Abstract
It is generally accepted that organic–inorganic interactions involving H-rich fluids (i.e., H2O and H2) contribute significantly to hydrocarbon (HC) generation in sedimentary basins. However, the effects of two hydrogenation processes involving H2O and H2 on the generation and C/H isotope fractionation of HC gases from organic matter (OM) remain unclear. In this study, two groups of hydrothermal experiments involving low-mature kerogen without (Group 1) and with FeS (Group 2) at 330–420 °C and 50 MPa were conducted to simulate the hydrogenation of OM by H2O and H2, respectively. The experimental results show that the redox reactions between H2O and FeS lead to the generation of considerable amounts of H2 in the Group 2 experiments. HC gas yield in the Group 2 experiments reaches 1.8–3.6 times that in the Group 1 experiments at Easy%Ro of 1.05–2.50%. In addition, indirect hydrogenation via H2O-derived H2 generates HC gases with smaller 13C fractionation and more negative δ2H compared with direct hydrogenation via H2O. On this basis, the mechanisms for HC gas generation from two hydrogenations were addressed. Additionally, it is demonstrated that the equilibrium isotope effect (EIE) is responsible for the 13C and 2H isotope fractionation in the hydrogenation of OM by H2.
1. Introduction
The thermal maturation of sedimentary organic matter (OM) generating oil and hydrocarbon (HC) gases occurs in complex inorganic surroundings [1]. Essentially, water (H2O) and minerals may act as reactants or catalysts in this process [2,3,4,5,6,7,8,9,10,11,12]. Numerous hydrothermal experiments have been conducted to investigate the effects of H2O on HC generation [4,5,6,7,8,9,10,11,12,13]. These studies have demonstrated that H2O can react with OM at elevated temperatures and alter the yields and chemical compositions of HC products [7,8,9,10,11]. In addition, H transfer from H2O to OM and HCs has been extensively documented through the use of hydrothermal experiments [6,7,8,9,10,11,12,13]. Considering the contribution of extra H from H2O, Seewald [2] implied a greater yield and a higher maturity deadline for HC gas generation in sedimentary basins.
In summary, the hydrogenation of OM by H2O contributing to HC generation mainly includes two pathways. One is the direct H2O-OM reaction involving H2O and OM with/without catalysts (“the direct hydrogenation”) [2,7,9]. Till now, the mechanisms for H2O-OM reactions have been well understood due to experiments [6,7,8]. Direct hydrogenation was interpreted as having either a free radical or ionic mechanism [2,5,6,7,8,9,10,11,12]. For the free radical mechanism, the capture of alkyl free radicals from the homo-cleavage of OM by •H from H2O is responsible for the release of lower-molecular-weight HCs [7,8]. Meanwhile, the incorporation of •OH with alkyl groups generates carbon dioxide (CO2). Alternatively, the ionic reaction between intermediate alkenes and H+ from H2O dissociation also occurs in H2O-OM reactions, leading to isomeric HC generation [10]. Particular minerals (i.e., clay) may act as favorable catalysts for ionic reactions [8,9,10]. In addition, C/H isotope fractionation for HC gas generation from H2O-OM reactions has been discussed in previous studies [9,10,11,12,13]. H exchange or transfer from H2O to OM and HCs could occur in H2O-OM reactions [9,13,14,15,16,17]. It has been suggested that the H transfer from H2O accounts for the 2H isotope rollover of CH4 (i.e., δ2H becomes more negative with maturity or depth increasing) in natural gas at high maturity [18].
Another hydrogenation pathway is the indirect reactions between H2O-derived H2 and OM (“the indirect hydrogenation by H2”). In this process, H2 can be generated from the redox reactions between H2O and Fe(II)-bearing minerals in sedimentary rocks (i.e., pyrrohotite FeS, pyrite FeS2, magnetite Fe3O4, and siderite FeCO3) [19,20,21]. It is thermodynamically feasible for Reactions (1)–(4) to form a redox buffer and H2 [19,22]. In addition, H2 can also originate from serpentinization in ultrabasic rocks and during the radiolysis of H2O in U-rich rocks, and migrate into sedimentary strata via deep faults [23,24,25]. Under these conditions, the contact of H2 with OM and the indirect hydrogenation of OM by H2O-derived H2 in sedimentary rocks may become available [21].
1.5 FeS(pyrrhotite) + H2O = 0.75 FeS2(pyrite) + 0.25 Fe3O4(magnetite) + H2
2 Fe3O4(magnetite) + H2O = 3 Fe2O3(hematite) + H2
FeS2(pyrite) + 2 FeS(pyrrhotite) + 4 H2O = Fe3O4(magnetite) + 4 H2S
FeS2(pyrite) + Fe3O4(magnetite) + 2 H2O = 2 Fe2O3(hematite) + 2 H2S
It has been revealed that the presence of H2 can alter the thermal decompositions of organics [19,20]. Pyrolysis experiments have shown that H2 evidently promotes HC yields via the thermal maturation of OM [26]. In addition, hydrogenation via H2O and H2 may have different effects on the generation and C/H isotope fractionation of HC gas [8,9,27]. Geochemists have implied that hydrogenation by H2 is responsible for the 13C isotope reversal of HC gases (i.e., δ13C1 < δ13C2 < δ13C3) in deep sedimentary formations [28,29]. Unfortunately, the effects of the indirect hydrogenation of OM by H2O-derived H2 on the chemical and C/H isotopic compositions of gas products are little understood.
