1. Introduction
Coal tar naphtha is the naphtha range (typically C
5–175 °C boiling range) material obtained from coal pyrolysis. Sources of this product are low- and high-temperature coal pyrolysis in coke ovens to produce coke [
1,
2,
3], moving bed gasification of coal to produce synthesis gas [
4,
5], and direct thermal liquefaction of coal [
6,
7]. The composition of the coal tar naphtha depends on the process and coal used as feed material, but it comprises a mixture of aromatics, paraffins, naphthenes, olefins, and heteroatom-containing species.
On account of its aromatic-rich nature, the coal tar naphtha can be refined to produce aromatic commodity chemicals [
8,
9]. There is also the possibility of refining specialty aromatics for smaller markets. Alternatively, coal tar naphtha can be refined to produce transport fuel, which is the application of interest in this study.
Producing motor gasoline from coal tar naphtha is faced with one particular challenge, namely, to retain octane number in the final product. In terms of aromatic and olefinic content, coal tar naphtha from different sources can contain up to 60% aromatics and 15% olefins, as is the case with coal tar naphtha from moving bed gasification. One would expect the octane number of such a product to be high. However, coal tar naphtha must also be hydrotreated to reduce the heteroatom content and herein lies the problem.
Coal tar naphtha has nitrogen-, oxygen-, and sulfur-containing compounds, such as pyridines, phenols, and thiophenes. Isostructurally, sulfur is usually the easiest heteroatom to remove by hydrotreating [
10,
11,
12]. Sulfur is also the only heteroatom with a specific concentration limit imposed on the final product by fuel specifications (typically 10–15 μg∙g
−1). The nitrogen content is not directly regulated, but it is indirectly regulated through storage-stability-related specifications. Motor gasoline may contain oxygen, but phenol is problematic due to its acidity. The phenol content of the fuel is indirectly regulated through the total acid number specification.
When the heteroatom content in the coal tar naphtha is reduced by hydrotreating, the olefins are also hydrotreated to produce paraffins. The olefins typically have high octane numbers, but unless paraffins are highly branched, they have low octane numbers [
13]. It is therefore found that, in practice, hydrotreated coal tar naphtha has a poor octane number rating despite its high aromatic content but as a consequence of the high paraffin content. To illustrate the point, two examples are given. The hydrotreated coal tar naphtha from moving bed coal gasification has a research octane number (RON) of 80–85 and a motor octane number (MON) of 70–76 [
5]. The hydrotreated coal tar naphtha from high-temperature coal coking has a RON of 79–81 and a MON of 68–76 [
14].
The use of alkylated phenols to improve storage stability is well known and industrially practiced [
15,
16]. At the same time, alkylated phenols and phenol ethers have good octane number ratings [
17,
18,
19]. Retaining some phenolic compounds in the naphtha would therefore be acceptable, as long as the total amount of phenols did not cause the fuel to exceed the total acid number. The same restriction did not apply to phenyl ethers, which had high octane numbers and no acidity. At the same time the argument is academic because it is unlikely that the alkylated products would remain in the naphtha boiling range.
It was reasoned that the olefin content and the phenol content of coal tar naphtha could be decreased, with less loss in the octane number, if the phenol was alkylated with olefins to produce alkylated phenols and phenol ethers. In principle, the same reaction could produce alkylated aromatic hydrocarbons. This means that it would be possible to substantially reduce the olefin content of the coal tar naphtha prior to hydrotreating and thereby limit the octane number loss during hydrotreating.
The primary objective of performing phenol alkylation is to reduce the olefin content before hydrotreating since the main octane number loss is associated with olefin hydrogenation to paraffins. Alkylation will cause a change in the boiling point of the alkylated products, which will distill in the kerosene or atmospheric gas oil boiling range. It may therefore seem as if the octane number benefit is lost due to the loss of aromatics, but this is not the case.
To illustrate this point, consider the following example. An arbitrary naphtha is employed that contains cresol and 1-hexene in a mixture of hydrocarbons that will not be affected by hydrogenation. The concentration of cresol and 1-hexene is 10 and 12 vol%, respectively, i.e., an equimolar mixture of phenolic and olefinic species. The naphtha has the same octane number as the equimolar mixture of cresol and 1-hexene, RON = 96 and MON = 82. If the cresol and 1-hexene are hydrogenated to toluene and n-hexane, the octane number of the naphtha decreases to RON = 89 and MON = 77, because an equimolar mixture of toluene and n-hexane has a RON = 68 and a MON = 61. When cresol and 1-hexene are completely converted to hexyl cresol and removed by distillation, the naphtha retains its octane number.
