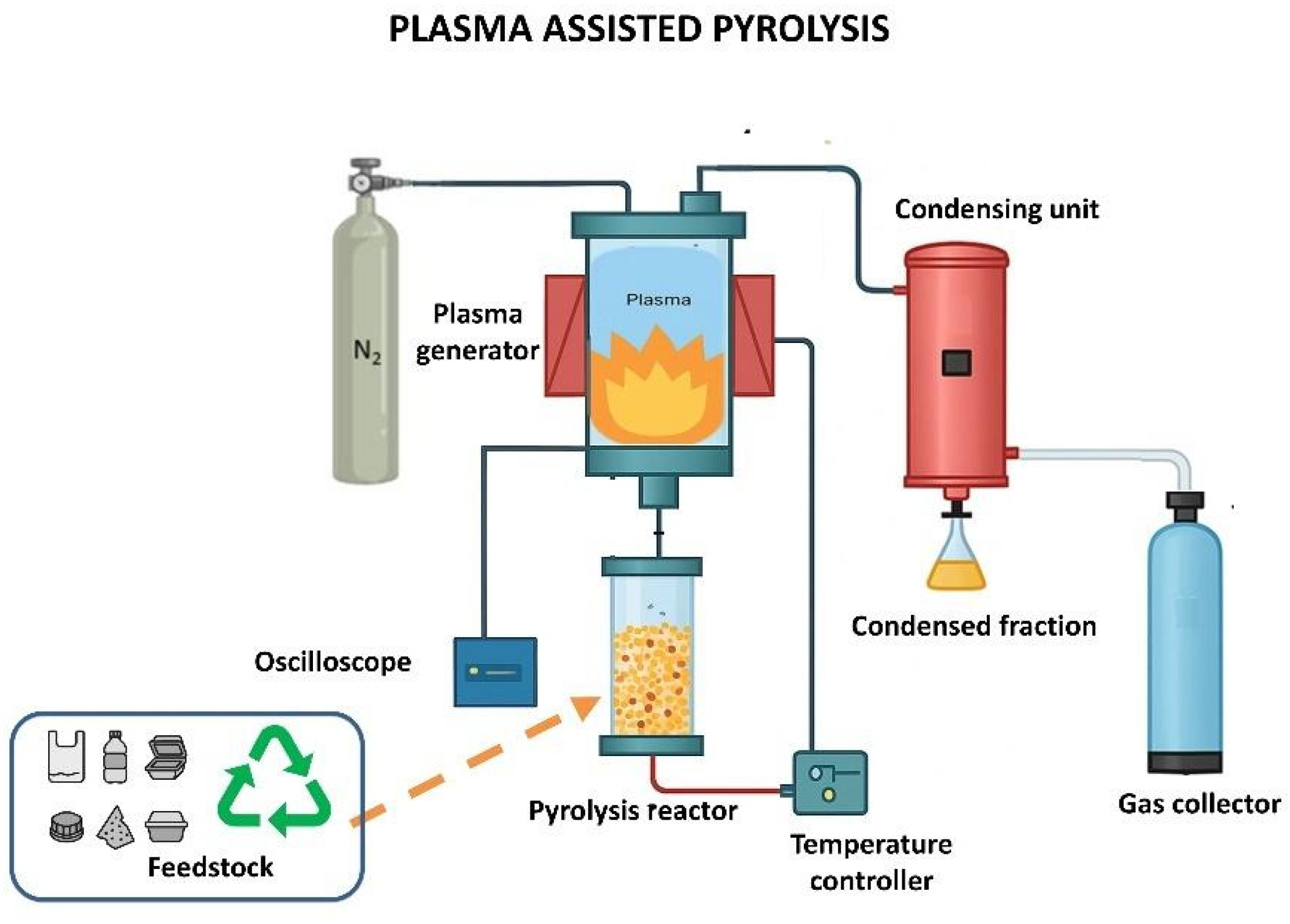
3. Plasma-Assisted Pyrolysis
3.1. Fundamentals of Plasma-Assisted Pyrolysis
3.2. Previous Works of Plasma-Assisted Pyrolysis
3.3. Perspectives on Plasma-Assisted Pyrolysis
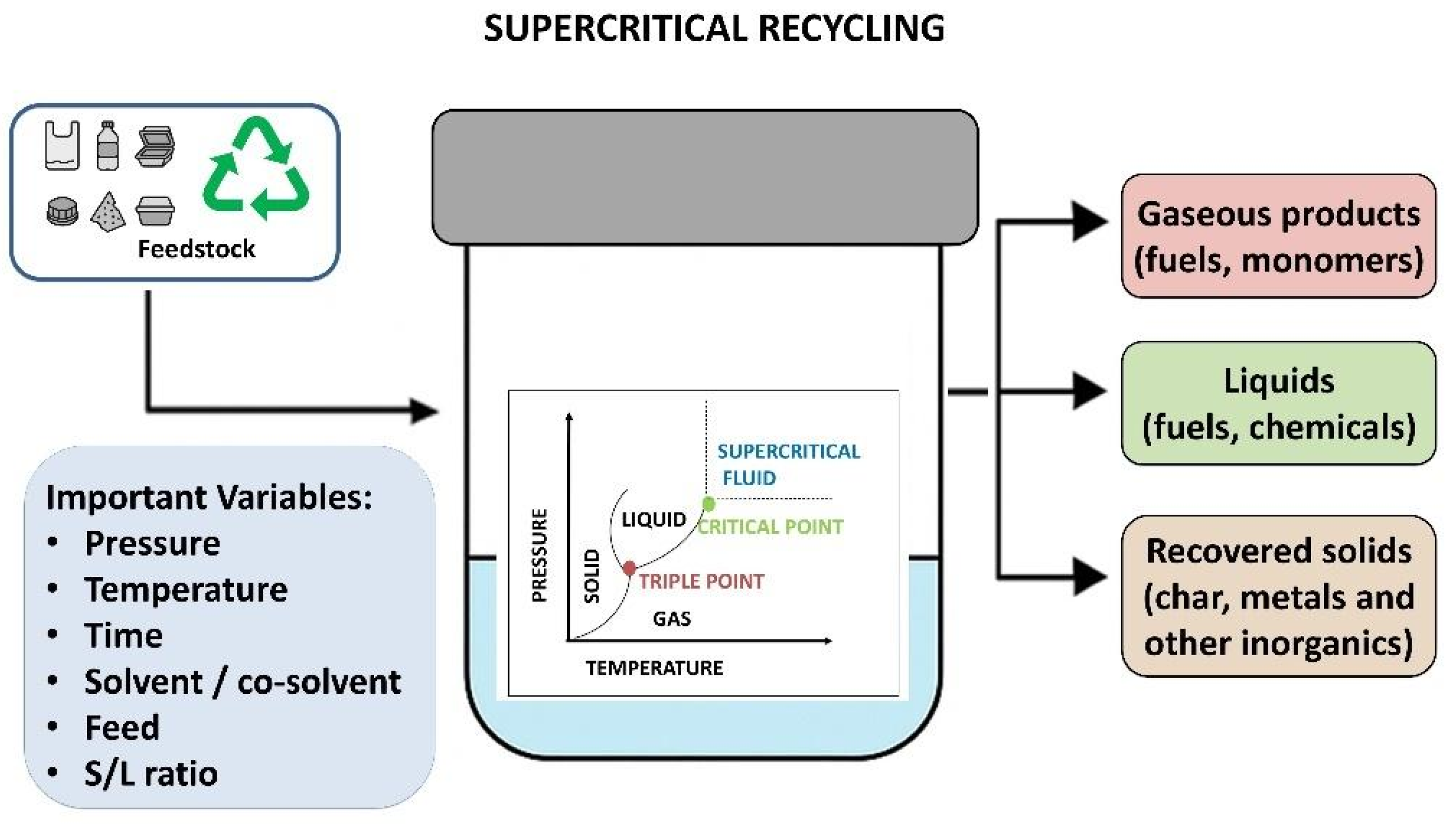
4. Chemical Recycling Through Supercritical Technology
4.1. Fundamentals of Chemical Recycling Through Supercritical Technology
4.2. Previous Works of Chemical Recycling Through Supercritical Technology
4.3. Perspectives on Chemical Recycling Through Supercritical Technology
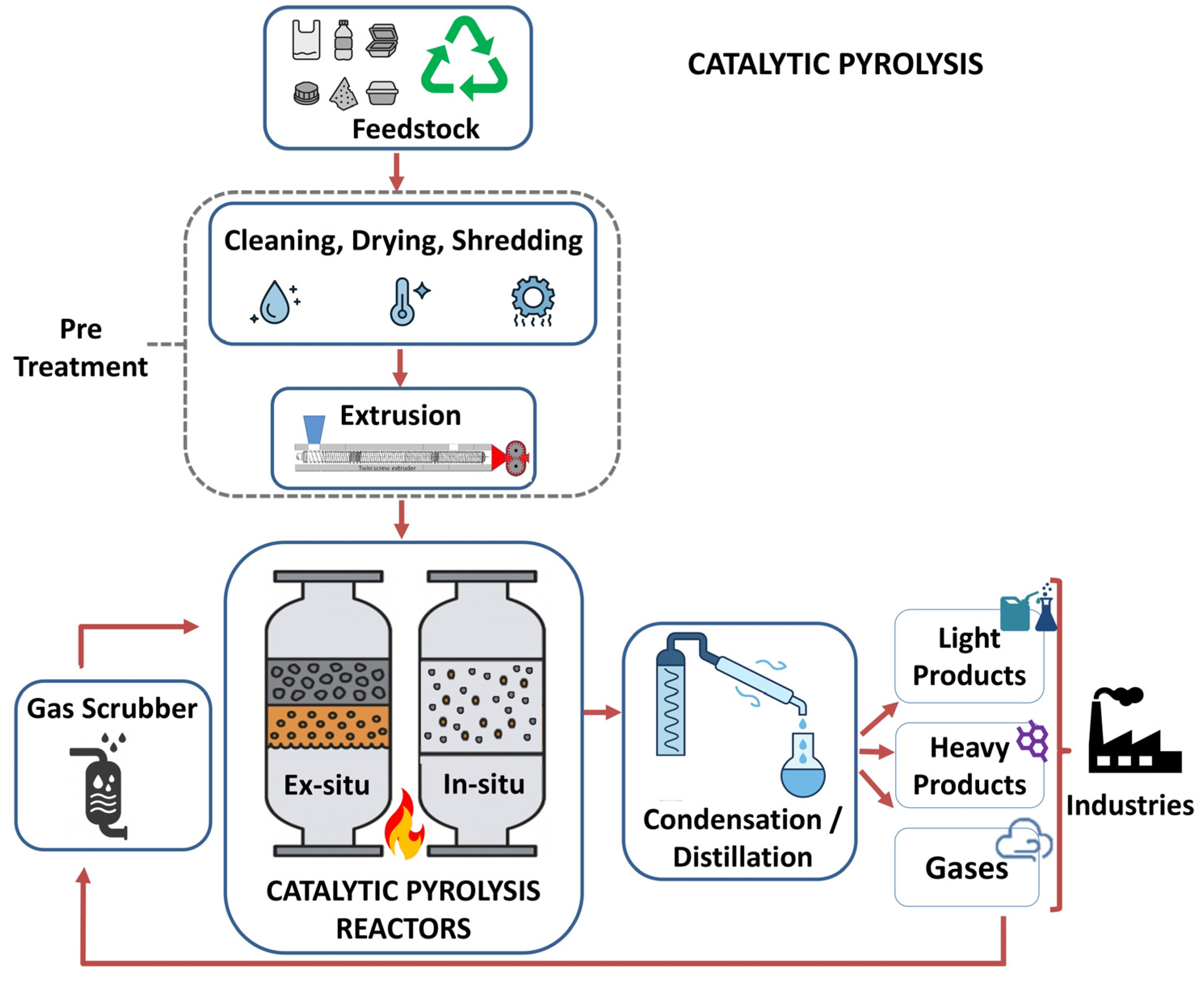
5. Chemical Recycling Through Catalytic Pyrolysis
5.1. Fundamentals of Catalytic Pyrolysis
5.2. Previous Works of Catalytic Pyrolysis
5.3. Perspectives on Catalytic Pyrolysis
6. Chemical Recycling Through Electrochemical and Oxidative Degradation
6.1. Fundamentals of Electrochemical and Oxidative Degradation
6.2. Previous Works of Electrochemical and Oxidative Degradation
6.3. Perspectives on Electrochemical and Oxidative Degradation
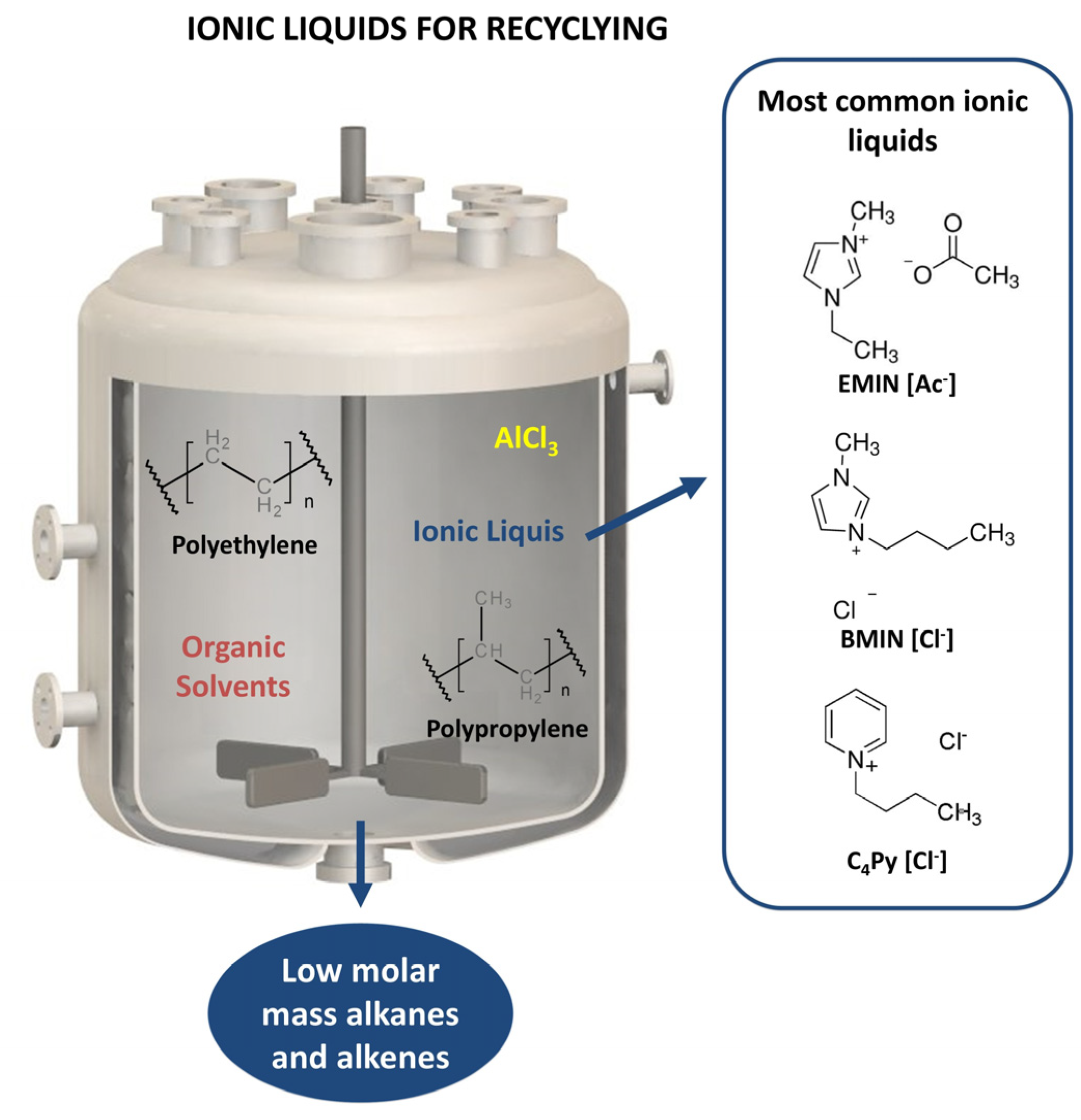
7. Chemical Recycling with Ionic Liquids
7.1. Fundamentals of Chemical Recycling with Ionic Liquids
7.2. Previous Works of Chemical Recycling with Ionic Liquids
7.3. Perspectives on Chemical Recycling with Ionic Liquids
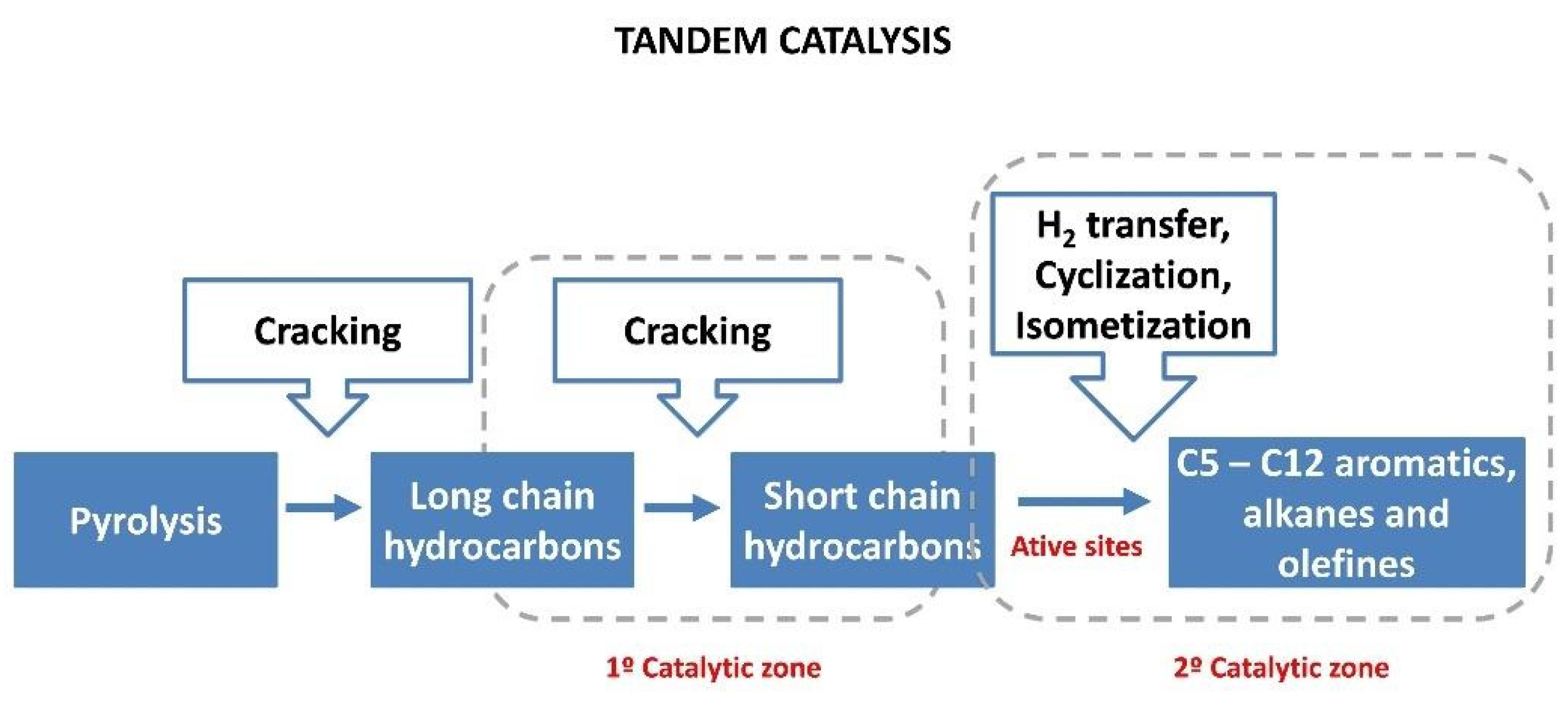
8. Chemical Recycling Through Tandem Catalysis
8.1. Fundamentals of Tandem Catalysis
8.2. Previous Works of Tandem Catalysis
8.3. Perspectives on Tandem Catalysis
9. Some Environmental Aspects
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| BMIM | 1-Butyl-3-methylimidazolium |
| BMIM [PF6] | 1-Butyl-3-methylimidazolium hexafluorophosphate |
| C4Py | 1-butylpyridinium |
| CNT | Carbon nanotube |