Gold tubes, which have excellent flexibility and chemical inertness, are usually used as ideal materials for hydrous pyrolysis [8,19]. In this study, the isothermal pyrolysis of kerogen with H2O and H2O-FeS was conducted using a gold tube system. The yields and chemical compositions of gas products were determined to ascertain the effects of direct and indirect hydrogenation on HC gas generation. In addition, the mechanisms and C/H isotope fractionation for HC gas generation in two hydrogenations were also discussed. Our experimental results are greatly beneficial for understanding the effects of two hydrogenation processes on the generation and C/H isotope fractionation of HC gases, as well as their mechanisms. More importantly, we first observed that the hydrogenation of OM by H2 can generate HC gases with small 13C fractionation, providing new insights into the origin of natural gas with abnormal isotopes in deep formations.
2. Materials and Methods
2.1. Samples and Reagents
Shale was collected from the Mesoproterozoic Xiamaling (XML) Formation in northern China, which was deposited in an anoxic environment with a water body of oxygen in the minimum zone. Tmax and hydrogen index (HI) of the shale are 437 °C and 330.1 mg/g TOC, respectively. Prior to the pyrolysis experiments, the shale sample was prepared as kerogen according to procedures described previously [30]. The detected vitrinite reflectance (Ro) for XML kerogen is about 0.60%, with a H/C ratio of 0.98 (Table 1). The carbon (δ13C) and hydrogen (δ2H) isotopic ratios of the kerogen are −28.5‰ and −111‰. Pyrrhotite (FeS), which was commercially available from J&K Company, Beijing, China, has a purity > 90 wt%. Distilled water was used, with a δ2H value of −64‰.

Table 1.
The essential geochemical characteristics of XML kerogen.
2.2. Hydrothermal Experiments
All of the pyrolysis experiments in this study were conducted using a gold tube pyrolysis system [8]. The tubes used were 60 mm in length with an inner diameter of 5.5 mm and a wall thickness of 0.5 mm. In a typical procedure, the solid or liquid reactants were sampled and loaded into the tube with one end sealed. The air in the tube was evacuated via flushing with inert argon. Then, the other end of the tube was crimped and sealed using an argon arc welder, with most of the sealed end submerged in liquid nitrogen. Finally, the tube with the sample loaded was placed inside the autoclaves. A constant pressure of 50 MPa was maintained, and isothermal pyrolysis at 330–420 °C was carried out. Here, the hydrothermal experiments with FeS were used to represent indirect hydrogenation via water-derived H2 (Reaction (1)). Two groups of hydrothermal experiments were conducted, including (1) XML kerogen and H2O and (2) XML kerogen, FeS and H2O (Table S1). The amounts of kerogen, H2O, and FeS loaded were about 50, 100, and/or 200 mg, respectively. When the set reaction time was reached, the gold tubes were withdrawn. Before the analysis, the tubes were weighed to ensure that no leakage occurred during pyrolysis.
2.3. Gas Determination
The collection of gas in each gold tube was performed through the use of a custom-made unit that was connected to a vacuum pump. Prior to piercing the tube, the unit was pumped to a residual pressure of <0.1 kPa. The detailed volume and molar determination of the gas products method was carried out according to Zhang et al. [31]. The identification and quantification of the individual HCs and non-HC gas components were conducted using a two-channel Agilent 7890 Series Gas Chromatograph (GC) integrated with an auxiliary oven, which was custom-configured by Wasson-ECE instrumentation (Fort Collins, CO, USA).
Stable carbon isotopic ratios (δ13C) of the gas products were analyzed using Thermo Delta V Advantage isotope ratio mass spectrometry (IRMS). The ratios of the stable hydrogen isotope (δ2H) were measured using a Thermo Mat253 mass spectrometer composed of Agilent 6890N GC and Mat253 IRMS (Thermo Fisher Scientific Inc., Waltham, MA, USA). The δ13C and δ2H values were relative to those of Vienna Peedee Belemnite (VPDB) and Vienna Standard Mean Ocean Water (VSMOW), respectively. Three international references, including NG1, NG2, and NG3, were used for a multi-point calibration. The measurement precision was within ±0.5‰ for δ13C and ±5.0‰ for δ2H. For detailed methods used in the calibration and analysis of the C/H isotope, refer to He et al. [9].
3. Results
3.1. Gas Yields and Compositions
Figure 1 shows the yields of the gas products in the hydrothermal experiments. For comparison, the units used for the gas yields were converted to millilitres (mL) per gram of kerogen (mL/g). The equivalent vitrinite reflectance (Easy%Ro) for each sample was calculated based on the EASY%Ro kinetic model of Sweeney and Burnham [32]. With Easy%Ro from 1.05% to 2.50%, total HC gas (C1–5) yields in the pyrolysis of XML kerogen involving H2O (Group 1) and involving FeS and H2O (Group 2) gradually increased from 1.25 to 40.88 mL/g and from 4.08 to 72.87 mL/g, respectively (Figure 1a). Notably, the C2+ HC gas (C2–5) yield in the Group 2 experiments also becomes much higher than that in Group 1 (Table S1). For instance, the C2–5 yield in the Group 2 experiments reaches 1.22–21.82 mL/g, which is 6.3–11.4 times that in the Group 1 experiments (0.19–1.91 mL/g). In addition, gas dryness (C1/C1–5) in the Group 2 experiments is apparently lower compared with that in Group 1 (Figure 1b).
