Abstract
The discharge of dye-laden effluents remains an environmental challenge since conventional treatments remove color but not the organic load. This study systematically compared anodic oxidation (AO), electro-Fenton (EF), and photoelectro-Fenton (PEF) processes for three representative industrial dyes, such as Coriasol Red CB, Brown RBH, and Blue VT, and their ternary mixture, using boron-doped diamond (BDD) and Ti/IrO2–SnO2–Sb2O5 (MMO) anodes. Experiments were conducted in a batch reactor with 50 mM Na2SO4 at pH = 3.0 and current densities of 20–60 mA cm−2. Kinetic analysis showed that AO-BDD was most effective at low pollutant loads, EF-BDD became superior at medium loads due to efficient H2O2 electrogeneration, and PEF-MMO dominated at higher loads by fast UVA photolysis of surface Fe(OH)2+ complexes. In a ternary mixture of 120 mg L−1 of dyes, EF-BDD and PEF-MMO achieved >98% decolorization in 22–23 min with pseudo-first-order rate constants of 0.111–0.136 min−1, whereas AO processes remained slower. COD assays revealed partial mineralization of 60–80%, with EF-BDD providing the most consistent reduction and PEF-MMO minimizing treatment time. These findings confirm that decolorization overestimates efficiency, and electrode selection must be tailored to dye structure and effluent composition. Process selection rules allow us to conclude that EF-BDD is the best robust dark option, and PEF-MMO, when UVA is available, offers practical guidelines for cost-effective electrochemical treatment of textile wastewater.
1. Introduction
The fast and sustained growth of the textile and dyeing industry has led to the discharge of substantial volumes of wastewater containing synthetic dyes. These effluents are typically characterized by intense coloration, chemical recalcitrance, and a complex matrix of auxiliary chemicals, including salts, surfactants, and organic additives [1,2]. The most commonly used dyes, like azo, anthraquinone, and phthalocyanine derivatives, are designed to be highly stable, a property that, while desirable in their application, makes them notoriously resistant to conventional biodegradation pathways [3,4,5]. As a result, the discharge of untreated or partially treated dye-laden wastewater into aquatic environments causes severe ecological disturbances, including inhibition of photosynthesis due to light attenuation, disruption of aquatic food chains, and bioaccumulation of mutagenic or carcinogenic compounds [2].
Traditional treatment strategies such as coagulation–flocculation, activated sludge systems, adsorption on activated carbon, and membrane-based separation processes have demonstrated varying degrees of success but share common limitations [6,7,8]. These methods often focus on phase transfer rather than degradation, producing concentrated sludge or secondary waste streams that require further handling. Moreover, membrane fouling, high operational costs, and limited capacity to treat recalcitrant dyes under extreme conditions (e.g., high salinity or low biodegradability) compromise their long-term sustainability [9]. These limitations have intensified the search for advanced oxidation processes (AOPs), which are capable of achieving mineralization of a wide range of organic pollutants through the in situ generation of highly reactive species, primarily hydroxyl radicals (•OH) that can non-selectively attack complex molecular structures [10,11]. Hybrid electrochemical trains, e.g., sulfate-radical, Electrochemical Advanced Oxidation Processes (EAOPs) coupled to electrocoagulation, are also being explored for dyeing wastewater [12].
Recent studies have expanded the scope of electrochemical and photocatalytic oxidation systems by developing multifunctional materials and optimized process configurations for dye degradation. For instance, Thomas et al. [13] reported a bifunctional NiFe2O4@C/PPy composite capable of simultaneous electrochemical detection and catalytic oxidation of Rhodamine B, illustrating the kinetic advantages of hybrid catalysts. Similarly, Bonthula et al. [14] demonstrated efficient photodegradation of mixed dyes using Pd-doped CuO–ZnO composites under mild conditions, highlighting the importance of simultaneous treatment of anionic and neutral species. In parallel, Malitha et al. [15] optimized chloride-mediated Electrochemical Advanced Oxidation Processes (EAOPs) for the treatment of commercial azo dyes and real dyeing effluents, underscoring the practical relevance of tailoring electrolyte composition and current density. These recent advances further motivate systematic comparisons of AO, EF, and PEF processes under identical electrochemical conditions to delineate their relative efficiencies and mechanistic domains.
In this context, electrochemical advanced oxidation processes (EAOPs) have emerged as particularly attractive technologies due to their environmental compatibility, operational simplicity, and compatibility with renewable energy sources [12,16]. These processes operate under ambient temperature and pressure without the need for external chemical dosing, making them suitable for decentralized applications. Among them, anodic oxidation (AO) and electro-Fenton (EF) are the most extensively studied. AO generates hydroxyl radicals at the anode surface, providing boron-doped diamond (BDD) with the highest flux of physisorbed BDD(•OH) due to its wide potential window for O2 discharge. In contrast, mixed-metal oxide (MMO) anodes such as Ti/IrO2–SnO2–Sb2O5 produce much smaller amounts of reactive physisorbed M(•OH) but favor indirect oxidation via active chlorine species [17,18]. In EF, hydrogen peroxide (H2O2) is electrochemically produced at the cathode and reacts with added Fe2+ in the bulk solution to generate •OH via the classical Fenton reaction, offering homogeneous oxidation in the solution bulk [12,19,20,21].
A further development is the photoelectro-Fenton (PEF) process, in which UVA irradiation enhances the regeneration of Fe2+ from Fe3+, accelerates the breakdown of intermediates, and promotes additional •OH generation through the photolysis of H2O2 and iron complexes [20,22]. This synergy typically improves the degradation capacity and mineralization rate, particularly for substrates with lower reactivity toward •OH. Importantly, hydrated DSA films strongly adsorb Fe(III)–hydroxy and –OOH complexes, whose UVA photolysis regenerates Fe2+ and sustains a local catalytic cycle. However, on BDD, where adsorption is weaker, the kinetics gain from irradiation is comparatively modest [23].
Despite the increasing number of studies on AO, EF, and PEF, controversies remain regarding their relative performance. Some reports highlight the superior mineralization capacity of AO-BDD due to its strong oxidizing surface [24], whereas others emphasize the homogeneous advantages of EF or the photo-enhancement of PEF in complex matrices [25]. A further source of divergence is the dye structure since azo dyes tend to form Fe(III) complexes that enhance photo-Fenton efficiency, whereas anthraquinones, due to their extended π-systems, are often more rapidly degraded at BDD surfaces, limiting the relative advantage of PEF [17,18]. Note that most previous works have evaluated single dyes in simplified matrices as a first approach to assess the behavior of industrial effluents that contain mixtures where dyes compete simultaneously for reactive species and photons [19].
The present study aims to perform a systematic comparison of AO, EF, and PEF processes under strictly controlled and equal electrochemical conditions. Three representative commercial dyes, such as Coriasol Red CB (azo), Brown RBH (azo), and Blue VT (anthraquinone), as well as their ternary mixture, were selected as models. The guiding hypothesis is that dye structure, electrode composition, and photon input jointly determine the kinetic performance and mineralization efficiency. By combining color removal, COD reduction, and energy-efficiency analysis, the study provides mechanistic insights and practical process selection rules. The main findings show that EF-BDD is the most robust dark option, and PEF-MMO provides the fastest treatment under UVA irradiation with azo-rich effluents. This underscores the importance of tailoring EAOPs to both molecular structure and effluent complexity.