| CSBR | Conical spouted bed reactor |
| DBD | Dielectric barrier discharge |
| EG | Ethylene glycol |
| EMIM | 1-Ethyl-3-methylimidazolium |
| FCC | Fluid catalytic cracking |
| FTO | Fluorine-doped tin oxide |
| HDPE | High-density polyethylene |
| HTP | Hydrothermal processing |
| IBGE | Instituto Brasileiro de Geografia e Estatística |
| KFD | Potassium diformate |
| LDPE | Low-density polyethylene |
| LLDPE | Linear low-density polyethylene |
| PC | Polycarbonate |
| PE | Polyethylene |
| PET | Poly(ethylene terephthalate) |
| PIP | Polyisoprene |
| PP | Polypropylene |
| PS | Polystyrene |
| scCO2 | Supercritical carbon dioxide |
| SCF | Supercritical Fluid |
| TEA | Techno-economic analysis |
| THF | Tetrahydrofuran |
| TPA | Terephthalic acid |
| TRL | Technology Readiness Level |
| VGO | Vacuum gas oil |
| XLPE | Crosslinked polyethylene |
References
- Mancini, S.D.; Bezerra, M.N.; Zanin, M. Reciclagem de PET Advindo de Garrafas de Refrigerante Pós-Consumo. Polímeros 1998, 8, 68–75. [Google Scholar] [CrossRef]
- Oliveira, E.J.G. Estudo Sobre a Processabilidade de PET Reciclado Obtido Pelo Processo de Reciclagem Mecânica. Master’s Thesis, Universidade do Minho, Minho, Portugal, 2016. [Google Scholar]
- Ahamed, A.; Vallam, P.; Iyer, N.S.; Veksha, A.; Bobacka, J.; Lisak, G. Life Cycle Assessment of Plastic Grocery Bags and Their Alternatives in Cities with Confined Waste Management Structure: A Singapore Case Study. J. Clean. Prod. 2021, 278, 123956. [Google Scholar] [CrossRef]
- 2024 Plastics Europe AISBL Plastics—The Fast Facts 2024. Available online: https://plasticseurope.org/knowledge-hub/plastics-the-fast-facts-2024/ (accessed on 24 March 2025).