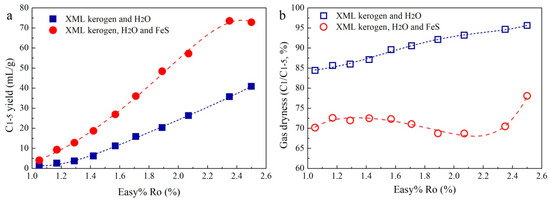
Figure 1.
Total HC gas (C1–5) yield (a) and gas dryness (b) in the hydrothermal experiments.
H2 yields in Group 1 and Group 2 experiments are 0.006–0.23 mL/g and 1.38–14.27 mL/g, respectively (Figure 2a). The yields of carbon dioxide (CO2) in the Group 1 experiment are 24.19–54.05 mL/g (Figure 2b). The decomposition of the C-O groups in the OM structures and H2O–OM reactions at elevated temperatures may collectively contribute to the generation of CO2 during the pyrolysis of kerogen with H2O [7]. Certain amounts of H2S were also generated at Easy%Ro > 1.42% in the Group 1 experiments (Figure 2c). However, CO2 yield became extremely low, and there was no detectable H2S in the Group 2 experiments.
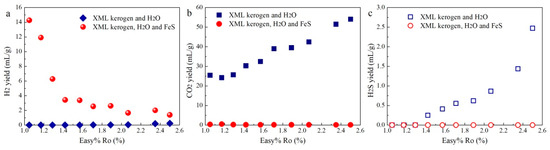
Figure 2.
Yields of H2 (a), CO2 (b), and H2S (c) in the hydrothermal experiments.
3.2. Carbon Isotopic Compositions
The δ13C values of CH4 (δ13C1) in the Group 1 experiments first decrease from −36.9‰ to −37.4‰ and then gradually increase to −29.2‰ with Easy%Ro increasing (Figure 3a). δ13C1 in the Group 2 experiments ranges from −35.5‰ to −30.5‰ (Figure 3b). The δ13C of C2H6 (δ13C2) and δ13C of C3H8 (δ13C3) in the Group 1 experiments range from −29.1‰ to −18.8‰ and from −26.6‰ to −13.8‰, respectively. C2H6 and C3H8 in the Group 2 experiments are more depleted in 13C compared with those in Group 1. In addition, the 13C isotope fractionation of individual HC gases in Group 2 is apparently smaller. For instance, δ13C2 and δ13C3 in the Group 2 experiments vary in narrow ranges from −32.0‰ to −29.8‰ and from −29.6‰ to −28.5‰ with Easy%Ro at 1.05%–2.35%. δ13C values of CO2 (δ13CCO2) in Group 1 and Group 2 experiments are from −23.4‰ to −21.1‰ and from −16.9‰ to −11.5‰, respectively (Table S1).
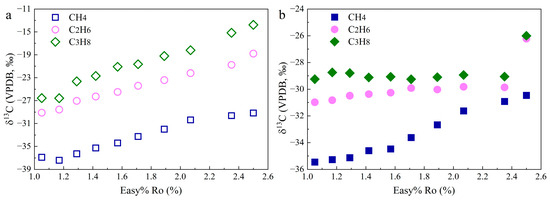
Figure 3.
δ13C of CH4, C2H6, and C3H8 in the hydrothermal experiments without (a) and with FeS (b).
3.3. Hydrogen Isotopic Compositions
The δ2H of CH4 (δ2H1) in the Group 1 experiments first decreases from −256‰ to −323‰ and then increases to −215‰ (Figure 4a). Although δ2H1 in the Group 2 experiments shows a similar evolution trend to that in Group 1, it becomes more negative. The δ2H of C2H6 (δ2H2) in the Group 1 experiments gradually increases from −182‰ to −101‰, and δ2H2 in Group 2 is almost constant, ranging from −224‰ to −213‰ (Figure 4b). The δ2H of H2 in Group 2 ranges from −476‰ to −340‰ (Table S1).
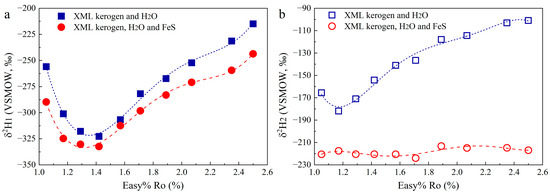
Figure 4.
δ2H of CH4 (a) and C2H6 (b) in the hydrothermal experiments.