However, there was still a challenge in the implementation because olefin–aromatic alkylation is an acid-catalyzed process and coal tar naphtha usually contains pyridines. Pyridines are basic compounds and would strongly adsorb on acid sites of acid catalysts, potentially poisoning the catalyst.
The working hypothesis in this study was that nitrogen bases are not necessarily catalyst poisons but inhibitors of acid catalysis. It should therefore be possible to perform target conversion, acid-catalyzed olefin–aromatic alkylation, despite the presence of basic nitrogen compounds in the feed. There are two equilibria that can potentially be manipulated for this purpose. The first is the liquid–solid equilibrium and the second is the acid–base equilibrium. Both equilibria are temperature-dependent. The acid–base equilibrium is also dependent on the relative strength of the acids (catalyst acid sites) and the bases (nitrogen bases in coal tar naphtha).
The temperature-dependent equilibria favored a reaction at higher temperatures, but the onset of dealkylation and cracking placed an upper limit on temperature. An operating temperature of 315 °C was selected for this study as a compromise.
The approach that was taken was to experimentally explore acid-catalyzed olefin–aromatic alkylation using a model feed comprising phenol and 1-hexene in the presence and absence of pyridine as a nitrogen base. Although the aim is to ultimately apply the knowledge gained to coal tar naphtha, this study did not include coal tar naphtha as feed material.
2. Experimental Section
2.1. Materials
Phenol (>99%), 1-hexene (97%), and pyridine (>99%) were commercially obtained from Sigma-Aldrich and used without further purification. Additionally, n-pentane (99.5%) solvent for preparing samples for gas chromatography was commercially obtained from Acros Organics.
The alumina and amorphous silica–alumina catalyst types were commercially obtained from Sasol, Germany (former Condea GmbH). These were Siral series catalysts (silica-doped silica–alumina, with the silica content varying from 5 to 40 wt% over the series, e.g., Siral 5 contains 5 wt% SiO
2). All amorphous silica–alumina catalysts were calcined in air at 550 °C for 6 h before use. The average particle size of the catalysts, as provided by the supplier, was 50 μm. Catalyst characterization is presented as part of the results in
Section 3.1.
2.2. Equipment and Procedure
Batch reactor conversion of phenol and 1-hexene. These experiments were conducted using 15 mL micro-batch reactors manufactured from stainless steel Swagelok tubing and fittings with a diameter of 1.3 cm (½ inch). The reactors were externally heated by submerging them in a hot fluidized sand bath heater. The reaction temperature was controlled indirectly by adjusting the temperature of the sand bath heater. In a typical experiment, the micro-batch reactor was loaded with approximately 5.2 g phenol, 4.3 g 1-hexene and 0.5 g catalyst. This corresponded to a molar ratio phenol/1-hexene of 1:1 in the feed. The reactions were performed under autogenous pressure at the indicated temperatures (in the range of 220–350 °C) for a duration of 1 h, equivalent to a weight hourly space velocity (WHSV) of 19 h−1. These conditions were selected to limit overall conversion to better study the impact of pyridine deactivation.
Five sets of experiments were performed using batch reactor conversion phenol and 1-hexene:
- (i)
Catalytic conversion at 315 °C, using the Siral 5, 10, 20, 30 and 40 catalysts.
- (ii)
Catalytic conversion at 315 °C, using the Siral 5, 10, 20, 30 and 40 catalysts pretreated with pyridine, i.e., catalyst exposed to flowing He saturated with pyridine vapor for 1 h, followed by purging with He only for 1 h.
- (iii)
Catalytic conversion as in (ii), but adding 6 mg (0.05 wt%) pyridine to the feed mixture.
- (iv)
Thermal conversion at 220, 250, 315, and 350 °C, i.e., no catalyst added.
- (v)
Thermal conversion as in (iv), with 0.05 wt% pyridine added to the feed.