- Pinto, J.C.; Magrini, A.; Melo, C.K.; Castor, C.A., Jr.; Gaioto, C.C.; dos Santos, D.P.; Borges, G.; Rosa, I.d.S.; Delgado, J.J.S.; de Souza, M.N.; et al. Impactos Ambientais Causados Pelos Plásticos, 1st ed.; E-papers Serviços Editoriais Ltda.: Rio de Janeiro, Brazil, 2012. [Google Scholar]
- Saibuatrong, W.; Cheroennet, N.; Suwanmanee, U. Life Cycle Assessment Focusing on the Waste Management of Conventional and Bio-Based Garbage Bags. J. Clean. Prod. 2017, 158, 319–334. [Google Scholar] [CrossRef]
- Coates, G.W.; Getzler, Y.D.Y.L. Chemical Recycling to Monomer for an Ideal, Circular Polymer Economy. Nat. Rev. Mater. 2020, 5, 501–516. [Google Scholar] [CrossRef]
- Diário Oficial da União: Brasília, Brazil. LEI No 12.305, DE 2 DE AGOSTO DE 2010. Available online: https://www.planalto.gov.br/ccivil_03/_ato2007-2010/2010/lei/l12305.htm (accessed on 3 June 2025).
- dos Santos, B.C.M. Análise Do Ciclo de Vida Da Embalagem de Polietileno Tereftalato. In Proceedings of the VII congresso Nacional de Excelência em Gestão, Rio de Janeiro, Brazil, 12–13 August 2011. [Google Scholar]
- Romão, W.; Spinacé, M.A.S.; Paoli, M.-A. De Poli(Tereftalato de Etileno), PET: Uma Revisão Sobre Os Processos de Síntese, Mecanismos de Degradação e Sua Reciclagem. Polímeros 2009, 19, 121–132. [Google Scholar] [CrossRef]
- Jorge, F.E.; Neves, M.A.F.S. Comparação Entre Técnicas Simples e a Análise de Espectroscopia No Infravermelho Na Caracterização de Polímeros Recicláveis. Perspect. Ciência Tecnol. 2016, 8, 47. [Google Scholar]
- OECD Plastic Pollution Is Growing Relentlessly as Waste Management and Recycling Fall Short, Says OECD. Available online: https://www.oecd.org/environment/plastic-pollution-is-growing-relentlessly-as-waste-management-and-recycling-fall-short.htm (accessed on 9 July 2023).