4. Discussion
4.1. Mechanisms for Gas Generation in Two Hydrogenation Reactions
It has been demonstrated that the thermal maturation of OM and H2O-OM reactions collectively contribute to gas generation in hydrothermal experiments [6,9,13]. That is, the H sources for HC generation in hydrous conditions include H2 from kerogen condensation and H from H2O (Figure 5). Hence, HC gas in the Group 1 experiments should be generated by two pathways: (1) the cleavage and hydrogenation of alkyl radicals by H2, which can be previously released via the condensation or aromatization of a kerogen structure [33], and (2) the direct reaction between H2O and alkyl groups, generating alcohols, which would be finally oxidized by H2O to form lower-weight HCs (i.e., HC gases), CO2 and H2 [7,8,34].
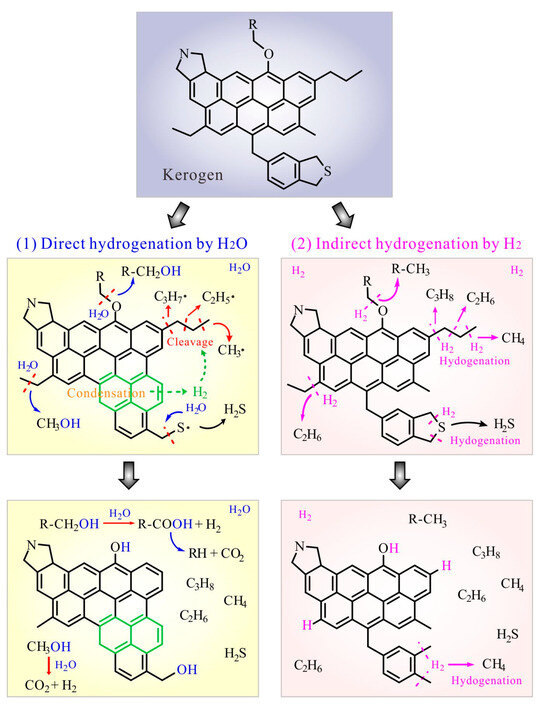
Figure 5.
The reaction pathways for the direct and indirect hydrogenation of kerogen.
In the Group 2 experiments, the redox reaction between FeS and H2O (Reaction (1)) should occur and account for H2 generation under hydrothermal conditions [19,20,21,22]. The evident higher HC gas yield in the Group 2 experiments (Figure 1a) suggests that the indirect hydrogenation of OM via H2O-derived H2 promotes HC generation. Actually, extra H2 can inhibit the condensation or cross-linking reactions of OM and enhance the hydrogenation of alkyl chains to form HCs (Reactions (5)–(7)) [7].
ARO-CH3 + H2 = ARO-H + CH4
ARO-C2H5 + H2 = ARO-H + C2H6
ARO-C2H5 + H2 = ARO-H + C2H6
This mechanism may also account for the lower gas dryness in the Group 2 experiments (Figure 1b). Thermodynamic calculations revealed that the Gibbs energies for Reactions (5)–(7) at 100–500 °C range from −10.3 to −10.6 kcal/mol, from −10.9 to −12.4 kcal/mol, and from −12.2 to −14.9 kcal/mol, respectively. This fact means that the generation of C2H6 and C3H8 from hydrogenation via H2 is more thermodynamically favorable than CH4. The yield of individual HC gases from hydrogenation via H2 should be governed by the content of the corresponding alkyl chain (i.e., -CH3, -C2H5, or -C3H7) in OM.
The extremely low H2S and CO2 yields in Group 2 may be attributed to the precipitation of pyrite (FeS2) and siderite (FeCO3) via reactions between H2S/CO2 and FeS/Fe3O4 (Reactions (8) and (9)) [19,22]. Alternatively, the hydrogenation of R-O groups by H2 preferentially forming R-CH3 rather than CO2 may be another possible reason for the low CO2 yield (Figure 5). In addition, CO2 may also be reduced by H2 to generate HC gases at elevated temperatures [20,24,31].
FeS + H2S = FeS2 + H2
Fe3O4 + 3 CO2 + H2 = 3 FeCO3 + H2O
4.2. Carbon Isotope Fractionation for HC Gas Generation
Numerous pyrolysis experiments and theoretical calculations have been conducted to understand 13C isotope fractionation for gas generation from OM maturation [35,36,37]. It is generally accepted that 13C isotope fractionation for HC gas generation from OM cracking obeys the kinetic isotope effect (KIE) [35,36]. That is, the δ13C of individual HC gas (i.e., CH4) is dominated by that of its precursor and the KIE factor (αKIE), which refers to the ratio of the rate constant with a heavier isotope versus that with a lighter isotope [36]. Because the cleavage of alkyl radicals with 13C substituted has a higher energy barrier than that with 12C, the αKIE value is reasonably lower than 1. KIE would result in the enrichment of the δ13C of individual HC gas during its generation from a certain precursor [36,37]. The gradual increase in the δ13C of CH4, C2H6, and C3H8 with their yields in Group 1 (Figure 3) demonstrates that KIE dominates 13C isotope fractionation. Essentially, αKIE for the generation of CH4 is lower than those for C2H6 and C3H8 [36,37,38], and the δ13C of HC gases due to KIE always show a positive correlation with the carbon number. Although the presence of H2O-OM reactions may alter the gas generation pathways, it shows a limited effect on δ13C1 and a certain depletion of 13C for C2+ HC gases [9,11]. The 13C isotope of HC gases in the Group 1 experiments still has a normal trend (Figure 6a), similar to those during the anhydrous pyrolysis of OM [38]. Hence, the direct hydrogenation of OM by H2O may alter the gas generation pathway but not the 13C isotope patterns of HC gases unless there are particular catalysts.