Flow reactor conversion of phenol and 1-hexene. These experiments were conducted using a packed bed reactor, with a diameter of 1.3 cm, with a central thermowell. The reactor was loaded with 1 g catalyst dilute with an equivalent amount of silicon carbide (38 μm particle diameter). The total bed volume was 5 mL. The catalyst bed was kept in place using glass wool. The feed was a 1:1 molar ratio of phenol and 1-hexene and was delivered to the reactor using a positive displacement pump at a rate of 0.15 mL∙min−1, corresponding to a WHSV of 17 h−1. To reduce the risk of poor catalyst wetting, the flow direction was from the bottom to the top and the catalyst was pre-wetted by feeding at a high flow rate beforehand. The temperature inside the reactor was controlled by adjusting the temperature of an external heater element. Conversion was performed at 315 °C and near atmospheric pressure using Siral 30 and Siral 40 catalysts only.
In a typical experiment, conversion was performed using phenol and 1-hexene for a predetermined time period (5 h), after which pyridine was added to the feed, with the pyridine concentration being increased hourly with steps at 0.05, 0.07, 0.10, 0.15, 0.20, and 0.25%. At the end of this period, the catalyst was rejuvenated by switching to clean feed again and increasing the temperature briefly to 450 °C to assist with pyridine desorption, before reducing it to 315 °C again. The sequence was repeated once.
2.3. Analyses
Catalyst pore volume and surface area were determined by mercury porisometry with a Quantachrome PoreMaster® Hg intrusion porosimeter. BET surface area was provided by the catalyst supplier.
The composition of the catalysts was determined by energy-dispersive X-ray fluorescence (XRF) analysis with a Bruker S2 Ranger, with a silicon drift detector. The X-ray source uses a Pd target. The analyses were performed at 10 kV with a total count rate in the order of 20,000 counts per second; no filters or secondary targets were employed.
Total acid concentration and the acid strength of the catalysts were determined by ammonia temperature-programmed desorption (NH3-TPD). The NH3-TPD analysis was performed using a Quantachrome ChemBET TPR/TPD Chemisorption Flow Analyzer equipped with a thermal conductivity detector (TCD). For each analysis, about 60 mg of catalyst was pretreated under flowing helium (50 mL∙min−1) at 550 °C for 6 h and then cooled down to room temperature. Ammonia adsorption was then performed at room temperature by exposing the catalyst to an NH3 flow of 70 mL∙min−1 for 1 h. Physisorbed NH3 was removed by keeping the sample in a He flow for 100 min. For the TPD experiments, the sample was heated from room temperature to 650 °C at a heating rate of 15 °C∙min−1 under a He flow of 50 mL∙min−1.
The nature of the acid sites was determined using Fourier-transform infrared (FTIR) analysis. The spectra were collected on an ABB MB3000 FTIR with a Pike DiffusIR attachment. The DiffusIR has an environmental chamber that is temperature-controlled and can heat up to 500 °C. The analytical procedure was as follows. The catalyst (2 mg) was pressed into a ceramic cup. The catalyst was then exposed to flowing He saturated with pyridine vapor for 1 h, followed by purging with clean He flow for about 1 h. Purging with clean He also removed physisorbed pyridine from the surface, leaving only the stronger chemisorbed species. This cleaning process could be followed by FTIR and was continued until no further spectral changes were observed. The analysis of the catalyst with adsorbed pyridine started at ambient temperature and the ceramic cup was heated at a rate of 10 °C∙min−1 under a He flow from 25 to 500 °C. The temperature was held constant during spectral acquisition. The spectra were recorded at 50 °C intervals at a resolution of 16 cm−1 and as the average of 120 scans.
The reaction products from the batch and continuous-flow reactor experiments were identified using an Agilent 7890 GC with 5975C mass selective detector (GC-MS). The products were quantified by analysis with an Agilent 7890 gas chromatograph with flame ionization detector (GC-FID). The peak areas from the GC-FID chromatograms were related to product mass by employing appropriate FID response factors. A 50 m HP-PONA column was employed for product separation. The temperature program started at 40 °C, with a hold of time of 5 min, whereafter the temperature was increased by 4 °C∙min−1 up to 120 °C, and then at 20 °C∙min−1 to 300 °C and holding for 5 min.