- Dai, L.; Zhou, N.; Lv, Y.; Cheng, Y.; Wang, Y.; Liu, Y.; Cobb, K.; Chen, P.; Lei, H.; Ruan, R. Pyrolysis Technology for Plastic Waste Recycling: A State-of-the-Art Review. Prog. Energy Combust. Sci. 2022, 93, 101021. [Google Scholar] [CrossRef]
- Spadetti, C.; Silva Filho, E.A.d.; Sena, G.L.d.; Melo, C.V.P. de Propriedades Térmicas e Mecânicas Dos Compósitos de Polipropileno Pós-Consumo Reforçados Com Fibras de Celulose. Polímeros 2017, 27, 84–90. [Google Scholar] [CrossRef]
- Duailibe, A.; Pinto, J.C.; Delgado, J.J.S. Reciclagem Química de Resíduos Plásticos, 1st ed.; E-papers Serviços Editoriais Ltda.: Rio de Janeiro, Brazil, 2019. [Google Scholar]
- Silva, E.A.d.; Moita Neto, J.M. Possibilidades de Melhorias Ambientais No Processo de Reciclagem Do Polietileno. Polímeros 2016, 26, 49–54. [Google Scholar] [CrossRef]
- Genuino, H.C.; Ruiz, M.P.; Heeres, H.J.; Kersten, S.R.A. Pyrolysis of Mixed Plastic Waste (DKR-350): Effect of Washing Pre-Treatment and Fate of Chlorine. Fuel Process. Technol. 2022, 233, 107304. [Google Scholar] [CrossRef]
- Hasan, M.M.; Haque, R.; Jahirul, M.I.; Rasul, M.G. Pyrolysis of Plastic Waste for Sustainable Energy Recovery: Technological Advancements and Environmental Impacts. Energy Convers. Manag. 2025, 326, 119511. [Google Scholar] [CrossRef]
- Simpson, D.M.; Vaughan, G.A. Ethylene Polymers, LLDPE. In Encyclopedia of Polymer Science and Technology; Wiley: Hoboken, NJ, USA, 2003. [Google Scholar]
- Aboulkas, A.; El harfi, K.; El Bouadili, A. Thermal Degradation Behaviors of Polyethylene and Polypropylene. Part I: Pyrolysis Kinetics and Mechanisms. Energy Convers. Manag. 2010, 51, 1363–1369. [Google Scholar] [CrossRef]
- Siqueira, Y.F.; Miranda, D.M.V.; Carvalho, L.L.A.; Sitton, N.K.; de Azeredo, A.P.; Nonemacher, R.F.; Mauler, R.S.; Borges, L.E.P.; Pinto, J.C. Impact of Extrusion Pre-Treatment on Polyethylene Waste Pyrolysis. Polym. Eng. Sci. 2025, 65, 1–15. [Google Scholar] [CrossRef]
- Frączak, D. Chemical Recycling of Polyolefins (PE, PP): Modern Technologies and Products. In Waste Material Recycling in the Circular Economy—Challenges and Developments; IntechOpen: London, UK, 2022. [Google Scholar]
- Putra, P.H.M.; Rozali, S.; Patah, M.F.A.; Idris, A. A Review of Microwave Pyrolysis as a Sustainable Plastic Waste Management Technique. J. Environ. Manag. 2022, 303, 114240. [Google Scholar] [CrossRef]
- Nasreen, S.A.A.N.; Sundarrajan, S.; Nizar, S.A.S.; Wei, H.; Xuecheng, D.; Ramakrishna, S. Pyrolysis, Microwave, Chemical and Biodegradation Methodology in Recycling of Plastic Waste: A Circular Economy Concept. Circ. Econ. Sustain. 2022, 2, 609–632. [Google Scholar] [CrossRef]
- Suriapparao, D.V.; Vinu, R. Resource Recovery from Synthetic Polymers via Microwave Pyrolysis Using Different Susceptors. J. Anal. Appl. Pyrolysis 2015, 113, 701–712. [Google Scholar] [CrossRef]
- Hussain, A.R.J.; Alahyari, A.A.; Eastman, S.A.; Thibaud-Erkey, C.; Johnston, S.; Sobkowicz, M.J. Review of Polymers for Heat Exchanger Applications: Factors Concerning Thermal Conductivity. Appl. Therm. Eng. 2017, 113, 1118–1127. [Google Scholar] [CrossRef]
- Zhang, X.; Lei, H.; Yadavalli, G.; Zhu, L.; Wei, Y.; Liu, Y. Gasoline-Range Hydrocarbons Produced from Microwave-Induced Pyrolysis of Low-Density Polyethylene over ZSM-5. Fuel 2015, 144, 33–42. [Google Scholar] [CrossRef]
- Undri, A.; Rosi, L.; Frediani, M.; Frediani, P. Efficient Disposal of Waste Polyolefins through Microwave Assisted Pyrolysis. Fuel 2014, 116, 662–671. [Google Scholar] [CrossRef]
- Jing, X.; Zhao, Y.; Wen, H.; Xu, Z. High Olefin Yield in Pyrolysis of Heavier Hydrocarbon Liquids Using Microwave as Heat Supplier. Energy Fuels 2017, 31, 2052–2062. [Google Scholar] [CrossRef]
- Suriapparao, D.V.; Hemanth Kumar, T.; Reddy, B.R.; Yerrayya, A.; Srinivas, B.A.; Sivakumar, P.; Prakash, S.R.; Sankar Rao, C.; Sridevi, V.; Desinghu, J. Role of ZSM5 Catalyst and Char Susceptor on the Synthesis of Chemicals and Hydrocarbons from Microwave-Assisted in-Situ Catalytic Co-Pyrolysis of Algae and Plastic Wastes. Renew. Energy 2022, 181, 990–999. [Google Scholar] [CrossRef]
- Chen, Z.; Monzavi, M.; Latifi, M.; Samih, S.; Chaouki, J. Microwave-Responsive SiC Foam@zeolite Core-Shell Structured Catalyst for Catalytic Pyrolysis of Plastics. Environ. Pollut. 2022, 307, 119573. [Google Scholar] [CrossRef] [PubMed]
- Cao, Q.; Dai, H.-C.; He, J.-H.; Wang, C.-L.; Zhou, C.; Cheng, X.-F.; Lu, J.-M. Microwave-Initiated MAX Ti3AlC2-Catalyzed Upcycling of Polyolefin Plastic Wastes: Selective Conversion to Hydrogen and Carbon Nanofibers for Sodium-Ion Battery. Appl. Catal. B 2022, 318, 121828. [Google Scholar] [CrossRef]
- Arshad, H.; Sulaiman, S.A.; Hussain, Z.; Naz, M.Y.; Moni, M.N.Z. Effect of Input Power and Process Time on Conversion of Pure and Mixed Plastics into Fuels Through Microwave-Metal Interaction Pyrolysis. Waste Biomass Valorization 2021, 12, 3443–3457. [Google Scholar] [CrossRef]
- Jing, X.; Wen, H.; Gong, X.; Xu, Z. Heating Strategies for the System of PP and Spherical Activated Carbon during Microwave Cracking for Obtaining Value-Added Products. Fuel Process. Technol. 2020, 199, 106265. [Google Scholar] [CrossRef]
- Dai, L.; Zhou, N.; Li, H.; Wang, Y.; Liu, Y.; Cobb, K.; Cheng, Y.; Lei, H.; Chen, P.; Ruan, R. Catalytic Fast Pyrolysis of Low Density Polyethylene into Naphtha with High Selectivity by Dual-Catalyst Tandem Catalysis. Sci. Total Environ. 2021, 771, 144995. [Google Scholar] [CrossRef]
- Arshad, H.; Sulaiman, S.A.; Hussain, Z.; Moni, M.N.Z. Effect of Reaction Time and Microwave Power on Coil Temperature during Microwave-Metal Interaction Pyrolysis of Plastics. IOP Conf. Ser. Mater. Sci. Eng. 2020, 863, 012007. [Google Scholar] [CrossRef]
- Ramzan, F.; Shoukat, B.; Naz, M.Y.; Shukrullah, S.; Ahmad, F.; Naz, I.; Makhlouf, M.M.; Farooq, M.U.; Kamran, K. Single Step Microwaves Assisted Catalytic Conversion of Plastic Waste into Valuable Fuel and Carbon Nanotubes. Thermochim. Acta 2022, 715, 179294. [Google Scholar] [CrossRef]
- Wang, C.; Lei, H.; Qian, M.; Huo, E.; Zhao, Y.; Zhang, Q.; Mateo, W.; Lin, X.; Kong, X.; Zou, R.; et al. Application of Highly Stable Biochar Catalysts for Efficient Pyrolysis of Plastics: A Readily Accessible Potential Solution to a Global Waste Crisis. Sustain. Energy Fuels 2020, 4, 4614–4624. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, Y.; Duan, D.; Ruan, R.; Fan, L.; Zhou, Y.; Dai, L.; Lv, J.; Liu, Y. Fast Microwave-Assisted Ex-Catalytic Co-Pyrolysis of Bamboo and Polypropylene for Bio-Oil Production. Bioresour. Technol. 2018, 249, 69–75. [Google Scholar] [CrossRef]
- Solis, M.; Silveira, S. Technologies for Chemical Recycling of Household Plastics—A Technical Review and TRL Assessment. Waste Manag. 2020, 105, 128–138. [Google Scholar] [CrossRef] [PubMed]