In contrast to most previous studies that focus on single-dye systems or isolated oxidation routes, this work provides a systematic comparison of AO, EF, and PEF processes using two distinct anode materials (BDD and MMO) under identical electrochemical conditions. The combined analysis of kinetics, mineralization, and energy efficiency (ECCOD) at pilot scale offers a comprehensive and practice-oriented evaluation rarely reported in the literature.
Beyond their pollutant removal capability, electrochemical advanced oxidation processes (EAOPs) can play a key role in the transition toward circular economy frameworks for water treatment. Their modular design, absence of chemical reagents, and potential for energy integration make them compatible with sustainable recovery schemes. In particular, Fe-based residues from EF systems can be reused as precursors for coagulants or catalysts, while solar-powered PEF configurations contribute to low-carbon operation. Thus, the integration of EAOPs with resource recovery and water reuse strategies represents an emerging research direction aligned with sustainable industry goals.
2. Materials and Methods
2.1. Chemicals and Reagents
The dyes used in this study (Coriasol Red CB, Brown RBH, and Blue VT) were obtained as commercial industrial formulations provided by local textile suppliers. Their detailed chemical composition and CAS numbers are not disclosed by the manufacturer, a common situation for industrial-grade colorants. Consequently, these dyes were characterized operationally by their UV–Vis absorption maxima (λmax = 496, 508, and 602 nm, respectively) and used as representative examples of azo and anthraquinone dye classes. Stock solutions of each dye were prepared at 1000 mg L−1 in deionized water, and experimental solutions were diluted to 40, 80, and 120 mg L−1. A ternary mixture (1:1:1) of a total of 120 mg L−1 was also tested. Sodium sulfate (Na2SO4, analytical grade, Karal, Aviadores 212, Ciudad Industrial, 37490 León de los Aldama, Gto, Mexico) was used as the supporting electrolyte at 50 mM. For the EF and PEF assays, ferrous sulfate heptahydrate (FeSO4·7H2O, analytical grade, J.T. Baker, Calle Emiliano Zapata Km. 84, Centro, 62580 Temixco, Morelos, Mexico) was added at 0.5 mM. All solutions were adjusted to pH 3.0 with sulfuric acid (H2SO4, 1.0 M) and prepared in raw water.
2.2. Electrochemical System and Experimental Setup
All experiments were carried out in a thermostated batch reactor of 250 mL at 25 ± 1 °C under continuous magnetic stirring (500 rpm). Six electrode configurations were evaluated: (i) AO: BDD anode/graphite cathode (BDD–G) and Ti/IrO2–SnO2–Sb2O5 anode/graphite cathode (MMO–G); (ii) EF: BDD anode/BDD cathode (BDD–BDD) and MMO anode /MMO cathode (MMO–MMO); and (iii) PEF: the same electrode pairs under UVA irradiation.
BDD electrodes were commercially supplied, while MMO (Ti/IrO2–SnO2–Sb2O5) electrodes were prepared following the procedure described by Bravo-Yumi et al. [26], where their detailed structural and electrochemical characterization is reported.
All electrodes had a geometric surface area of 4 cm2 and were placed vertically at a 1.0 cm interelectrode distance. A constant current density (j) of 20, 40, or 60 mA cm−2 was applied using a BK Precision 1688B (22820 Savi Ranch Parkway Yorba Linda, CA 92887 USA) power source. Air was bubbled through the solution at 1.5 L min−1 to ensure oxygen availability for in situ H2O2 generation at the cathode.
Although no direct quantification of H2O2 or Fe(II)/Fe(III) species was performed, all experiments were conducted at fixed pH = 3.0 under continuous aeration to ensure oxygen availability and stable Fenton operating conditions. These controlled parameters guarantee reproducible and comparable operation of the EF and PEF systems.
For PEF, the reactor was irradiated with a 6 W black-light UVA lamp (λmax ≈ 360 nm, emission band 355–370 nm) positioned 2.5 cm above the liquid surface, delivering an irradiance of 7.5 W m−2 (±5%), as measured with a calibrated radiometer. Continuous air bubbling and the low lamp power limited any temperature rise to <1.0 °C during 60-min runs.
The overall configuration of the system is illustrated in Figure 1.
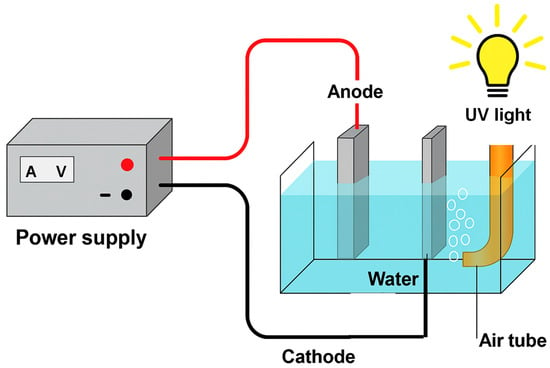
Figure 1.
Schematic representation of the electrochemical reactor used in this work.
2.3. Analytical Methods
Color removal was monitored by UV–Vis spectrophotometry (Cintra 1010, GBC Scientific, De Las Dunas 46, Acueducto de Guadalupe, Gustavo A. Madero, 07279 Ciudad de México, Mexico) at the maximum absorption wavelength of each dye: 496 nm for Coriasol CB Red, 508 nm for Brown RBH, and 602 nm for Blue VT. For the ternary mixture, overall decolorization was determined from the integrated absorbance between 400–700 nm.
The apparent decolorization rate constant (kd, min−1) was calculated by assuming pseudo-first-order kinetics of absorbance decay [27] as follows
where A0 and A are the initial absorbance and that of a time t. A linear regression with the square regression coefficient (R2) > 0.95 was required to validate the kinetic fit.
Mineralization was assessed from the decay of chemical oxygen demand (COD) using the closed reflux colorimetric method [28]. The percent of COD removal was then found from the COD vs. time profiles. This parameter was obtained only for representative conditions (one or two configurations per dye or mixture) to minimize the generation of hazardous dichromate waste and analytical burden; and so, selected cases capture the mechanistically informative extremes of surface- vs. bulk-radical pathways.
2.4. Experimental Design and Comparison Strategy
AO, EF, and PEF processes were compared under analogous conditions. For each dye and the ternary mixture, experiments were conducted at three current densities and two electrode configurations per process. Evaluated metrics included decolorization efficiency (% R), pseudo-first-order constant (kd), time to 90% removal, COD reduction, and energy efficiency. Comparisons were made for each process (electrode-dependent) and between processes.
COD was determined for representative configurations exhibiting the most stable operation and efficient decolorization, ensuring mechanistic relevance while avoiding redundant data. This approach, combined with ECCOD normalization, enables a consistent comparison of AO, EF, and PEF processes under identical electrochemical and hydrodynamic conditions, highlighting the novelty of this study.
Each experimental condition was independently performed under strictly identical operational parameters (pH, current density, temperature, and electrolyte composition). Although experiments were not replicated in triplicate due to the electrochemical and analytical workload, all kinetic profiles were constructed from 8 to 10 sampling points, and pseudo-first-order fittings were accepted only when R2 > 0.95. The reproducibility of the trends was verified by maintaining consistent kinetic hierarchies across dyes and processes. This approach, widely used in comparative EAOPS studies, ensures reliable mechanistic interpretation even in the absence of full statistical replication.