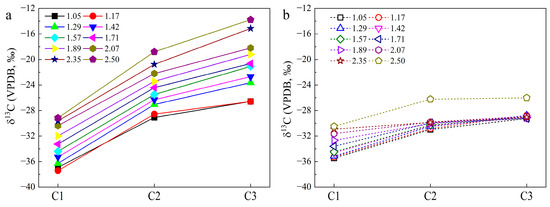
Figure 6.
δ13C distribution for C1, C2, and C3 at different Easy%Ro in the hydrothermal experiments without (a) and with FeS (b).
As shown in Figure 6b, the δ13C of the HC gases in the Group 2 experiments are more depleted compared with those in Group 1, and the difference in δ13C between CH4, C2H6, and C3H8 becomes much smaller. Hydrogenation via H2O-derived H2 not only alters the gas generation pathway but also 13C isotope fractionation. The δ13C values of C2H6 and C3H8 almost keep constant even with increasing yield (Figure 3b). This result implies that the equilibrium isotope effect (EIE) rather than KIE may govern 13C fractionation for HC gas generation in the Group 2 experiments. Assuming the higher yield of individual HC gases (Ci) in Group 2 compared with that in Group 1 (Table S1) were derived from the hydrogenation of kerogen by H2, the δ13C of Ci from hydrogenation by H2 (δ13Ci,H) can be obtained based on mass balance (Equation (10)).
where M1 and M2 refer to the yields of Ci in group 1 and group 2 experiments. δ13Ci,1 and δ13Ci,2 are the δ13C of Ci in the Group 1 and Group 2 experiments.
δ13Ci,H × (M2 + M1) = M2 × δ13Ci,1 − M1 × δ13Ci,2
The calculated δ13C1,H, δ13C2,H, and δ13C3,H from hydrogenation by H2 range from −34.6‰ to −33.3‰, from −31.7‰ to −31.3‰, and from −29.7‰ to −29.0‰, respectively (Figure 7a). For equilibrium 13C fractionation in Reactions (5)–(7), the difference in δ13C (Δδ13C) between the precursors of alkyl chains in OM (δ13Cal) and HC gases (δ13Ci) can be addressed through the application of Equation (11).
where αEIE refers to the EIE factor for 13C exchange between alkyl chains in OM and the corresponding HC gases.
Δδ13Ci (VPDB, ‰) = δ13Cal − δ13Ci = (αEIE − 1) × 1000
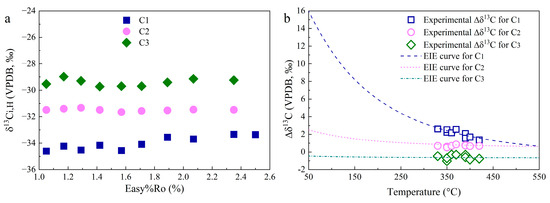
Figure 7.
δ13Ci,H for CH4, C2H6, and C3H8 generated from hydrogenation by H2 (a), and Δδ13C between alkyl groups in OM and HC gases from the experimental data and EIE calculations (b).
Based on the ab initio quantum calculation using the B3LYP-631G** flavor of DFT, the αEIE for the 13C exchange involving CH4, C2H6, and C3H8 at 50–550 °C were addressed as 1.0158–1.0004, 1.0025–1.0006, and 0.9995–0.9993, respectively. According to Equation (10), the Δδ13C values are calculated from 15.7‰ to 0.7‰, from 2.5‰ to 0.6‰, and from −0.5‰ to −0.7‰ (Figure 7b). Intramolecular equilibrium 13C fractionation suggests that the alkyl groups in OM are more depleted in 13C compared with the aromatic C and C-O groups [39]. In addition, the EIE factor of 13C between the -CH2- groups and CH3 is higher than 1. This means that the δ13C of the alkyl chains should be more negative than bulk kerogen and increase with chain length. Taking the δ13C of methyl, ethyl, and propyl in XML kerogen as −32.0‰, −31.0‰, and −30‰, the calculated Δδ13C for CH4, C2H6, and C3H8 from the experimental data fit well with the theoretical curves. Therefore, EIE in the hydrogenation of OM by H2 may account for the small fractionation of δ13C between CH4, C2H6, and C3H8 in the Group 2 experiments.