- Ruj, B.; Ghosh, S. Technological Aspects for Thermal Plasma Treatment of Municipal Solid Waste—A Review. Fuel Process. Technol. 2014, 126, 298–308. [Google Scholar] [CrossRef]
- Dave, P.N.; Joshi, A.K. Plasma Pyrolysis and Gasification of Plastics Waste—A Review. J. Sci. Ind. Res. (JSIR) 2010, 69, 177–179. [Google Scholar]
- Bogaerts, A.; Neyts, E.; Gijbels, R.; van der Mullen, J. Gas Discharge Plasmas and Their Applications. Spectrochim. Acta Part. B At. Spectrosc. 2002, 57, 609–658. [Google Scholar] [CrossRef]
- Varshney, R.; Singh, P.; Yadav, D. Hazardous Wastes Treatment, Storage, and Disposal Facilities. In Hazardous Waste Management; Elsevier: Amsterdam, The Netherlands, 2022; pp. 33–64. [Google Scholar]
- Xiao, H.; Harding, J.; Lei, S.; Chen, W.; Xia, S.; Cai, N.; Chen, X.; Hu, J.; Chen, Y.; Wang, X.; et al. Hydrogen and Aromatics Recovery through Plasma-Catalytic Pyrolysis of Waste Polypropylene. J. Clean. Prod. 2022, 350, 131467. [Google Scholar] [CrossRef]
- Gabbar, H.; Aboughaly, M.; Stoute, C.A. DC Thermal Plasma Design and Utilization for the Low Density Polyethylene to Diesel Oil Pyrolysis Reaction. Energies 2017, 10, 784. [Google Scholar] [CrossRef]
- Diaz-Silvarrey, L.S.; Zhang, K.; Phan, A.N. Monomer Recovery through Advanced Pyrolysis of Waste High Density Polyethylene (HDPE). Green. Chem. 2018, 20, 1813–1823. [Google Scholar] [CrossRef]
- Huang, H.; Tang, L. Treatment of Organic Waste Using Thermal Plasma Pyrolysis Technology. Energy Convers. Manag. 2007, 48, 1331–1337. [Google Scholar] [CrossRef]
- Tang, L.; Huang, H.; Zhao, Z.; Wu, C.Z.; Chen, Y. Pyrolysis of Polypropylene in a Nitrogen Plasma Reactor. Ind. Eng. Chem. Res. 2003, 42, 1145–1150. [Google Scholar] [CrossRef]
- Guddeti, R.R.; Knight, R.; Grossmann, E.D. Depolymerization of Polypropylene in an Induction-Coupled Plasma (ICP) Reactor. Ind. Eng. Chem. Res. 2000, 39, 1171–1176. [Google Scholar] [CrossRef]
- Mohsenian, S.; Esmaili, M.S.; Fathi, J.; Shokri, B. Hydrogen and Carbon Black Nano-Spheres Production via Thermal Plasma Pyrolysis of Polymers. Int. J. Hydrogen Energy 2016, 41, 16656–16663. [Google Scholar] [CrossRef]
- Yao, L.; King, J.; Wu, D.; Chuang, S.S.C.; Peng, Z. Non-Thermal Plasma-Assisted Hydrogenolysis of Polyethylene to Light Hydrocarbons. Catal. Commun. 2021, 150, 106274. [Google Scholar] [CrossRef]
- Matuszewska, A.; Owczuk, M.; Biernat, K. Current Trends in Waste Plastics’ Liquefaction into Fuel Fraction: A Review. Energies 2022, 15, 2719. [Google Scholar] [CrossRef]
- Fortunati, E.; Luzi, F.; Puglia, D.; Torre, L. Extraction of Lignocellulosic Materials From Waste Products. In Multifunctional Polymeric Nanocomposites Based on Cellulosic Reinforcements; Elsevier: Amsterdam, The Netherlands, 2016; pp. 1–38. [Google Scholar]
- Li, K.; Xu, Z. A Review of Current Progress of Supercritical Fluid Technologies for E-Waste Treatment. J. Clean. Prod. 2019, 227, 794–809. [Google Scholar] [CrossRef]
- Cheng, Y.; Xue, F.; Yu, S.; Du, S.; Yang, Y. Subcritical Water Extraction of Natural Products. Molecules 2021, 26, 4004. [Google Scholar] [CrossRef]
- Karki, S.; Sanjel, N.; Poudel, J.; Choi, J.H.; Oh, S.C. Supercritical Transesterification of Waste Vegetable Oil: Characteristic Comparison of Ethanol and Methanol as Solvents. Appl. Sci. 2017, 7, 632. [Google Scholar] [CrossRef]
- Gómez-Hernández, J.; Grimes, R.; Briongos, J.V.; Marugán-Cruz, C.; Santana, D. Carbon Dioxide and Acetone Mixtures as Refrigerants for Industry Heat Pumps to Supply Temperature in the Range 150–220 OC. Energy 2023, 269, 126821. [Google Scholar] [CrossRef]
- Moriya, T.; Enomoto, H. Characteristics of Polyethylene Cracking in Supercritical Water Compared to Thermal Cracking. Polym. Degrad. Stab. 1999, 65, 373–386. [Google Scholar] [CrossRef]
- Genta, M.; Iwaya, T.; Sasaki, M.; Goto, M. Supercritical Methanol for Polyethylene Terephthalate Depolymerization: Observation Using Simulator. Waste Manag. 2007, 27, 1167–1177. [Google Scholar] [CrossRef]
- Watanabe, M.; Adschiri, T.; Arai, K. Supercritical Fluid in Polymer Science and Technology. II. Polyethylene Decomposition via Pyrolysis and Partial Oxidation in Supercritical Water. Kobunshi Ronbunshu 2001, 58, 631–641. [Google Scholar] [CrossRef]
- Čolnik, M.; Kotnik, P.; Knez, Ž.; Škerget, M. Chemical Recycling of Polyolefins Waste Materials Using Supercritical Water. Polymers 2022, 14, 4415. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.-T.; Jin, K.; Linda Wang, N.-H. Use of Supercritical Water for the Liquefaction of Polypropylene into Oil. ACS Sustain. Chem. Eng. 2019, 7, 3749–3758. [Google Scholar] [CrossRef]
- Seshasayee, M.S.; Savage, P.E. Oil from Plastic via Hydrothermal Liquefaction: Production and Characterization. Appl. Energy 2020, 278, 115673. [Google Scholar] [CrossRef]
- Su, X.; Zhao, Y.; Zhang, R.; Bi, J. Investigation on Degradation of Polyethylene to Oils in Supercritical Water. Fuel Process. Technol. 2004, 85, 1249–1258. [Google Scholar] [CrossRef]
- Lu, T.; Jan, K.; Chen, W.-T. Hydrothermal Liquefaction of Pretreated Polyethylene-Based Ocean-Bound Plastic Waste in Supercritical Water. J. Energy Inst. 2022, 105, 282–292. [Google Scholar] [CrossRef]
- Liu, Y.; Chandra Akula, K.; Phani Raj Dandamudi, K.; Liu, Y.; Xu, M.; Sanchez, A.; Zhu, D.; Deng, S. Effective Depolymerization of Polyethylene Plastic Wastes under Hydrothermal and Solvothermal Liquefaction Conditions. Chem. Eng. J. 2022, 446, 137238. [Google Scholar] [CrossRef]
- Zhang, H.; Su, X.; Sun, D.; Zhang, R.; Bi, J. Investigation on Degradation of Polyethylene to Oil in a Continuous Supercritical Water Reactor. J. Fuel Chem. Technol. 2007, 35, 487–491. [Google Scholar] [CrossRef]
- Jin, K.; Vozka, P.; Kilaz, G.; Chen, W.-T.; Wang, N.-H.L. Conversion of Polyethylene Waste into Clean Fuels and Waxes via Hydrothermal Processing (HTP). Fuel 2020, 273, 117726. [Google Scholar] [CrossRef]
- Mura Technology. The Next Generation of Advanced Plastic. Available online: https://muratechnology.com/app/uploads/2025/04/Mura-Technology-2025-Brochure.pdf (accessed on 24 June 2025).
- Mura Technology. Dow and Mura Technology Announce Partnership to Scale Game-Changing New Advanced Recycling Solution for Plastics. Available online: https://corporate.dow.com/en-us/news/press-releases/dow-and-mura-technology-announce-partnership-to-scale-game-chang.html (accessed on 24 June 2025).
- Mura Technology. Hydrothermal Plastic Recycling Technology. Available online: https://muratechnology.com/hydroprt/#:~:text=HYDROTHERMAL%20PLASTIC%20RECYCLING%20TECHNOLOGY,of%20stakeholders%20working%20with%20polymers (accessed on 24 June 2025).
- Stopford. Redefining the Management of Waste Plastics. Available online: https://www.stopford.co.uk/capabilities-circuplast (accessed on 24 June 2025).