4. Discussion
4.1. Phenol Alkylation in the Absence of a Catalyst
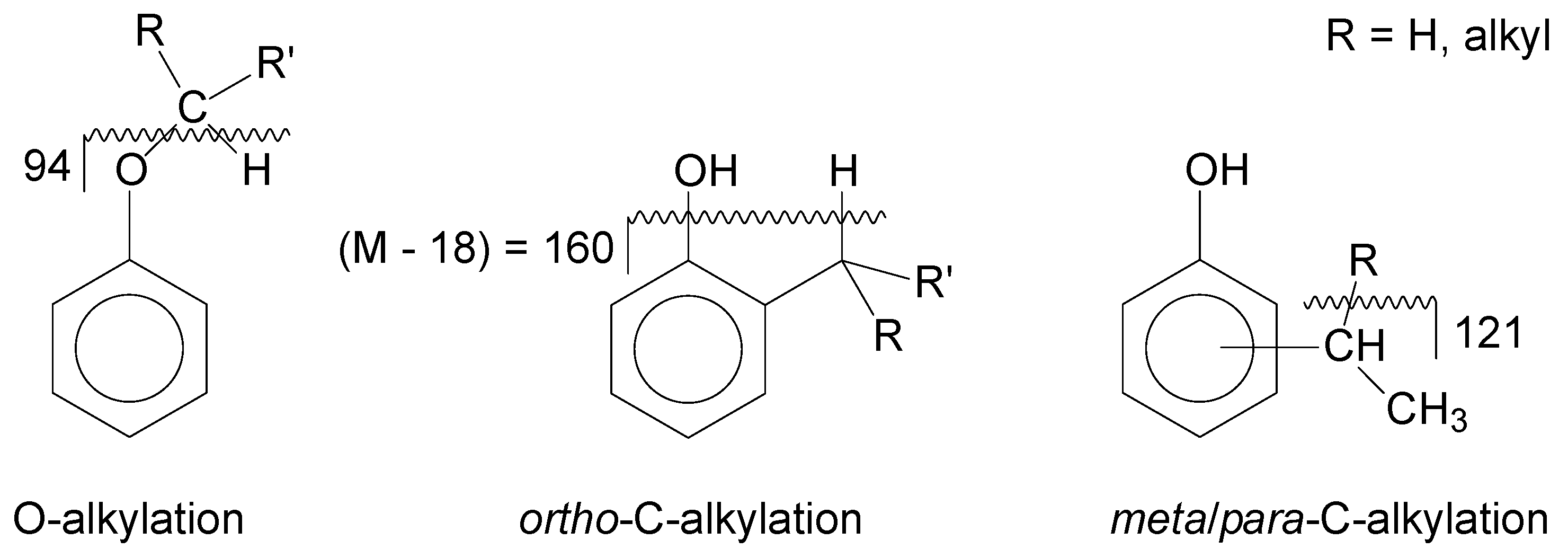
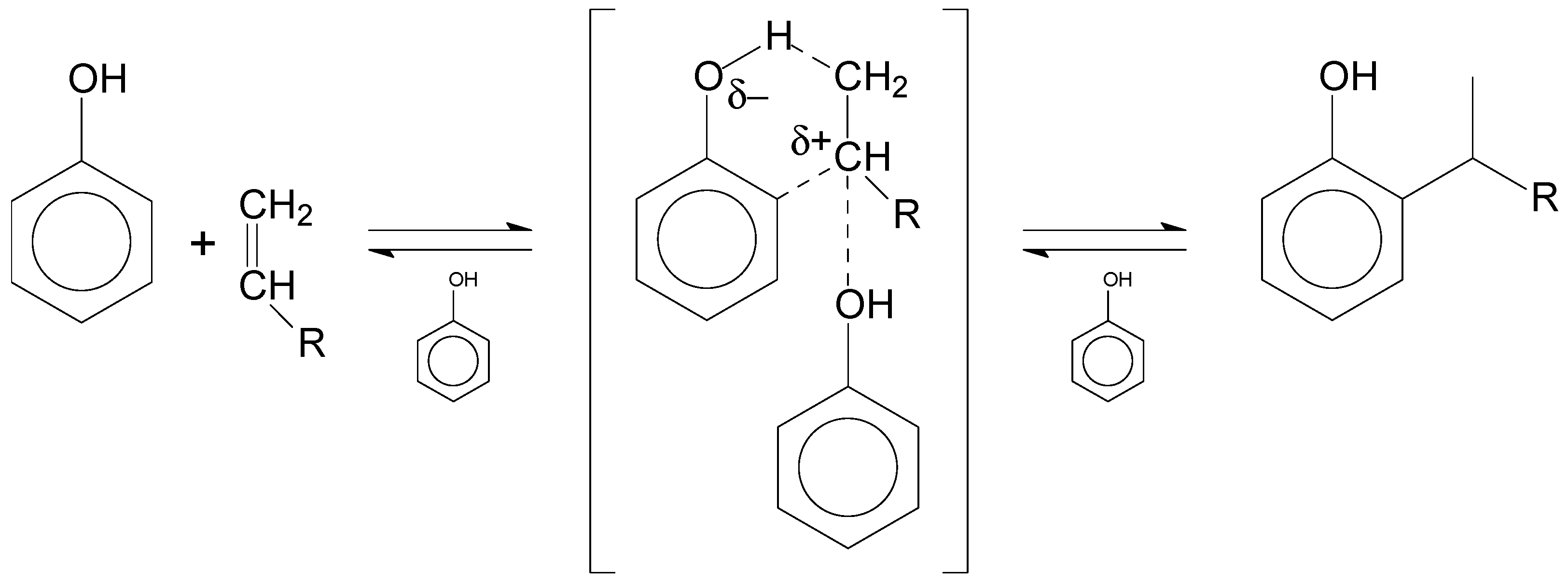
The study of phenol alkylation is somewhat complicated by the contribution of phenol alkylation in the absence of a catalyst, as shown in
Table 8. The reaction is described in terms of the formation of a six-membered ring as a transition state, which is stabilized by additional phenol in proximity (
Figure 3) and leads to the formation of alkylated phenol as product. Considering this, it is appropriate to refer to phenol alkylation as self-catalyzed because it is not truly an uncatalyzed reaction.
Direct and indirect support for the pathway in
Figure 3 can be found from various sources. It offers a plausible explanation for the high
ortho-alkylation selectivity (
Table 9) observed in the present study and by Goldsmith et al. [
24]. Direct spectroscopic support for hydrogen bonding of olefins to phenol was presented by West [
25]. In the presence of olefins, the 3610 cm
−1 infrared absorption of unbonded phenol O–H decreases with an increase in the bonded phenol O–H infrared absorption at 3550–3510 cm
−1. The phenol conversion rate was reported to be strongly dependent (second- to sixth-order dependence) on phenol concentration [
24,
26], which is congruent with the stabilization of the transition state by additional phenol. Furthermore, it was reported that dilution in pentane [
24] and dioxane [
27] suppressed phenol alkylation as one would expect from the strong concentration dependence of the reaction.