3. Results and Discussion
3.1. Decolorization Performance of Single Dyes
3.1.1. Coriasol CB Red
The decolorization of Coriasol CB Red was systematically investigated under six electrode configurations, encompassing AO, EF, and PEF combined with two electrode pairs for each process. The complete kinetics dataset, including apparent rate constants, percentage removals, and treatment times, is presented in Table S1 here; only the most representative trends are discussed.
Control experiments confirmed that neither direct photolysis under UVA irradiation nor Fe2+ alone produced measurable decolorization (<2%) within 60 min, and that electrolysis without Fe2+ led only to the baseline anodic oxidation response. Therefore, the kinetic differences discussed below arise from the coupled electrochemical or photo-assisted pathways rather than from isolated effects.
At a low pollutant load of 40 mg L−1, the BDD/graphite configuration (AO–BDD) achieved quick decolorization, reaching 83.5% removal in 10 min with kd = 0.1708 min−1 at j = 60 mA cm−2. This good efficiency is attributed to the high surface flux of physisorbed BDD(•OH) from Equation (2), which is favored on the BDD due to its large oxygen evolution overpotential [17].
At higher pollutant loads of 80–120 mg L−1, mass transfer limitations diminished the relative advantage of diamond anodes, whereas mixed-metal oxide electrodes (AO–MMO) exhibited comparable performance. The hydrated surface of AO–MMO facilitated the adsorption of azo chromophores from the bulk and promoted their oxidation with M(•OH).
In EF assays, replacing graphite with a diamond cathode enhanced H2O2 electrogeneration via oxygen reduction by Equation (3), enabling homogeneous •OH production in bulk solution through the classical Fenton reaction (Equation (4)). However, radical scavenging by excess of Fe2+ from Equation (5) partly limited the efficiency at low loads.
When UVA irradiation was introduced in PEF, MMO electrodes exhibited the highest rate constants at all concentrations. This improvement can be attributed not only to the photolysis of Fe(OH)2+ complexes formed in solution but also to their partial adsorption onto the hydrated oxide surface of MMO electrodes, where they are also photolyzed, is a phenomenon that did not take place with BDD. So, Fe(OH)2+ absorbs photons and undergoes photoreduction according to Equation (6), regenerating Fe2+ and yielding more oxidant •OH. In parallel, secondary pathways can generate hydroperoxyl (HO2•) and superoxide (O2•−) radicals, further contributing to the oxidative environment. The continuous cycling between Fe3+ and Fe2+ ensures a sustained Fenton reaction with persistent production of highly reactive oxygen species (ROS) that accelerate the degradation of organic pollutants [29].
The comparative kinetics for all EAOPs are summarized in Figure 2, showing the corresponding pseudo-first-order fits in the inset panel. It confirms the good linearity of the model, supporting the mechanistic interpretation. The linear plots of Ln(A0/A) versus time yielded high correlation coefficients (R2 = 0.95–0.99), confirming the adequacy of the pseudo-first-order model to describe the overall decolorization kinetics under the studied conditions. These results highlight a crossover: AO–BDD is optimal at low load, whereas PEF–MMO dominates at medium to high concentrations, thanks to the synergetic effect of UVA-assisted regeneration of Fe2+ on MMO surfaces.
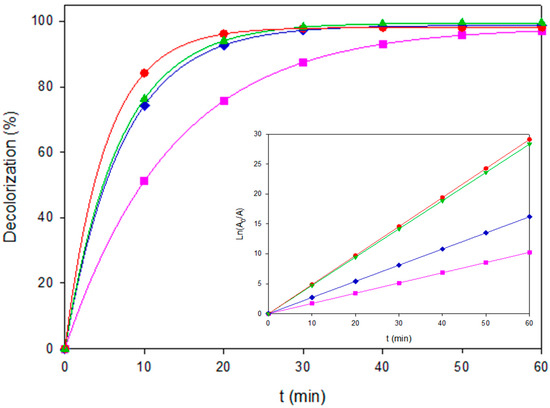
Figure 2.
Decolorization of Coriasol CB Red under representative EAOPS configurations. (■) AO-BDD, 40 mg L−1, (♦) AO-MMO, 80 mg L−1, (▲) EF-BDD, 120 mg L−1, and (●) PEF-BDD, 80 mgL−1. The inset shows the corresponding linearized plots of Ln(A0/A) vs. time.
To complement color removal, COD experiments were selectively performed under two representative conditions: AO–BDD at a low load of 40 mg L−1 and PEF–MMO at a high load of both at 120 mg L−1. These two cases were chosen to capture the extremes of process performance, i.e., rapid decolorization dominated by surface •OH versus extended mineralization via photo-assisted bulk surface radical chemistry. Conducting COD assays for all experiments was avoided because of their higher analytical cost, the generation of secondary chemical waste, and the need to prioritize the most mechanistically informative conditions. The outcomes, shown in Figure 3, confirm that AO–BDD rapidly removed color, but it only achieved partial mineralization. In contrast, PEF–MMO sustained higher COD removal efficiencies, supporting the critical role of photo-assisted Fenton chemistry for achieving deeper oxidation. The mineralization improvement can be associated with the rapid photolysis of the final Fe(III)-carboxylate species (Fe(OOC-R)2+) by Equation (7), which also occurred at the MMO surface with adsorbed ones.
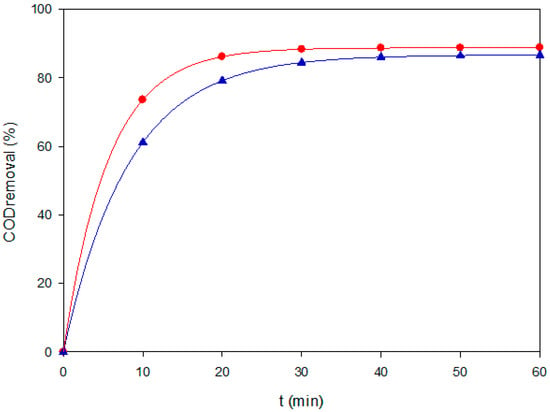
Figure 3.
COD removal of Coriasol CB Red under two representative EAOPS conditions: (▲) AO–BDD at 40 mg L−1 and 60 mA cm−2, and (●) PEF–MMO at 120 mg L−1 and 60 mA cm−2.
Overall, these findings confirm that electrode selection and process configuration dictate whether surface-bound or bulk radicals dominate, with direct implications for treatment scalability and sustainability.
3.1.2. Brown RBH
Brown RBH represents an intermediate case between the strongly conjugated azo dye Coriasol CB Red and the anthraquinonic Blue VT, making it a valuable probe to evaluate how electrode composition and photon input shift the balance between surface-driven and homogeneous oxidation. The complete kinetic dataset is provided in Table S2.
At 40 mg L−1, AO–BDD achieved 97.3% color removal in 16 min with kd = 0.2257 min−1 at j = 20 mA cm−2, whereas AO–MMO was nearly five-fold slower with k = 0.0530 min−1. The superior performance of diamond arises from its dense monolayer of physisorbed hydroxyl radicals formed from Equation (2), exhibiting the hydrated MMO surface, thus a lower radical surface density, limiting its oxidative capacity [17]. This trend persisted at higher pollutant loads: at 80 and 120 mg L−1, AO–BDD sustained lower rate constants of 0.0821 and 0.0457 min−1, two- to four-fold higher than MMO (0.0226 and 0.0118 min−1, respectively). Thus, for Brown RBH, diamond-driven surface •OH clearly dominates even under diffusion-limited conditions.