- Rahman, M.H.; Bhoi, P.R.; Menezes, P.L. Pyrolysis of Waste Plastics into Fuels and Chemicals: A Review. Renew. Sustain. Energy Rev. 2023, 188, 113799. [Google Scholar] [CrossRef]
- Al-Rumaihi, A.; Shahbaz, M.; Mckay, G.; Mackey, H.; Al-Ansari, T. A Review of Pyrolysis Technologies and Feedstock: A Blending Approach for Plastic and Biomass towards Optimum Biochar Yield. Renew. Sustain. Energy Rev. 2022, 167, 112715. [Google Scholar] [CrossRef]
- Chen, D.; Yin, L.; Wang, H.; He, P. Pyrolysis Technologies for Municipal Solid Waste: A Review. Waste Manag. 2014, 34, 2466–2486. [Google Scholar] [CrossRef] [PubMed]
- Teixeira Cardoso, A.R.; Conrado, N.M.; Krause, M.C.; Bjerk, T.R.; Krause, L.C.; Caramão, E.B. Chemical Characterization of the Bio-Oil Obtained by Catalytic Pyrolysis of Sugarcane Bagasse (Industrial Waste) from the Species Erianthus Arundinaceus. J. Environ. Chem. Eng. 2019, 7, 102970. [Google Scholar] [CrossRef]
- Kumar, R.; Strezov, V.; Lovell, E.; Kan, T.; Weldekidan, H.; He, J.; Dastjerdi, B.; Scott, J. Bio-Oil Upgrading with Catalytic Pyrolysis of Biomass Using Copper/Zeolite-Nickel/Zeolite and Copper-Nickel/Zeolite Catalysts. Bioresour. Technol. 2019, 279, 404–409. [Google Scholar] [CrossRef] [PubMed]
- Aguado, J.; Serrano, D.P.; San Miguel, G.; Castro, M.C.; Madrid, S. Feedstock Recycling of Polyethylene in a Two-Step Thermo-Catalytic Reaction System. J. Anal. Appl. Pyrolysis 2007, 79, 415–423. [Google Scholar] [CrossRef]
- Peng, Y.; Wang, Y.; Ke, L.; Dai, L.; Wu, Q.; Cobb, K.; Zeng, Y.; Zou, R.; Liu, Y.; Ruan, R. A Review on Catalytic Pyrolysis of Plastic Wastes to High-Value Products. Energy Convers. Manag. 2022, 254, 115243. [Google Scholar] [CrossRef]
- Jahirul, M.I.; Rasul, M.G.; Schaller, D.; Khan, M.M.K.; Hasan, M.M.; Hazrat, M.A. Transport Fuel from Waste Plastics Pyrolysis—A Review on Technologies, Challenges and Opportunities. Energy Convers. Manag. 2022, 258, 115451. [Google Scholar] [CrossRef]
- Harussani, M.M.; Sapuan, S.M.; Rashid, U.; Khalina, A.; Ilyas, R.A. Pyrolysis of Polypropylene Plastic Waste into Carbonaceous Char: Priority of Plastic Waste Management amidst COVID-19 Pandemic. Sci. Total Environ. 2022, 803, 149911. [Google Scholar] [CrossRef]
- Fadillah, G.; Fatimah, I.; Sahroni, I.; Musawwa, M.M.; Mahlia, T.M.I.; Muraza, O. Recent Progress in Low-Cost Catalysts for Pyrolysis of Plastic Waste to Fuels. Catalysts 2021, 11, 837. [Google Scholar] [CrossRef]
- Donaj, P.J.; Kaminsky, W.; Buzeto, F.; Yang, W. Pyrolysis of Polyolefins for Increasing the Yield of Monomers’ Recovery. Waste Manag. 2012, 32, 840–846. [Google Scholar] [CrossRef]
- Wei, T.-T.; Wu, K.-J.; Lee, S.-L.; Lin, Y.-H. Chemical Recycling of Post-Consumer Polymer Waste over Fluidizing Cracking Catalysts for Producing Chemicals and Hydrocarbon Fuels. Resour. Conserv. Recycl. 2010, 54, 952–961. [Google Scholar] [CrossRef]
- Lin, Y.-H.; Yen, H.-Y. Fluidised Bed Pyrolysis of Polypropylene over Cracking Catalysts for Producing Hydrocarbons. Polym. Degrad. Stab. 2005, 89, 101–108. [Google Scholar] [CrossRef]
- Lin, Y.-H.; Yang, M.-H. Tertiary Recycling of Polyethylene Waste by Fluidised-Bed Reactions in the Presence of Various Cracking Catalysts. J. Anal. Appl. Pyrolysis 2008, 83, 101–109. [Google Scholar] [CrossRef]
- Sharratt, P.N.; Lin, Y.-H.; Garforth, A.A.; Dwyer, J. Investigation of the Catalytic Pyrolysis of High-Density Polyethylene over a HZSM-5 Catalyst in a Laboratory Fluidized-Bed Reactor. Ind. Eng. Chem. Res. 1997, 36, 5118–5124. [Google Scholar] [CrossRef]
- Garcia-Nunez, J.A.; Pelaez-Samaniego, M.R.; Garcia-Perez, M.E.; Fonts, I.; Abrego, J.; Westerhof, R.J.M.; Garcia-Perez, M. Historical Developments of Pyrolysis Reactors: A Review. Energy Fuels 2017, 31, 5751–5775. [Google Scholar] [CrossRef]
- Miandad, R.; Barakat, M.A.; Aburiazaiza, A.S.; Rehan, M.; Nizami, A.S. Catalytic Pyrolysis of Plastic Waste: A Review. Process Saf. Environ. Prot. 2016, 102, 822–838. [Google Scholar] [CrossRef]
- Marcilla, A.; Gómez-Siurana, A.; Berenguer, D. Study of the Decomposition of Low Density Polyethylene Blends with Vacuum Gas Oils: Evolution of the Gases. Polym. Degrad. Stab. 2008, 93, 2204–2213. [Google Scholar] [CrossRef]
- Elordi, G.; Olazar, M.; Lopez, G.; Amutio, M.; Artetxe, M.; Aguado, R.; Bilbao, J. Catalytic Pyrolysis of HDPE in Continuous Mode over Zeolite Catalysts in a Conical Spouted Bed Reactor. J. Anal. Appl. Pyrolysis 2009, 85, 345–351. [Google Scholar] [CrossRef]
- Cai, N.; Li, X.; Xia, S.; Sun, L.; Hu, J.; Bartocci, P.; Fantozzi, F.; Williams, P.T.; Yang, H.; Chen, H. Pyrolysis-Catalysis of Different Waste Plastics over Fe/Al2O3 Catalyst: High-Value Hydrogen, Liquid Fuels, Carbon Nanotubes and Possible Reaction Mechanisms. Energy Convers. Manag. 2021, 229, 113794. [Google Scholar] [CrossRef]
- Li, Q.-L.; Shan, R.; Zhang, J.; Lei, M.; Yuan, H.-R.; Chen, Y. Enhancement of Hydrogen and Carbon Nanotubes Production from Hierarchical Ni/ZSM-5 Catalyzed Polyethylene Pyrolysis. J. Anal. Appl. Pyrolysis 2023, 169, 105829. [Google Scholar] [CrossRef]
- Lin, Y.-H.; Yang, M.-H. Catalytic Pyrolysis of Polyolefin Waste into Valuable Hydrocarbons over Reused Catalyst from Refinery FCC Units. Appl. Catal. A Gen. 2007, 328, 132–139. [Google Scholar] [CrossRef]
- Ali, S.; Garforth, A.A.; Harris, D.H.; Rawlence, D.J.; Uemichi, Y. Polymer Waste Recycling over “Used” Catalysts. Catal. Today 2002, 75, 247–255. [Google Scholar] [CrossRef]
- Kodera, Y.; Ishihara, Y.; Kuroki, T. Novel Process for Recycling Waste Plastics To Fuel Gas Using a Moving-Bed Reactor. Energy Fuels 2006, 20, 155–158. [Google Scholar] [CrossRef]
- Petersen, H.A.; Myren, T.H.T.; O’Sullivan, S.J.; Luca, O.R. Electrochemical Methods for Materials Recycling. Mater. Adv. 2021, 2, 1113–1138. [Google Scholar] [CrossRef]
- Jung, S.-H.; Cho, M.-H.; Kang, B.-S.; Kim, J.-S. Pyrolysis of a Fraction of Waste Polypropylene and Polyethylene for the Recovery of BTX Aromatics Using a Fluidized Bed Reactor. Fuel Process. Technol. 2010, 91, 277–284. [Google Scholar] [CrossRef]
- Park, K.-B.; Jeong, Y.-S.; Kim, J.-S. Activator-Assisted Pyrolysis of Polypropylene. Appl. Energy 2019, 253, 113558. [Google Scholar] [CrossRef]