The potential contribution of catalytic contribution by the reactor wall was not studied, but it was previously reported that there was little difference in phenol alkylation when conducted in either 304 stainless steel or in glass-lined vessels [
24]. Additionally, when comparing phenol conversion at 315 °C with catalysts with surface areas >300 m
2∙g
−1 (
Table 4) to that without a catalyst (
Table 8), it is improbable that the extent of phenol conversion without a catalyst can be explained in terms of the limited surface area of the reactor wall, even if it was catalytically active.
The inhibition of phenol alkylation in the absence of a catalyst by pyridine is thought to be primarily due to the acid–base reaction between pyridine (K
b = 2.3 × 10
−9) and phenol (K
a = 1 × 10
−10), which will convert a fraction of the pyridine and phenol to the corresponding acid–base pair. In poorly solvating solutions, the clustering of phenol–pyridine acid–base pairs may work [
28].
Several important conclusions can be drawn from the phenol alkylation of olefins in the absence of a catalyst. First, the reaction required only weak acidity. Phenol was sufficiently acidic to self-catalyze the alkylation reaction with olefins. Second, the olefin in the transition-state has carbocation character, but is not formed by protonation of the olefin. The carbocation character is a result of polarization through the action of more than one phenolic group. Third, a consequence of the geometry of olefin polarization by multiple phenolic groups is that ortho-alkylation is strongly favored.