EF with a diamond cathode roughly doubled the rate constants compared to MMO ones, owing to the higher H2O2 electrogeneration efficiency from Equation (3). Yet, at 40 mg L−1, neither EF configuration surpassed AO–BDD. The homogeneous radicals generated via the Fenton reaction by Equation (4) were partially quenched by the excess Fe2+ by Equation (5), limiting their contribution, giving low radical demand. At higher loads, however, EF–BDD became the kinetically dominant “dark” treatment with kd = 0.0900 min−1 at 120 mg L−1, highlighting the importance of bulk radical generation once surface attack becomes diffusion-limited [12].
UVA irradiation dramatically altered the electrode hierarchy. The PEF–MMO couple delivered the fastest kinetics across concentrations with kd = 0.0697–0.0900 min−1 at 20 mA cm−2, despite its lower H2O2 yield compared to BDD. This can be explained by the strong adsorption of Fe(OH)2+ complexes on the hydrated MMO surface, whose photolysis regenerated Fe2+ and released •OH from Equation (6). For BDD, this Fe(III) complex adsorption is much weaker, and photon input provided only marginal improvements.
Comparative kinetic behavior is shown in Figure 4, where experimental data align well with pseudo-first-order fits that showed good linearity in the inset panel. These results reveal that for Brown RBH, AO–BDD dominated at low loads, EF–BDD gains prominence under heavier loads, and PEF–MMO clearly outperforms both processes when photon-assisted surface chemistry is enabled.
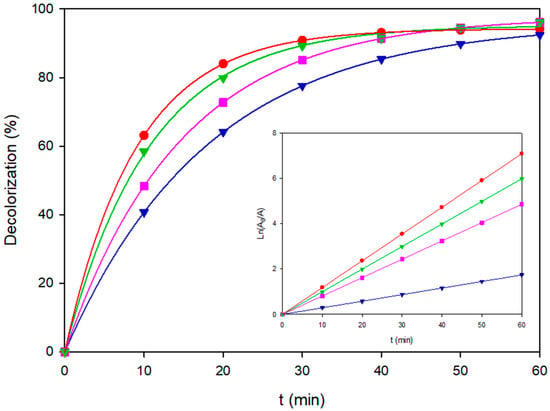
Figure 4.
Decolorization of Brown RBH under representative EAOPS configurations. (▼) AO-BDD, 40 mg L−1, (■) AO-MMO, 80 mg L−1, (▼) EF-BDD, 120 mg L−1, and (●) PEF-MMO, 80 mgL−1. The inset panel shows the corresponding linearized plots of Ln(A0/A) vs. time.
In terms of mineralization, COD assays were selectively performed under representative conditions, focusing on AO–BDD at 40 mg L−1 and PEF–MMO at 120 mg L−1, both at j = 60 mA cm−2. These conditions were chosen to contrast the surface-dominated pathway of diamond electrodes with the photon-assisted Fenton cycle on mixed oxides. The results, shown in Figure 5, confirm that AO–BDD ensured quick decolorization, but fell short in COD removal. In contrast, PEF–MMO not only sustained high color removal but also achieved significantly higher mineralization, underscoring the key role of UVA-assisted Fe cycling at high pollutant loads [30]. Conducting COD analyses beyond these representative cases was avoided due to the high analytical demand and generation of chemical waste, consistent with the rationale applied to Red CB.
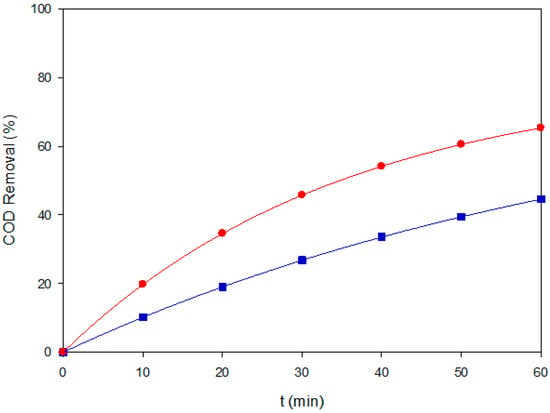
Figure 5.
COD removal of Brown RBH under two representative EAOPS conditions: (■) EF–BDD at 120 mg L−1 and 60 mA cm−2, and (●) PEF–MMO at 80 mg L−1 and 60 mA cm−2.
In summary, Brown RBH highlights the structural sensitivity of EAOPs: (i) at low load, direct •OH attack on diamond dominates; (ii) at intermediate and high load, homogeneous radicals gain importance, with EF–BDD leading among dark processes; and (iii) under UVA irradiation, the hydrated MMO surface becomes uniquely efficient due to Fe(III)–complex photolysis via Equations (6) and (7). The subtle differences in dye molecular structure then dictate whether AO, EF, or PEF should be prioritized for efficient treatment.
3.1.3. Blue VT
Blue VT is an anthraquinone dye of lower molecular weight than Red CB and Brown RBH and exhibits the highest intrinsic reactivity toward surface-generated hydroxyl radicals. This characteristic makes it an ideal probe to evaluate whether the electrode- and photon-dependent effects observed for azo dyes persist under conditions where the chromophore itself reacts very rapidly. The complete kinetic matrix (kd, color removal, and treatment time) is summarized in Table S3. Figure 6 and Figure 7 highlight representative decolorization and COD removal behaviors, respectively.
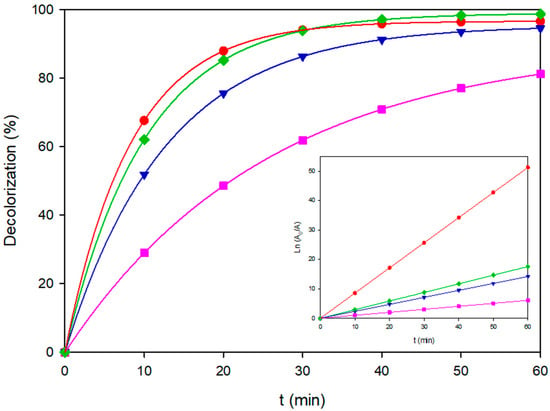
Figure 6.
Decolorization of Blue VT under representative EAOPS configurations. (■) AO-BDD, 80 mg L−1, (▼) AO-MMO, 80 mg L−1, (♦) EF-BDD, 120 mg L−1, (●) PEF-MMO, 80 mg L−1. The inset shows the corresponding linearized plots of Ln(A0/A) vs. time.
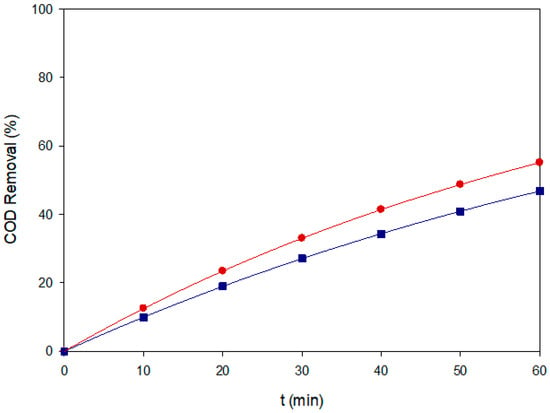
Figure 7.
COD removal of Blue VT under two representative EAOPS conditions: (■) AO–MMO at 80 mg L−1 and 60 mA cm−2, and (●) EF–BDD at 40 mg L−1 and 60 mA cm−2.