4.3. H Isotope Fractionation of H2O-H2-CH4 in Hydrothermal Conditions
It is generally accepted that H transfer could occur via H2O-OM reactions and affect the δ2H of OM and HC products at elevated temperatures [6,7,8,9,10,11]. The presence of deuterium water usually leads to the enrichment of 2H for OM and bitumen [6,15]. The hydrous pyrolysis of OM using distilled H2O (i.e., δ2H of −22‰ and −64‰) preferentially generates 2H-depleted HC gases compared with anhydrous pyrolysis [9,11]. Essentially, the final δ2H of HC gases that suffer from H exchange with H2O is governed by δ2H of H2O (δ2Hw) and H isotope fractionation via either EIE or KIE [9,14,15,16,17]. The EIE factor (αH2O-CH4) for H exchange/transfer from H2O to CH4 has been well addressed in the literature [16,40]. The δ2H of CH4 (δ2H1,e) after equilibrium 2H exchange with H2O (δ2Hw of −64‰) at 330–420 °C was calculated from −171‰ to −165‰ [9]. However, the measured δ2H of CH4 in the Group 1 experiments deviates significantly from the equilibrium δ2H1,e (Figure 4a). This is attributed to two possible reasons: (1) the thermal cracking of XML kerogen in addition to H2O-OM reactions, which collectively contributes to CH4 generation, and (2) the fact that the H transfer from H2O to CH4 did not reach equilibrium in the Group 1 experiments [12,17]. Indeed, kinetic calculations revealed that the equilibrium time (i.e., with conversion of 99.9%) for the H transfer from H2O to CH4 at 330–420 °C should be much longer than the pyrolysis time in our experiments [9,41].
As mentioned above, H2 was first generated from redox reactions involving H2O-FeS and then participated in the hydrogenation of OM in the Group 2 experiments. In this process, there is H isotope fractionation for the H2O-H2-CH4 system [16]. The EIE factors for H fractionation of H2O-H2 (αH2O-H2) and CH4-H2 (αCH4-H2) are expressed through the use of Equations (12) and (13).
αH2O-H2 = (δ2HH2O + 1000)/(δ2HH2 + 1000)
αCH4-H2 = (δ2HCH4 + 1000)/(δ2HH2 + 1000)
Previous experiments and theoretical calculations have addressed αH2O-H2 and αCH4-H2 as 4.54–1.36 and 3.68–1.21 at 0–500 °C [16,41,42]. Based on Equations (12) and (13), αH2O-H2 and αCH4-H2 in terms of measured δ2H of CH4 and H2 in Group 2 experiments were calculated as 1.79–1.42 and 1.35–1.10 (Figure 8). In hydrothermal systems, the occurrence of H2-rich fluids/gases has been extensively documented [16,23,40,41,42,43,44,45]. The redox reaction involving Fe(II)-minerals and H2O is considered as the most important pathway for natural H2 generation [22,23,24]. Kinetic calculations have revealed that the 2H exchange between H2O and H2O-derived H2 can reach equilibrium in a short time at high T [42]. This may be the reason that αH2O-H2 obtained from our experimental data fit well with the theoretical curves. Actually, the T-dependent H isotope equilibrium involving H2-H2O has usually been used as a temperature indicator for H2 generation in geological settings [40,42,44]. Considering the equilibrium H isotope exchange, previous studies have established a diagram of 1000lnαH2-H2O versus 1000lnαCH4-H2O to distinguish CH4 generation from the direct hydrogenation via H2O or from indirect hydrogenation via H2 [27,40]. Noticeably, when using this diagram to identify the origin of CH4 from hydrogenation via H2, two critical issues should be firstly addressed: (1) H2 is the dominant H source for CH4, and (2) the H isotope fractionation of H2-CH4 reaches equilibrium. As shown in Figure 8b, the experimental αCH4-H2 from the Group 2 experiments and some geological αCH4-H2 values deviate from the theoretical curves. Such a discrepancy may be interpreted by other H sources (i.e., OM and H2O) in addition to H2, which significantly contributes to CH4 generation.
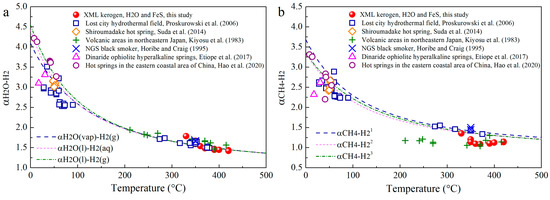
Figure 8.
The αH2O-H2 (a) and αCH4-H2 (b) from the theoretical calculations and experimental data. αH2O(vap)-H2(g), αH2O(l)-H2(aq), αH2O(l)-H2(g) refer to the EIE factors between vapor H2O and gaseous H2, liquid H2O and aqueous H2, liquid H2O and gaseous H2, respectively [23,40,42,43,44,45]. 1,2,3 Theoretical curves of αCH4-H2 versus T were obtained from equations from [16].
4.4. Geological Implications for GAS Generation in Deep Formations
As known, conventional or shale gas discovered in sedimentary basins is mostly derived from the thermal cracking of crude oil or kerogen [1]. The cracking of OM is governed by kinetic processes and preferentially generates HC gases with a normal 13C isotope [36,37,38]. For instance, in the anhydrous pyrolysis of kerogen or oil in closed systems, 13C fractionation between C2H6 and CH4 reaching > 10‰ has been observed [37]. However, kinetic 13C isotope fractionation has difficulty in explaining the δ13C features of high over-mature natural gas. Typically, the δ13C of HC gases in shale gas usually evolve from a normal trend to a reversed trend, finally becoming almost equal with increasing maturity [46]. A similar phenomenon has also been observed for HC gases in carbonate reservoirs [18]. Gas mixing with different genesis or maturity has been introduced to interpret the 13C reversal of HC gases in sedimentary basins [18,46,47]. However, the mixing of HC gases with different genesis or maturity in shale or carbonate reservoirs is sometimes unconvincing. The indirect hydrogenation of OM via H2 may provide an alternative interpretation for the occurrence of HC gases with an abnormal 13C isotope. Actually, the 2H rollover of CH4 observed in natural gas with high maturity may be important evidence of the hydrogenation of OM via inorganic H [9,18,28].