4.2. Phenol Alkylation with Amorphous Silica–Alumina Catalysts
Brønsted acid-catalyzed phenol alkylation with olefins is a type of Friedel–Crafts alkylation, but it differs from aromatic hydrocarbon alkylation in some respects. Alkylation of aromatic hydrocarbons with olefins proceeds by protonation of the olefin by the catalyst, which is followed by a nucleophilic attack from the bulk fluid phase, i.e., an Eley–Rideal type of reaction [
29]. Since both aromatics and olefins are nucleophiles, olefin oligomerization is an important side-reaction and, in industrial processes, the selectivity to alkylation is improved by operating at a high aromatic-to-olefin ratio [
9]. Conversely, when the aromatic hydrocarbon is adsorbed on the acid catalyst, the aromatic is not protonated at typical alkylation reaction conditions, although it is reactive for ring protonation at more severe conditions.
The present study employed a 1:1 molar ratio of phenol and 1-hexene; yet, hexene oligomerization was negligible (
Section 3.2). This was consistent with previous studies of phenol alkylation over a range of acid catalysts, which found that selectivity to oligomerization was low (<5%) or negligible [
30,
31,
32,
33,
34,
35]. The alkylation of phenolic compounds with very reactive olefins, such as isobutene, found a higher proportion of oligomerization [
36], but even so, selectivity to oligomerization was low (<10%) compared to alkylation.
There are likely two contributing factors explaining these observations. The first is that phenol alkylation does not require protonation of the olefin by the catalyst to proceed, but this is a requirement for olefin oligomerization. The second is that phenol is a more polar molecular than olefins, and surface coverage of the polar catalyst surface by phenol is likely higher than the bulk fluid concentration, i.e., competitive adsorption.
Although these observations could explain the low oligomerization selectivity, they do not fully explain the high phenol conversion to alkyl phenols in combination with low oligomerization selectivity. To explain this, one should consider the reactivity of adsorbed phenol. There are multiple reaction pathways for phenols and olefins [
37,
38,
39], and not all proceed by adsorption and protonation of the olefin. Unlike adsorbed aromatic hydrocarbons that are unreactive for alkylation by olefins at alkylation reaction conditions, adsorbed phenol appears to be reactive for alkylation. In this case, the reactions that are possible involve adsorbed olefins that can react with either phenol or olefins, but importantly, adsorbed phenol can react with olefins. This would offer a plausible explanation for the high phenol alkylation selectivity at high phenol conversion in equimolar feed mixtures, such as studied in this work.
Further support for this interpretation of the results is found the inhibitory effect of pyridine (compare
Table 5 with
Table 6 and
Table 7). There was a noticeable increase in
ortho-alkylation selectivity when pyridine inhibited the catalysis, although it did not reach the
ortho-alkylation selectivity of the uncatalyzed reaction by the pathway, as shown in
Figure 3. Pyridine decreased the availability of stronger Brønsted acid sites, and weaker acid sites were still able to provide additional polarization analogous to phenol in the bulk fluid (as shown in
Figure 3), causing the observed increase in
ortho-alkylation. Differently put, the inhibition of the acid catalyst by pyridine caused a proportionately lower reaction to proceed by protonation of the olefin, and a proportionately greater reaction to proceed by nucleophilic attack of the olefin on the adsorbed phenol.
Comparable observations can be found in the literature. For example, poisoning of Beta-zeolite by potassium exchange resulted in a decrease in phenol alkylation conversion at a constant temperature but an increase in
ortho-alkylation selectivity [
33]. Deprotonation of an industrial silica–alumina catalyst likewise resulted in a decrease in phenol conversion at a constant temperature but an increase in
ortho-alkylation selectivity [
40].
In conclusion, this study presented evidence to support that acid-catalyzed phenol alkylation took place by at least two pathways. The one pathway is typical of olefin–aromatic alkylation, which involves protonation of the olefin by the catalyst, followed by nucleophilic attack by phenol from the bulk fluid. The other pathway involves the adsorption of phenol on the acid catalyst, with polarization of the olefin, which is responsible for the nucleophilic attack.