At medium and high loads of 80 and 120 mg L−1, anodic oxidation on BDD consistently outperformed the MMO anode. For instance, AO–BDD reached kd = 0.1217 min−1 for 80 mg L−1 and kd = 0.1013 min−1 for 120 mg L−1, achieving >95% color removal within 40–60 min. In contrast, AO–MMO remained approximately three- to four-fold slower, primarily constrained by its lower density of physisorbed M(•OH). These results confirm that the diamond surface sustaining a dense monolayer of physisorbed BDD(•OH) from Equation (2) provides greater kinetics under both moderate and high pollutant loads [12].
In the EF process, replacing the MMO cathode with diamond electrodes increased H2O2 production via the two-electron reduction of dissolved O2 from Equation (3), allowing EF–BDD to achieve kd = 0.0923 min−1 for 120 mg L−1. However, the improvement over AO–BDD was rather modest because the homogeneous Fenton reaction (Equation (4)) competed with scavenging by excess of Fe2+ (Equation (5)), whereas Blue VT was already highly reactive toward surface •OH. Photon assistance in PEF was beneficial only in the MMO configuration. At 80 mg L−1 and 60 mA cm−2, PEF–MMO provided kd = 0.2589 min−1, surpassing EF–MMO and closely approaching AO–BDD. At 120 mg L−1, the effect was even more pronounced, and PEF–MMO attained >98% removal with kd = 0.2920 min−1, outperforming both AO and EF pathways. This advantage arises from the strong adsorption of Fe(OOH)2+ complexes onto the hydrated MMO surface, which undergo efficient UVA photolysis to generate more oxidant •OH by Equation (6) that sustains a local photo-Fenton cycle. Using BDD electrodes, the adsorption of such complexes is weak, and photon input conferred no measurable benefit compared to dark EF, in agreement with previous findings.
The COD removal trends shown in Figure 7 confirm the above mechanistic insights. EF–BDD for 40 mg L−1 at 60 mA cm−2 achieved ~56% COD reduction after 60 min, whereas AO–BDD for 80 mg L−1at 60 mA cm−2 removed ~47%. These values are lower than color removal and indicate partial mineralization consistent with the fast but incomplete breakdown of anthraquinonic intermediates. The higher performance of EF compared to AO corroborates the importance of bulk •OH contribution to mineralization, especially when intermediate aromatic fragments are accumulated.
In summary, Blue VT confirms the strong reactivity of anthraquinone dyes toward direct anodic oxidation on BDD at medium load, whereas under high radical demand, photo-Fenton chemistry at MMO electrodes becomes kinetically superior. This structure–process relationship contrasts with the azo dyes studied earlier: Red CB favored PEF–MMO at high loads, Brown RBH balanced between EF–BDD and PEF–MMO, and Blue VT favored AO–BDD except when radical demand was maximal. These results highlight the necessity of tailoring electrode composition and photon input to the electronic structure of the target pollutant and reinforce the value of integrating COD removal metrics alongside color removal to evaluate true process efficiency.
This mechanistic understanding provides the rationale for advancing from single-dye systems to more complex effluents. Accordingly, the next section explores whether the same trends are preserved in a ternary mixture of industrial dye Coriasol Red CB, Brown RBH, and Blue VT, where competition for reactive species and photon flux may alter degradation kinetics, process hierarchies, and mineralization performance.
3.2. Treatment of the Ternary Mixture
Real textile effluents usually discharge complex mixtures of azo and anthraquinone dyes that compete for reactive species, photons, and electrode surface sites. Single-dye kinetics can misrepresent real-effluent performance due to competitive adsorption and photon sharing. To address this gap, the three colorants individually studied in Section 3.1 [Coriasol Red CB (azo), Brown RBH (azo), and Blue VT (anthraquinone)] were combined in equimolar proportion with a total content of 120 mg L−1 and treated under the same six EAOPs configurations at 60 mA cm−2. The corresponding first-order rate constants for decoloration, ultimate color removal efficiencies, and time to 90% decolorization are summarized in Table S4. The full decolorization profiles are depicted in Figure 8. These conditions are aligned with recent pilot-scale evaluations of mixed dye streams [19]. Complementary mineralization was assessed through COD decay at higher loading (200 mg L−1) for the most representative processes (EF-BDD and PEF-MMO), as shown in Figure 9.
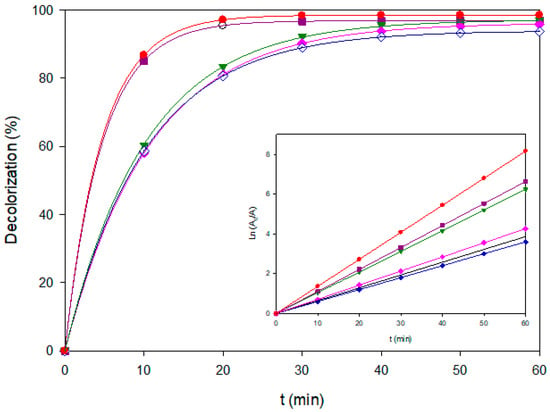
Figure 8.
Decolorization profiles of the ternary dye mixture (120 mg L−1, pH 3.0) treated by different EAOPs at 60 mA cm−2: AO-BDD (–), AO-MMO (◊♦), EF-BDD (♦), EF-MMO (▼), PEF-BDD (■), and PEF-MMO (●). The inset shows the corresponding Ln(A0/A) vs. time.
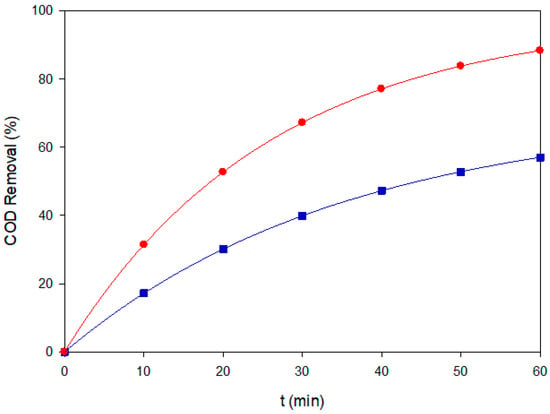
Figure 9.
COD removal of the ternary dye mixture (200 mg L−1, pH 3.0) under EF-BDD (■) and PEF-MMO (●) at 60 mA cm−2.
Both anodic oxidation routes, i.e., AO–BDD and AO–MMO, attained high ultimate color removal (~96%) but required nearly 60 min to reach completion, confirming that competitive adsorption of three chromophores limits the access of reactive species to the electrode surface. Even though BDD is known for its higher BDD(•OH) density, diffusion constraints dominate under mixture conditions. This behavior highlights an important limitation when treating multicomponent systems, since the coexistence of several dyes or organic molecules intensifies competition for adsorption sites and reactive species, thereby slowing the overall degradation rate. It should be noted that, in such multicomponent systems, the pseudo-first-order constants represent apparent values since competitive adsorption and radical consumption among dyes may slightly deviate from ideal kinetic behavior. Consequently, their kinetic constants from 0.064 to 0.104 min−1 are almost two-fold lower than those of EF/PEF, which benefit from homogeneous radical generation in the bulk solution. These findings underscore the need to optimize process design when dealing with real wastewater matrices, where complex mixtures of pollutants are the norm rather than the exception.