Free H2 (with its content from several ppm to over 10%) is commonly present in shale and conventional reservoirs [25], providing the possibility for the hydrogenation of OM via H2. The evident increase in HC gas yield in the Group 2 experiments implies that gas potential from OM in deep formations, when considering hydrogenation via H2, may be much higher than traditional concepts [2]. In the low maturation stage, hydrogenation via H2 mainly occurs via the hydrogenation of alkyl chains, and the generated HC gases still show a normal trend in 13C. However, in this process, EIE results in a smaller difference in 13C between individual HC gases, which is more comparable with that observed in natural gas. In the high over-maturation stage, the content of alkyl chains in OM becomes extremely low or only methyl chains remain [37]. Hydrogenation via H2 preferentially generates CH4, which would be converted to C2+ HC gases. Meanwhile, CO2 released via the decarboxylation of OM at high maturity could be reduced by H2 via FFT to form HC gases [28,29]. These two hydrogenation processes by H2 may both generate HC gases with 13C isotope reversal or nearly equal.
Indeed, more work is required to understand the possible hydrogenation of OM by H2 in deep to ultra-deep formations. Specifically, the supply and origin of H2 in oil and gas reservoirs should be first clarified. However, the identification of H2 origin in geological settings remains challenging, and traditional analysis of the content and δ2H of H2 cannot provide unambiguous evidence [25]. New isotope techniques (i.e., clumped H isotopes) may enable the determination of the formation temperature and origin of H2 in sedimentary basins [48]. In addition, more experiments and analysis of geological samples should be conducted to identify the contribution of the hydrogenation of OM with different maturity by H2 to HC gas generation in the subsurface.
5. Conclusions
Based on hydrothermal experiments involving CL kerogen without and with FeS at 330–450 °C, the effects of the hydrogenation of OM by H2O and H2O-derived H2 on HC gas generation were addressed. The yields of H2 and HC gas in the hydrothermal experiments with FeS are evidently higher than those without FeS. This result means that indirect hydrogenation via H2 is more efficient for HC generation from OM than direct hydrogenation via H2O. Meanwhile, gas dryness in the hydrothermal experiments with FeS is evidently lower compared with that without FeS. Moreover, the difference in δ13C between CH4, C2H6, and C3H8 in the hydrothermal experiments with FeS is smaller, and the δ2H of CH4 is more depleted.
On this basis, two different mechanisms were proposed for the hydrogenation of OM by H2O and H2. It is suggested that the presence of H2 can inhibit the condensation of kerogen and thus enable the hydrogenation of alkyl chains to form HC products. In this process, the hydrogenation of C2+ alkyl chains in OM is more thermodynamically favorable than that of the methyl chain. This fact can reasonably interpret the higher yield and low dryness of HC gases in the hydrothermal experiments with FeS. In addition, thermodynamic calculations reveal that EIE rather than KIE should govern the 13C isotope fractionation for HC gas generation from the hydrogenation of OM by H2. The H isotope fractionation factor of H2O-H2 (αH2O-H2) in the hydrothermal experiments with FeS fits well with the equilibrium curves. However, the contributions of multiple H sources (i.e., kerogen, H2O and H2) for CH4 result in the deviation of αCH4-H2 with EIE values.
The universal presence of H2 in conventional and shale reservoirs may imply that the hydrogenation of OM by H2 has contributed to HC gas generation. More importantly, our experiment results may provide new insights for the understanding of HC gas potential as well as the origin of HC gases with an abnormal 13C isotope in sedimentary basins.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/pr12030458/s1, Table S1: The experimental conditions, yields, and isotopic compositions of the gas products in the hydrothermal experiments.
Author Contributions
Conceptualization, K.H. and S.Z.; methodology, K.H. and X.W.; validation, K.H., X.W., C.Y. and S.Z.; formal analysis, K.H.; investigation, K.H., X.W., C.Y. and S.Z.; data curation, K.H.; writing—original draft preparation, K.H. and X.W.; writing—review and editing, K.H., X.W., C.Y. and S.Z.; visualization, K.H.; supervision, X.W. and S.Z.; funding acquisition, K.H. and X.W. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Natural Science Foundation of China, grant number 41973068, the Research Projects from China National Petroleum Corporation, grant numbers 2023ZZ0203 and 2020D-5008-001.
Data Availability Statement
The data will be available when requested.