4.3. Acid Catalyst Properties and Impact of Pyridine
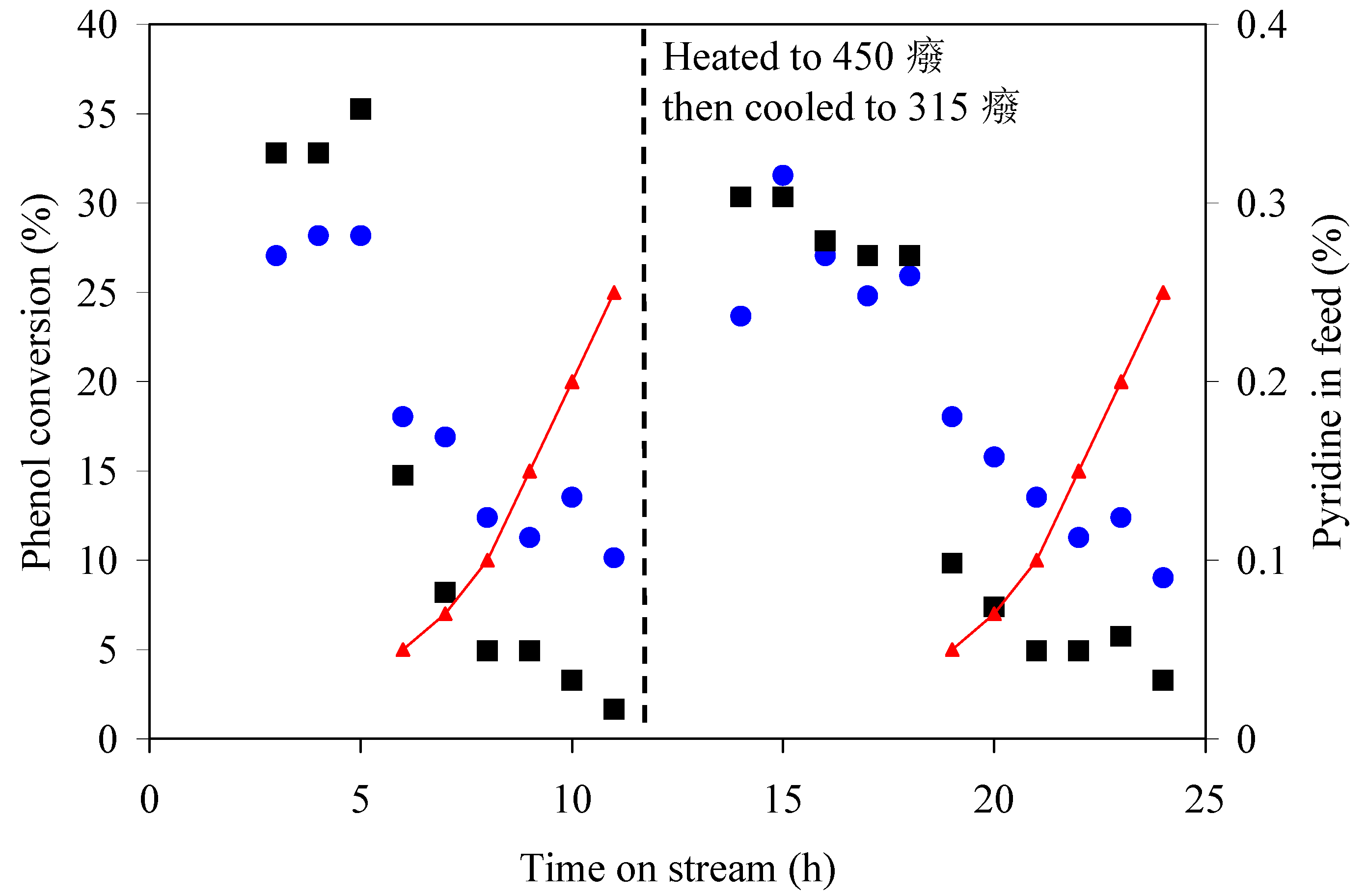
Pyridine inhibited conversion and there is a relationship between pyridine concentration in the feed and the extent of inhibition (
Figure 2). At the same time, pyridine was not a catalyst poison and catalyst activity could be recovered by desorbing pyridine at a higher temperature, as one would expect from the TPD results (
Table 2).