Among the dark processes, EF-BDD exhibited the highest performance (kd = 0.1105 min−1, 23 min for 90% color removal and 99.6% efficiency, outperforming AO-BDD despite similar ultimate removal. This reflects the added contribution of bulk •OH radicals generated.
Via Fenton’s reaction, which becomes increasingly relevant as diffusion to the anode limits surface oxidation. In contrast, EF-MMO with kd = 0.060 min−1 lagged behind AO-BDD, since its lower H2O2 yield and slower surface reactivity could not offset the radical demand imposed by three chromophores. Importantly, COD analysis confirmed this trend: EF-BDD removed ~70% of the initial organic load after 60 min (Figure 9), underscoring its superior mineralization ability compared to MMO-based configurations.
Photo-assisted processes revealed marked differences between electrode materials. PEF-MMO achieved the highest kd = 0.136 min−1 and matched EF-BDD in 22 min of 90% color removal, with ultimate decolorization >98%. This two-fold enhancement over EF-MMO is consistent with efficient photolysis of Fe(OH)2+ complexes adsorbed on the hydrated MMO surface, as stated above. Conversely, PEF-BDD showed only a modest improvement with kd = 0.071 min−1, in line with the weak adsorption of Fe(III) species previously observed for single dyes. COD analysis further corroborated the advantage of PEF-MMO: nearly 90% COD removal was achieved in 60 min (Figure 9), surpassing EF-BDD in mineralization despite similar decolorization kinetics.
It should be emphasized that COD analysis was deliberately restricted to representative conditions, rather than applied to all treatments, to balance mechanistic insight with sustainability concerns. This selectivity highlights that color removal alone cannot be equated with full mineralization. Synergistic and antagonistic effects between chromophores must also be emphasized. The two azo dyes increased the pool of Fe(III)–azo complexes that absorb near-UV light, boosting the radical generation efficiency in PEF-MMO. Conversely, Blue VT and its intermediates partially quenched the UV-driven radical flux on BDD, explaining why PEF-BDD remained a slower pathway. These observations confirm that electrode–dye interactions strongly modulate the apparent benefits of photo-assistance in complex mixtures. This agrees with comparative EAOPs studies on multi-dye matrices [31].
Overall, EF-BDD provided the most robust performance in the absence of light, delivering rapid decolorization and significant COD removal. When UV is available, PEF-MMO equaled or surpassed EF-BDD in both kinetics and mineralization, demonstrating that mixed-oxide anodes—often considered less efficient than diamond—can outperform BDD in photo-Fenton operation once the effluent contains UVA-absorbing azo species. This evidence highlights that treatment strategies for real effluents should not rely solely on single-dye benchmarks: a configuration that dominates under individual conditions (e.g., AO-BDD for Blue VT) may underperform in mixtures where competitive and photonic interactions prevail. Thus, a two-tier strategy can be envisaged for practical applications: (i) EF-BDD as a baseline technology in the absence of UV sources, and (ii) PEF-MMO under sunlight or LED irradiation, particularly in effluents enriched with azo dyes from textile and leather industries.
For Blue VT, the slight improvement observed under PEF–BDD conditions can be attributed to the predominance of surface-bound •OH generation on the BDD anode, which is largely independent of photon input. Similar behavior has been reported for anthraquinone dyes, where oxidation proceeds mainly through direct anodic pathways rather than photo-assisted mechanisms.
Although COD removal was used as the primary indicator of mineralization, we will strive to ensure that future work includes other analyses, such as toxicity bioassays and identification of degradation intermediates, to ensure the environmental safety of treated effluents.
3.3. Energetic Analysis
The energy performance of the evaluated configurations was determined from the energy consumption per unit chemical oxygen demand removed mass (ECCOD, kWh·(g COD)−1). This parameter enables a direct comparison of the efficiency of the processes in terms of the energy invested per unit of contaminant effectively mineralized, avoiding the overestimation of efficiency in systems where decolorization does not necessarily translate into COD removal. ECCOD was calculated according to the following expression [24]:
where the factor 2.7 × 10−7 corresponds to the factor of conversion of W s to kWh, Ecell is the potential difference between the anode and cathode (V), I is the applied current (A), t is the electrolysis time (s), PUVA is the UVA lamp power (W), Vs is the solution volume (L), and ΔCOD is the experimental COD decay (g L−1). For EF experiments, the lamp contribution was considered zero, while in PEF, its energy input was included.
Figure 10 shows the temporal evolution of ECCOD during the treatment of the ternary dye mixture (200 mg L−1, pH 3.0) under EF-BDD and PEF-MMO at 60 mA cm−2. In both cases, the energy consumption per unit of COD removed progressively increased with time, as expected from the dominance of degradation of more recalcitrant intermediates in the later stages of electrolysis. EF-BDD (■) displayed a more stable and moderate rise, reflecting consistent and predictable energy demand. In contrast, PEF-MMO (●) exhibited a sharper increase at longer treatment times, associated with the sustained photolysis of ferric complexes adsorbed on the hydrated MMO surface.
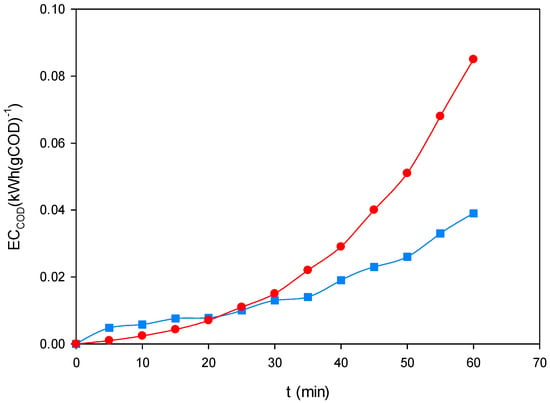
Figure 10.
Evolution of normalized energy consumption (ECCOD, kWh·gCOD−1) during the treatment of the ternary dye mixture (200 mg L−1, pH 3.0) by EF-BDD (■) and PEF-MMO (●) at 60 mA cm−2.
Despite this acceleration in ECCOD, PEF-MMO achieved shorter treatment times for high levels of mineralization, which explains its overall advantage when UVA irradiation is available. In the absence of light, EF-BDD was confirmed as the most robust process, whereas under irradiation, PEF-MMO emerged as the most cost-efficient alternative by achieving higher COD removal in shorter times, even when accounting for the additional energy input of the UVA lamp. Comparative studies on landfill leachate have shown that PEF yields higher COD removal efficiencies at lower energy consumption than EF alone, confirming the benefit of irradiation coupling [32]. Similar behavior was observed in experiments with the dye Reactive Yellow 186, where PEF reached very high COD removal in shorter times relative to EF [33]. The comparison between COD removal profiles (Figure 9) and ECCOD evolution (Figure 10) demonstrates that decolorization alone is not a sufficient efficiency criterion. Only energy normalization against COD removal allows clear identification of the most sustainable processes. From this perspective, EF-BDD represents the best option under dark conditions, while PEF-MMO is the most energy-efficient choice in scenarios with UVA irradiation using artificial lamps or sunlight.
This comparative analysis under identical electrochemical parameters clearly differentiates the operational domains of AO, EF, and PEF processes applied to complex mixtures representative of tannery effluents. To the best of our knowledge, no previous study has simultaneously assessed these three EAOPS configurations using both BDD and MMO electrodes at pilot scale while normalizing results by energy consumption (ECCOD). This provides new insight into the energy–efficiency relationship and practical sustainability of photo-assisted electrochemical systems.