- Elordi, G.; Olazar, M.; Lopez, G.; Artetxe, M.; Bilbao, J. Product Yields and Compositions in the Continuous Pyrolysis of High-Density Polyethylene in a Conical Spouted Bed Reactor. Ind. Eng. Chem. Res. 2011, 50, 6650–6659. [Google Scholar] [CrossRef]
- Zhang, W.; Killian, L.; Thevenon, A. Electrochemical Recycling of Polymeric Materials. Chem. Sci. 2024, 15, 8606–8624. [Google Scholar] [CrossRef]
- Weber, R.S. Electrochemical Upvaluing of Waste Plastic. Curr. Opin. Electrochem. 2024, 46, 101493. [Google Scholar] [CrossRef]
- Zhou, H.; Wang, Y.; Ren, Y.; Li, Z.; Kong, X.; Shao, M.; Duan, H. Plastic Waste Valorization by Leveraging Multidisciplinary Catalytic Technologies. ACS Catal. 2022, 12, 9307–9324. [Google Scholar] [CrossRef]
- Wang, J.; Li, X.; Wang, M.; Zhang, T.; Chai, X.; Lu, J.; Wang, T.; Zhao, Y.; Ma, D. Electrocatalytic Valorization of Poly(Ethylene Terephthalate) Plastic and CO2 for Simultaneous Production of Formic Acid. ACS Catal. 2022, 12, 6722–6728. [Google Scholar] [CrossRef]
- Pichler, C.M.; Bhattacharjee, S.; Rahaman, M.; Uekert, T.; Reisner, E. Conversion of Polyethylene Waste into Gaseous Hydrocarbons via Integrated Tandem Chemical–Photo/Electrocatalytic Processes. ACS Catal. 2021, 11, 9159–9167. [Google Scholar] [CrossRef] [PubMed]
- Partenheimer, W. Valuable Oxygenates by Aerobic Oxidation of Polymers Using Metal/Bromide Homogeneous Catalysts. Catal. Today 2003, 81, 117–135. [Google Scholar] [CrossRef]
- Pifer, A.; Sen, A. Chemical Recycling of Plastics to Useful Organic Compounds by Oxidative Degradation. Angew. Chem. Int. Ed. 1998, 37, 3306–3308. [Google Scholar] [CrossRef]
- Jiao, X.; Zheng, K.; Chen, Q.; Li, X.; Li, Y.; Shao, W.; Xu, J.; Zhu, J.; Pan, Y.; Sun, Y.; et al. Photocatalytic Conversion of Waste Plastics into C2 Fuels under Simulated Natural Environment Conditions. Angew. Chem. Int. Ed. 2020, 59, 15497–15501. [Google Scholar] [CrossRef]
- Jiang, T.; Zhao, X.; Gu, D.; Yan, C.; Jiang, H.; Wu, H.; Wang, B.; Wang, X. STEP Polymer Degradation: Solar Thermo-Coupled Electrochemical Depolymerization of Plastics to Generate Useful Fuel plus Abundant Hydrogen. Sol. Energy Mater. Sol. Cells 2020, 204, 110208. [Google Scholar] [CrossRef]
- Bäckström, E.; Odelius, K.; Hakkarainen, M. Trash to Treasure: Microwave-Assisted Conversion of Polyethylene to Functional Chemicals. Ind. Eng. Chem. Res. 2017, 56, 14814–14821. [Google Scholar] [CrossRef]
- Bäckström, E.; Odelius, K.; Hakkarainen, M. Designed from Recycled: Turning Polyethylene Waste to Covalently Attached Polylactide Plasticizers. ACS Sustain. Chem. Eng. 2019, 7, 11004–11013. [Google Scholar] [CrossRef]
- MacFarlane, D.R.; Tachikawa, N.; Forsyth, M.; Pringle, J.M.; Howlett, P.C.; Elliott, G.D.; Davis, J.H.; Watanabe, M.; Simon, P.; Angell, C.A. Energy Applications of Ionic Liquids. Energy Environ. Sci. 2014, 7, 232–250. [Google Scholar] [CrossRef]
- Plechkova, N.V.; Seddon, K.R. Applications of Ionic Liquids in the Chemical Industry. Chem. Soc. Rev. 2008, 37, 123–150. [Google Scholar] [CrossRef]
- Zhang, W.; Kim, S.; Wahl, L.; Khare, R.; Hale, L.; Hu, J.; Camaioni, D.M.; Gutiérrez, O.Y.; Liu, Y.; Lercher, J.A. Low-Temperature Upcycling of Polyolefins into Liquid Alkanes via Tandem Cracking-Alkylation. Science 2023, 379, 807–811. [Google Scholar] [CrossRef]
- Adams, C.J.; Earle, M.J.; Seddon, K.R. Catalytic Cracking Reactions of Polyethylene to Light Alkanes. Green. Chem. 2000, 2, 21–24. [Google Scholar] [CrossRef]
- Jia, X.; Qin, C.; Friedberger, T.; Guan, Z.; Huang, Z. Efficient and Selective Degradation of Polyethylenes into Liquid Fuels and Waxes under Mild Conditions. Sci. Adv. 2016, 2, e1501591. [Google Scholar] [CrossRef] [PubMed]
- Goldman, A.S.; Roy, A.H.; Huang, Z.; Ahuja, R.; Schinski, W.; Brookhart, M. Catalytic Alkane Metathesis by Tandem Alkane Dehydrogenation-Olefin Metathesis. Science 2006, 312, 257–261. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wu, K.; Wang, S.; Yu, J.; Tao, Y.; Zhang, H. Preparation of Monocyclic Aromatic Hydrocarbons from Chlorine-Containing Mixed Waste Plastics via Tandem Catalysis Coupled with Hydrothermal Pretreatment. Chem. Eng. J. 2023, 465, 142885. [Google Scholar] [CrossRef]
- Zhang, F.; Zeng, M.; Yappert, R.D.; Sun, J.; Lee, Y.-H.; LaPointe, A.M.; Peters, B.; Abu-Omar, M.M.; Scott, S.L. Polyethylene Upcycling to Long-Chain Alkylaromatics by Tandem Hydrogenolysis/Aromatization. Science 2020, 370, 437–441. [Google Scholar] [CrossRef]
- Dai, L.; Zhou, N.; Lv, Y.; Cobb, K.; Cheng, Y.; Wang, Y.; Liu, Y.; Chen, P.; Zou, R.; Lei, H.; et al. Pyrolysis-Catalysis for Waste Polyolefin Conversion into Low Aromatic Naphtha. Energy Convers. Manag. 2021, 245, 114578. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Authors | Feedstock | Catalyst | Temperature (°C) | Maximum Gaseous Yield (wt%) | Gas Composition |
|---|---|---|---|---|---|
| Undri et al. (2014) [28] | HDPE | - | 25–250 | ~34 | - |
| Zhang et al. (2015) [27] | LDPE | ZSM-5 | 300–500 | ~70 | H2, methane, ethane, ethylene, and others |
| Suriapparao et al. (2015) [25] | PP | (Susceptors) graphite, aluminum, silicon carbide, activated carbon, lignin and fly ash | 260 | ~87 | 0–12, 68 wt% of C3 |
| Suriapparao et al. (2022) [30] | PP, PE, EPS | ZSM-5 | 600 | ~50 | - |
| Zhao et al. (2018) [39] | PP | ZSM-5 | 550 | ~30 | - |
| Chen et al. (2022) [31] | LDPE | ZSM-5 | 450 | ~75 | ~50 wt% of olefins C2, C3, and C4 |
| Cao et al. (2022) [32] | LDPE, HDPE | MAX (Ti3AlC2) | 500–1000 | >60 | High purity hydrogen, and value-added graphitic carbon nanofibers |
| Jing et al. (2017) [29] | LDPE, HDPE, PP | - | zone 1:1000 zone 2:550 | ~85 | ~55 wt% of olefins C2, C3 and C4 |
| Authors | Feedstock | Power Input | Temperature (°C) | Maximum Gaseous Yield (wt%) | Gas Composition (mol%) |
|---|---|---|---|---|---|
| Tang et al. (2003) [49] | PP | 35.2 kVA | Not informed | ~96 | H2 (~54%), acetylene (~17%), methane (~5.0%), CO (~2%), and CxHy and unknown (~19%) |
| Guddeti et al. (2000) [50] | PP | 10–20 kVA | 2727–7727 | ~78 | Propylene (93.7%,) methane (2.6%), ethylene (1.7%), and butanes and butenes (1.3%) |
| Mohsenian et al. (2016) [51] | PE and PP | Not informed | 10,727–15,727 | - | H2 (up to 67.4%) and hydrocarbons (up to 47.4%) |
| Yao et al. (2021) [52] | HDPE | 60–90 W | Room temp. | ~95 | Methane (70%) and other hydrocarbons |