In a different study, it was found that some previously adsorbed pyridine could be removed from pyridine-treated silica–alumina catalysts by a clean feed at 100–250 °C [
41]. Analogous findings were reported for processing coal liquids over hydrotreating catalysts when minerals from coal liquefaction were present in the feed as a second solid phase [
42,
43], which caused re-partitioning of the nitrogen bases between the catalyst, coal liquid, and minerals in the coal liquid.
Nitrogen-base partitioning between different phases is related mainly to phase equilibrium. In thermodynamic terms, the chemical potential of each nitrogen base must be equal in each phase at equilibrium, which provides a driving force for desorption and re-partitioning of nitrogen bases when another ‘clean’ phase is introduced to the system. Although the partitioning cause by phase equilibrium is affected by acid–base equilibrium, the driving force is different.
When it comes to acid–base equilibrium, which is related to the strength of the interaction of nitrogen bases and acid sites of the catalyst, no simple relationship was found. Some trends were noted (
Section 3.2) between the acid site concentration, acid site strength, and Brønsted-to-Lewis acid ratio of the amorphous silica–alumina catalysts (
Table 2 and
Table 3) and phenol conversion in the absence and presence of pyridine (
Table 4). However, there were exceptions, which indicated that the relationship between catalytic activity and inhibition of catalytic activity by pyridine was more complex than a simple single-site acid–base reaction.
To offer an explanation for this, it is worthwhile keeping in mind that an important reaction pathway involved adsorbed phenol and cooperative polarization of the attacking olefin by the catalyst (
Section 4.2). Catalytic activity for such a reaction pathway is related to site pairs, analogous to the site pairs in catalysis by alumina [
44]. If this explanation is correct, then acid catalyst characterization, as reported in
Section 3.1, which did not consider site pairs, is insufficient to relate catalyst properties to catalytic activity.
4.4. Potential for Industrial Application
It was shown that pyridine, as a representative of the nitrogen bases in coal liquids, is not an acid catalyst poison, but that it inhibited acid-catalyzed conversion. The extent of inhibition reported in
Table 4 and
Figure 2 may seem severe but, considering that the evaluation was performed at high space velocity (WHSV 17–19 h
−1), there is scope for increasing phenol conversion in practical applications.
This study employed a comparatively high temperature (315 °C) for phenol alkylation, which was selected to facilitate the thermal desorption of pyridine. The practical upper threshold was not determined. At 330 °C, the onset of dealkylation of alkylphenol was reported in the absence of a catalyst [
24], but as the results in
Table 8 show, there was a net increase in phenol conversion as the temperature was increased from 315 to 350 °C despite competition from dealkylation. In acid-catalyzed phenol alkylation, both alkylation and dealkylation would be suppressed by pyridine, but with the expectation that dealkylation would be suppressed more due to its dependence on protonation of the aromatic, which requires strong Brønsted acid sites.
It was pointed out that there are several reaction pathways for phenol alkylation and that phenol alkylation was less dependent on protonation of the olefin to form carbocation than the alkylation of aromatic hydrocarbons. The findings about pyridine inhibition of phenol alkylation can therefore not be extended to Friedel–Crafts alkylation of aromatic hydrocarbons with olefins without considering this aspect. It was noted that the phenol alkylation products formed in the presence of pyridine were not exclusively those from a polarized intermediate and at least some of the products were those formed by Brønsted acid-catalyzed alkylation. It is expected that the use of acid catalysts for the alkylation of aromatic hydrocarbons in the presence of nitrogen bases would be less efficient than phenol alkylation but it remains plausible.
Nevertheless, the results presented suggest that the proposed use of acid-catalyzed alkylation in conjunction with coal tar naphtha, as suggested in the Introduction, may be viable. It is a speculative claim because the scope of this study did not extend to coal tar naphtha. At the same time, it can be mentioned that hydrotreated coal tar naphtha with a low pyridine content could be successfully alkylated [
45].
In terms of industrial applications, the primary benefits of olefin–aromatic and olefin–phenolic alkylation in feed materials containing nitrogen bases are in instances where the olefin content must be decreased without producing paraffins. This can be in a coal tar refinery context, where alkylation before hydrotreating would result in better octane number retention of the naphtha product. It can also be applied to other materials, such as cracked petroleum naphtha, to help reduce octane number loss resulting from hydrotreating.




{kind=link}
{kind=link}
{kind=link}