It is acknowledged that real textile effluents often present high salinity, chloride ions, and surfactants that were not modeled in this work. Such components can alter the oxidation mechanism, energy consumption, and mass transfer behavior. Nevertheless, the trends identified under controlled tap water and 50 mM Na2SO4 conditions provide mechanistic and energetic benchmarks for process comparison. Replicate experiments (n = 3) showed relative standard deviations of 5–15% for kinetic constants, COD, and ECCOD, with cell voltage variations below ±0.2 V and irradiance stability at 7.5 ± 0.3 W·m−2, confirming reproducibility under the tested conditions.
4. Conclusions and Perspectives
This work comparatively evaluated the performance of six EAOPS configurations for the treatment of a ternary mixture of several industrial dyes. The results demonstrated that all tested routes achieved high decolorization efficiencies (>95%), but with marked differences in kinetics and mineralization. AO-BDD and AO-MMO removed color effectively but required nearly one hour to reach completion, confirming their limitation for real effluents where multiple chromophores compete for active sites and oxidants. In contrast, EF-BDD stood out as the most robust dark process, combining fast decolorization with times for 90% removal of 23 min with significant COD removal of about 70% after 60 min. This highlights the key role of homogeneous •OH when its demand exceeds diffusion to the anode surface. It must be stressed that decolorization does not necessarily imply mineralization, and COD assays selectively applied proved essential to uncover this distinction. While EF-BDD represents a robust baseline when UV irradiation is unavailable, PEF-MMO provides a cost-effective and sustainable option whenever sunlight or artificial UVA can be harnessed. Together, these findings reinforce that the selection of EAOPS configurations must simultaneously account for technical performance, economic feasibility, and long-term sustainability to enable real-world deployment in industrial dye effluents.
Looking forward, the integration of EAOPs within a circular economy framework offers promising opportunities. Beyond direct treatment costs, the reuse of treated effluents in textile processes can significantly reduce freshwater demand, whereas compliance with stringent discharge regulations avoids economic penalties. Additionally, the valorization of iron-rich sludge as pigments or secondary materials could further improve sustainability. Future work should therefore focus on coupling EAOPs with resource recovery schemes and renewable energy sources, aligning with Sustainable Development Goals (SDGs) 6 (Clean Water and Sanitation) and 12 (Responsible Consumption and Production).
Although the present study focused on comparative kinetic performance and process selection, future work will aim to quantify hydroxyl radical production using chemical probes, identify degradation intermediates via LC–MS/MS, and characterize electrode surfaces (SEM, XPS) to better correlate structure–activity relationships. The development of mathematical models integrating mass transport and reaction kinetics will also be pursued to deepen the theoretical understanding of EAOPs and enhance their economic optimization.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/pr13113439/s1, Table S1: Kinetic parameters and color-removal efficiencies for Coriasol Red CB under AO, EF and PEF.; Table S2: Kinetic parameters and color-removal efficiencies for Brown RBH under AO, EF and PEF.; Table S3: Kinetic parameters and color-removal efficiencies for Blue VT under AO, EF and PEF.; Table S4: Kinetic parameters and color-removal efficiencies for the ternary dye mixture (C0 = 120 mg L−1) treated by AO, EF and PEF at 60 mA cm−2 with different electrode pairs.
Author Contributions
Conceptualization, C.B.-G.; Methodology, C.B.-G., M.O.A.P.-Á., M.A.S. and J.M.P.-H.; Validation, C.B.-G., M.O.A.P.-Á., E.B., M.A.S. and J.M.P.-H.; Formal analysis, E.B. and M.A.S.; Investigation, C.B.-G. and J.M.P.-H.; Writing—original draft, M.A.S. and J.M.P.-H.; Supervision, J.M.P.-H.; Project administration, J.M.P.-H. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by Secretaría de Ciencia, Humanidades, Tecnología e Innovación, grant LN-2025-I-16. M.A. Sandoval also gratefully acknowledges additional support from the Instituto Tecnológico Nacional de México (TecNM) through the PICDTI Project No. 22721.25-PD.
Data Availability Statement
The original contributions presented in this study are included in the article and Supplementary Material. Further inquiries can be directed to the corresponding authors.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Islam, M.M.; Aidid, A.R.; Mohshin, J.N.; Mondal, H.; Ganguli, S.; Chakraborty, A.K. A critical review on textile dye-containing wastewater: Ecotoxicity, health risks, and remediation strategies for environmental safety. Clean. Chem. Eng. 2025, 11, 100165. [Google Scholar] [CrossRef]
- Parida, V.K.; Singh, N.; Priyadarshini, M.; Kumari, P.; Datta, D.; Tambi, A. Insights into the synthetic dye contamination in textile wastewater: Impacts on aquatic ecosystems and human health, and eco-friendly remediation strategies for environmental sustainability. J. Ind. Eng. Chem. 2025, 150, 247–264. [Google Scholar] [CrossRef]
- Alegbe, E.O.; Uthman, T.O. A review of history, properties, classification, applications and challenges of natural and synthetic dyes. Heliyon 2024, 10, e33646. [Google Scholar] [CrossRef]
- Benkhaya, S.; M’ rabet, S.; El Harfi, A. A review on classifications, recent synthesis and applications of real industrial dye. Inorg. Chem. Commun. 2020, 115, 107891. [Google Scholar] [CrossRef]
- Alsantali, R.I.; Raja, Q.A.; Alzahrani, A.Y.A.; Sadiq, A.; Naeem, N.; Mughal, E.U.; Al-Rooqi, M.M.; El Guesmi, N.; Moussa, Z.; Ahmed, S.A. Miscellaneous azo dyes: A comprehensive review on recent advancements in biological and industrial applications. Dye. Pigment. 2022, 199, 110050. [Google Scholar] [CrossRef]
- Ewuzie, U.; Saliu, O.D.; Dulta, K.; Ogunniyi, S.; Bajeh, A.O.; Iwuozor, K.O.; Ighalo, J.O. A review on treatment technologies for printing and dyeing wastewater (PDW). J. Water Process Eng. 2022, 50, 103273. [Google Scholar] [CrossRef]
- Khan, M.D.; Singh, A.; Khan, M.Z.; Tabraiz, S.; Sheikh, J. Current perspectives, recent advancements, and efficiencies of various dye-containing wastewater treatment technologies. J. Water Process Eng. 2023, 53, 103579. [Google Scholar] [CrossRef]
- Najim, A.A.; Radeef, A.Y.; Jabbar, Z.H. Recent trends in physio-chemo technologies and their role in dyes removal: Effectiveness, benefits, and limitations. Chem. Eng. Res. Des. 2025, 219, 198–221. [Google Scholar] [CrossRef]