| Xiao et al. (2022) [45] | PP | 60–120 W | 200–500 | ~45 | H2 and hydrocarbons in different quantities |
| Gabbar et al. (2017) [46] | LDPE | 270 W | 550 | ~7 | - |
| Diaz-Silvarrey et al. (2018) [47] | HDPE | 30–60 W | 500–700 | ~60 | H2 and hydrocarbons in different quantities |
| Solvent | Critical Temperature (°C) | Critical Pressure (MPa) | Refs. |
|---|---|---|---|
| Water | 374.15 | 22.05 | [56] |
| Ethanol | 240.75 | 6.14 | [57] |
| Acetone | 234.95 | 4.70 | [58] |
| CO2 | 30.98 | 7.38 | [58] |
| Methanol | 239.45 | 8.10 | [58] |
| Authors | Feedstock | Pressure | Temperature (°C) | Maximum Gaseous Production | Gas Composition (mol%) |
|---|---|---|---|---|---|
| Watanabe et al. (2001) [61] | PE | Not informed | 420 | - | C1 to C4 molecules (~30%), 60% of CO, CO2, and H2 |
| Moriya et al. (1999) [59] | HDPE | 42 MPa | 420 | 6.5–13.2% | Methane (34.3%), ethane (28.5%), propane (14.6%), CO, CO2, H2, and other hydrocarbons (small amounts) |
| Čolnik et al. (2022) [62] | PP | 40 MPa | 425 and 450 | ~20% | C2 to C4 (70 to 80%), CO2, C1, and other hydrocarbons (small amounts) |
| Chen et al. (2019) [63] | PP | 23 MPa | 380 to 500 | ~20–30% | C3 (45%) |
| Jin et al. (2020) [69] | PE | 23 MPa | 380 to 500 | ~20% | Ethane (5.8%), propene (38.3%), C4 olefins (19.3%), other olefinic hydrocarbons for (5.5%), and alkanes (30.6%) |
| Seshasayee et al. (2020) [64] | PP | 25 MPa | 350 to 450 | ~80% | - |
| Su et al. (2004) [65] | HDPE | Not informed | 450 and 480 | ~30% | C2–C4 (74.40%) |
| Lu et al. (2022) [66] | HDPE | 23 MPa | 425 to 475 | ~20% | Paraffins and olefins |
| Liu et al. (2022) [67] | PE | 9 to 23 MPa | 300 to 375 | ~55% | H2 (6%), CH4 (94%) |
| Zhang et al. (2007) [68] | HDPE | 25 MPa | 500 to 550 | ~40% | C1 to >C5 (most C2 and C3), H2, and others |
| Authors | Feedstock | Reactor | Catalyst | Temperature (°C) | Time | Monomer (wt%) |
|---|---|---|---|---|---|---|
| Lin et al. (2008) [87] | HPDE/LDPE | Fluidized-bed | HUSY, HZSM-5, HMOR, SAHA, MCM-41 | 290, 330, 360, 390, 430 | 1–20 min | 4% C2; 23% C3 |
| Aguado et al. (2007) [79] | LDPE | Batch reactor—fixed bed reactor | HZSM-5, Al-MCM-41 | 425, 450, 475 | 120 min | >50% (C1–C4) |
| Marcilla et al. (2008) [91] | LDPE | Batch reactor—fixed bed reactor | FCC | 350–550 | 52 min | 4% C2; 15% C3, 20% C4 |
| Zhang et al. (2015) [27] | LDPE | Microwave + packed-bed reactor | ZSM-5 | 249–450 | - | 80% ethylene (375 °C) |
| Sharratt et al. (1997) [88] | HDPE | Fluidized-bed reactor | HZSM-5 | 290, 330, 360, 390, 430 | 30, 20, 15 min | 26.5% propene |
| Lin et al. (2005) [86] | PP | Fluidized-bed reactor | USY | 290, 330, 360, 390, 430 | 15 min | C1–C4 |
| Lin et al. (2007) [95] | PE/PP | Fluidized-bed reactor | RCat-c1 (FCC), USY, ZSM-5, SAHA, Silicalite | 330, 360, 390, 420, 450 | 30 min | C1–C4 |
| Wei et al. (2010) [85] | LDPE/HDPE/PP | Fluidized-bed reactor | USY | 290, 330, 360, 390, 420 | 20 min | C1–C4 |
| Donaj et al. (2012) [84] | LDPE/HDPE/PP | Fluidized quartz-bed reactor | TiCl4/MgCl2 | 500, 650 | 1.67, 2.5 h | 12% methane, 6% ethane, 13% ethene, 12% propane, and 12% propene |
| Jung et al. (2010) [99] | PE/PP | Fluidized-bed reactor | Quartz sand | 650, 750 | - | 34% methane, 7% ethane, 12% ethene, 1% propane, and 5% propene |
| Park et al. (2019) [100] | PP | Fluidized-bed reactors connected in series | Sand | 400 | - | 52% of ethene, propene, 1,3-butadiene, and butenes |
| Elordi et al. (2009) [92] | HDPE | Conical spouted bed reactor (CSBR) | HZSM-5, HY and Hβ zeolite | 500 | - | 70% (HZSM-5), 25% (HY), and 40% (Hβ) |
| Elordi et al. (2011) [101] | HDPE | Conical spouted bed reactor (CSBR) | HZSM-5, HY and Hβ zeolite | 500 | 15 h | 55% C2–C4 (HZSM-5), 20% C2–C4 (HY), and 25% C2–C4 (Hβ) |
| Lin et al. (2008) [87] | HDPE/LDPE | Fluidized-bed reactor | HZSM-5 | 290, 330, 360, 390, 430 | 20 min | C1–C4 |
| Ali et al. (2002) [96] | HDPE | Fluidized-bed reactor | ZSM-5, US-Y, ASA, Cat-A (FCC), E-Cat | 360, 450 | - | 72.6 (C1–C4) 68.6 (C1–C4) |
| Kodera et al. (2006) [97] | PP | Moving-bed reactor | Silica–alumina | 700 | 10 min | methane, 18.7 %; ethylene, 19.5 %, ethane, 9.7 %; propylene, 24.2 %; propane, 3.4 % |
| Cai et al. (2021) [93] | PP, HDPE and LDPE | Two stages fixed-bed reactor | Fe/Al2O3 | 500–800 | 30 min | Ethylene 3.7% |
| Li et al. (2023) [94] | PE | Two stages fixed-bed reactor | Ni/ZSM-5 | 500–800 | 40 min | Traces |
| Authors | Feedstock | Catalyst | Temperature (°C) | Maximum Gaseous Production | Gas Composition |
|---|---|---|---|---|---|
| Wang et al. (2022) [105] | PET | NiCo2O4 | - | - | - |
| Pichler et al. (2021) [106] | PE | TiO2, carbon nitride | 180 °C | 20% | H2, CO2, ethane, ethene, propane, and propylene |
| Partenheimer et al. (2003) [107] | PVC, PS, PP, PE, PET, PBT, PEN | Co/Mn/Br/Zr, Co, Co/Zr, Co/Mn, Co/Ce, Co/Ni, Ni, Co/NHPI | 150–220 °C | - | - |
| Pifer et al. (1998) [108] | PE, PP, PMMA, PS PAM | - | 170 °C | - | - |
| Jiao et al. (2020) [109] | PE, PP | Nb2O5 | 25 °C | - | - |
| Jiang et al. (2020) [110] | PP | - | 350 °C | 60.63% | C1–C5, H2 |
| Bäckström et al. (2017) [111] | LDPE | Nitric acid | 180 °C | - | - |
| Bäckström et al. (2019) [112] | HDPE | Nitric acid and crotonic acid | 180 °C | - | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carvalho, L.; Mattos, G.; Sitton, N.; Barros, J.; Miranda, D.; Luciano, R.; Pinto, J.C. A Survey on the Chemical Recycling of Polyolefins into Monomers. Processes 2025, 13, 2114. https://doi.org/10.3390/pr13072114
Carvalho L, Mattos G, Sitton N, Barros J, Miranda D, Luciano R, Pinto JC. A Survey on the Chemical Recycling of Polyolefins into Monomers. Processes. 2025; 13(7):2114. https://doi.org/10.3390/pr13072114
Chicago/Turabian StyleCarvalho, Larissa, Gabriela Mattos, Natasha Sitton, Jamilly Barros, Débora Miranda, Rodrigo Luciano, and José Carlos Pinto. 2025. "A Survey on the Chemical Recycling of Polyolefins into Monomers" Processes 13, no. 7: 2114. https://doi.org/10.3390/pr13072114
APA StyleCarvalho, L., Mattos, G., Sitton, N., Barros, J., Miranda, D., Luciano, R., & Pinto, J. C. (2025). A Survey on the Chemical Recycling of Polyolefins into Monomers. Processes, 13(7), 2114. https://doi.org/10.3390/pr13072114