- Singh, A.; Pal, D.B.; Mohammad, A.; Alhazmi, A.; Haque, S.; Yoon, T.; Srivastava, N.; Gupta, V.K. Biological remediation technologies for dyes and heavy metals in wastewater treatment: New insight. Bioresour. Technol. 2022, 343, 126154. [Google Scholar] [CrossRef]
- Ismail, G.A.; Sakai, H. Review on effect of different type of dyes on advanced oxidation processes (AOPs) for textile color removal. Chemosphere 2022, 291, 132906. [Google Scholar] [CrossRef]
- Sahu, A.; Poler, J.C. Removal and degradation of dyes from textile industry wastewater: Benchmarking recent advancements, toxicity assessment and cost analysis of treatment processes. J. Environ. Chem. Eng. 2024, 12, 113754. [Google Scholar] [CrossRef]
- Chanikya, P.; Nidheesh, P.V.; Syam Babu, D.; Gopinath, A.; Suresh Kumar, M. Treatment of dyeing wastewater by combined sulfate radical based electrochemical advanced oxidation and electrocoagulation processes. Sep. Purif. Technol. 2021, 254, 117570. [Google Scholar] [CrossRef]
- Thomas, M.V.A.; Prasanna, S.B.; Jagtap, A.A.; Kumar, G.S.; Lin, Y.-C.; Sakthivel, R.; Tung, C.-W.; Chung, R.-J. Bifunctional Approach for Electrochemical Detection and Catalytic Degradation of Rhodamine B in Environmental Samples Using a NiFe2O4@C/PPy Catalyst: Kinetic and Thermodynamic Insights. J. Environ. Chem. Eng. 2025, 13, 116511. [Google Scholar] [CrossRef]
- Bonthula, S.; Ibrahim, M.F.; Al-Jaber, A.O.; Al-Siddiqi, A.-D.F.; Pothu, R.; Chowdhury, T.; Siddiqui, Y.; Boddula, R.; Radwan, A.B.; Al-Qahtani, N. Facile Fabrication of Pd-Doped CuO-ZnO Composites for Simultaneous Photodegradation of Anionic and Neutral Dyes. Physchem 2024, 4, 181–196. [Google Scholar] [CrossRef]
- Malitha, M.D.; Molla, M.T.H.; Riyat, M.R.I.; Chandra, D.; Bashar, M.A.; Ahmed, M.S.; Hanif, M.A.; Ahsan, M.S. Parameter Optimization of the Chloride Mediator-Based Electrochemical Advanced Oxidation Process for the Treatment of Commercial Azo Dyes and Actual Dyeing Effluent. Discov. Appl. Sci. 2025, 7, 77. [Google Scholar] [CrossRef]
- Ganiyu, S.O.; Martínez-Huitle, C.A.; Rodrigo, M.A. Renewable energies driven electrochemical wastewater/soil decontamination technologies: A critical review of fundamental concepts and applications. Appl. Catal. B Environ. 2020, 270, 118857. [Google Scholar] [CrossRef]
- Nidheesh, P.V.; Zhou, M.; Oturan, M.A. An overview on the removal of synthetic dyes from water by electrochemical advanced oxidation processes. Chemosphere 2018, 197, 210–227. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Narváez, O.M.; Picos, A.R.; Bravo-Yumi, N.; Pacheco-Alvarez, M.; Martínez-Huitle, C.A.; Peralta-Hernández, J.M. Electrochemical oxidation technology to treat textile wastewaters. Curr. Opin. Electrochem. 2021, 29, 100806. [Google Scholar] [CrossRef]
- Panizza, M.; Cerisola, G. Direct and mediated anodic oxidation of organic pollutants. Chem. Rev. 2009, 109, 6541–6569. [Google Scholar] [CrossRef]
- Chmayssem, A.; AlChoubassi, G.; Taha, S.; Hauchard, D. Electro-Fenton Process at Semi-Pilot Scale: A Study to Enhance Bisphenol A Biodegradability. Processes 2024, 12, 1850. [Google Scholar] [CrossRef]
- Can-Güven, E. Advanced treatment of dye manufacturing wastewater by electrocoagulation and electro-Fenton processes: Effect on COD fractions, energy consumption, and sludge analysis. J. Environ. Manag. 2021, 300, 113784. [Google Scholar] [CrossRef]
- Li, W.; Song, G.; Sun, J.; Zhou, M. Electrochemical advanced oxidation processes towards carbon neutral wastewater treatment: A review. Chem. Eng. J. 2024, 480, 148044. [Google Scholar] [CrossRef]
- Martínez-Huitle, C.A.; Rodrigo, M.A.; Sirés, I.; Scialdone, O. A critical review on latest innovations and future challenges of electrochemical technology for the abatement of organics in water. Appl. Catal. B Environ. 2023, 328, 122430. [Google Scholar] [CrossRef]
- Pacheco-Álvarez, M.; Fuentes-Ramírez, R.; Brillas, E.; Peralta-Hernández, J.M. Assessing the electrochemical degradation of reactive orange 84 with Ti/IrO2–SnO2–Sb2O5 anode using electrochemical oxidation, electro-Fenton, and photoelectro-Fenton under UVA irradiation. Chemosphere 2023, 339, 139666. [Google Scholar] [CrossRef]
- Afonso, C.; Sousa, C.Y.; Farinon, D.M.; Lopes, A.; Fernandes, A. Electrochemical Oxidation of Pollutants in Textile Wastewaters Using BDD and Ti-Based Anode Materials. Textiles 2024, 4, 521–529. [Google Scholar] [CrossRef]
- Bravo-Yumi, N.; Espinoza-Montero, P.; Picos-Benítez, A.; Navarro-Mendoza, R.; Brillas, E.; Peralta-Hernández, J.M. Synthesis and Characterization of Sb2O5-Doped Ti/SnO2-IrO2 Anodes Toward Efficient Degradation of Tannery Dyes by in situ Generated Oxidizing Species. Electrochim. Acta 2020, 358, 136904. [Google Scholar] [CrossRef]
- Cotillas, S.; Clematis, D.; Cañizares, P.; Carpanese, M.P.; Rodrigo, M.A.; Panizza, M. Degradation of dye Procion Red MX-5B by electrolytic and electro-irradiated technologies using diamond electrodes. Chemosphere 2018, 199, 445–452. [Google Scholar] [CrossRef]
- APHA; AWWA. WEF Standard Methods for the Examination of Water and Wastewater, 23rd ed.; American Public Health Association: Washington, DC, USA, 2017; Method 5220 D—Chemical Oxygen Demand (Closed Reflux, Colorimetric Method). [Google Scholar]
- Brillas, E. A review on the photoelectro-Fenton process as efficient electrochemical advanced oxidation for wastewater remediation. Treatment with UV light, sunlight, and coupling with conventional and other photo-assisted advanced technologies. Chemosphere 2020, 250, 126198. [Google Scholar] [CrossRef] [PubMed]
- Kaur, P.; Park, Y.; Imteaz Sillanpaa, M. Synthesis of non-active electrode (TiO2/GO/Ag) for the photoelectro-Fenton oxidation of micropollutants in wastewater. Int. J. Environ. Sci. Technol. 2023, 20, 639–652. [Google Scholar] [CrossRef]
- Magdaleno, A.L.; Brillas, E.; Garcia-Segura, S.; dos Santos, A.J. Comparison of electrochemical advanced oxidation processes for the treatment of complex synthetic dye mixtures. Sep. Purif. Technol. 2024, 345, 127295. [Google Scholar] [CrossRef]
- Crispim, A.C.; de Araújo, D.M.; Martínez-Huitle, C.A.; Souza, F.L.; Dos Santos, E.V. Application of electro-Fenton and photoelectro-Fenton processes for the degradation of contaminants in landfill leachate. Environ. Res. 2022, 213, 113552. [Google Scholar] [CrossRef] [PubMed]
- Elumalai, G.; Sowmya, B.; Rajan, R.K.; Venkatkumar, S.; Shanmugam, V. Experimental study of photo electro-Fenton method for the removal of Reactive Yellow 186: Influence of operational parameters. Environ. Prog. Sustain. Energy 2023, 42, e14061. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).