Conflicts of Interest
Authors Kun He, Xiaomei Wang, Chunlong Yang and Shuichang Zhang were employed by the company PetroChina. All authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Tissot, B.P.; Welte, D.H. Petroleum Formation and Occurrence, 2nd ed.; Spring: Berlin/Heidelberg, Germany; New York, NY, USA, 1984. [Google Scholar]
- Seewald, J.S. Organic-inorganic interactions in petroleum-producing sedimentary basins. Nature 2003, 426, 327–333. [Google Scholar] [CrossRef]
- Pan, C.C.; Geng, A.S.; Zhong, N.; Liu, J.Z.; Yu, L. Kerogen pyrolysis in the presence and absence of water and minerals: Amounts and compositions of bitumen and liquid hydrocarbons. Fuel 2009, 88, 909–919. [Google Scholar] [CrossRef]
- Hoering, T.C. Thermal reactions of kerogen with added water, heavy water and pure organic substances. Org. Geochem. 1984, 21, 267–278. [Google Scholar] [CrossRef]
- Lewan, M.D.; Roy, S. Role of water in hydrocarbon generation from Type-I kerogen in Mahogany oil shale of the Green River Formation. Org. Geochem. 2011, 42, 31–41. [Google Scholar] [CrossRef]
- Schimmelmann, A.; Boudou, J.P.; Lewan, M.D.; Wintsch, R.P. Experimental controls on D/H and 13C/12C ratios of kerogen, bitumen and oil during hydrous pyrolysis. Org. Geochem. 2001, 32, 1009–1018. [Google Scholar] [CrossRef]
- Lewan, M.D. Experiments on the role of water in petroleum formation. Geochim. Cosmochim. Acta 1997, 61, 3691–3723. [Google Scholar] [CrossRef]
- He, K.; Zhang, S.C.; Mi, J.K.; Zhang, W.L. Pyrolysis involving n-hexadecane, water and minerals: Insight into the mechanisms and isotope fractionation for water-hydrocarbon reaction. J. Anal. Appl. Pyrol. 2018, 130, 198–208. [Google Scholar] [CrossRef]
- He, K.; Zhang, S.C.; Wang, X.M.; Mi, J.K.; Zhang, W.J.; Guo, J.H.; Zhang, W.L. Pyrolysis of 1-methylnaphthalene involving water: Effects of Fe-bearing minerals on the generation, C and H isotope fractionation of methane from H2O-hydrocarbon reaction. Org. Geochem. 2021, 152, 104151. [Google Scholar] [CrossRef]
- Leif, R.N.; Simoneit, B.R.T. The role of alkenes produced during hydrous pyrolysis of a shale. Org. Geochem. 2000, 31, 1189–1208. [Google Scholar] [CrossRef]
- Gao, L.; Schimmelmann, A.; Tang, Y.C.; Mastalerz, M. Isotope rollover in shale gas observed in laboratory pyrolysis experiments: Insight to the role of water in thermogenesis of mature gas. Org. Geochem. 2014, 68, 95–106. [Google Scholar] [CrossRef]
- Reeves, E.P.; Seewald, J.S.; Sylva, S.P. Hydrogen isotope exchange between n-alkanes and water under hydrothermal conditions. Geochim. Cosmochim. Acta 2012, 77, 582–599. [Google Scholar] [CrossRef]
- Wang, D.T.; Seewald, J.S.; Reeves, E.P.; Ono, S.; Sylva, S.P. Incorporation of water-derived hydrogen into methane during artificial maturation of source rock under hydrothermal conditions. Org. Geochem. 2022, 171, 104468. [Google Scholar] [CrossRef]
- Wang, Y.; Sessions, A.L.; Nielsen, R.J.; Goddard III, W.A. Equilibrium 2H/1H fractionations in organic molecules. II: Linear alkanes, alkenes, ketones, carboxylic acids, esters, alcohols and ethers. Geochim. Cosmochim. Acta 2009, 73, 7076–7086. [Google Scholar] [CrossRef]
- Schimmelmann, A.; Lewan, M.D.; Wintsch, R.P. D/H isotope ratios of kerogen, bitumen, oil, and water in hydrous pyrolysis of source rocks containing kerogen types I, II, IIS, and III. Geochim. Cosmochim. Acta 1999, 63, 3751–3766. [Google Scholar] [CrossRef]
- Horibe, Y.; Craig, H. D/H fractionation in the system methane-hydrogen-water. Geochim. Cosmochim. Acta 1995, 59, 5209–5217. [Google Scholar] [CrossRef]
- Sessions, A.L.; Sylva, S.P.; Summons, R.E.; Hayes, J.M. Isotopic exchange of carbon-bound hydrogen over geologic timescales. Geochim. Cosmochim. Acta 2004, 68, 1545–1559. [Google Scholar] [CrossRef]
- Zhang, S.C.; He, K.; Hu, G.Y.; Mi, J.K.; Ma, Q.S.; Liu, K.Y.; Tang, Y.C. Unique chemical and isotopic characteristics and origins of natural gases in the Paleozoic marine formations in the Sichuan Basin, SW China: Isotope fractionation of deep and high mature carbonate reservoir gases. Mar. Pet. Geol. 2018, 89, 68–82. [Google Scholar] [CrossRef]
- Seewald, J.S. Aqueous geochemistry of low molecular weight hydrocarbons at elevated temperatures and pressures: Constraints from mineral buffered laboratory experiments. Geochim. Cosmochim. Acta 2001, 65, 1641–1664. [Google Scholar] [CrossRef]
- McCollom, T.M.; Seewald, J.S. A reassessment of the potential for reduction of dissolved CO2 to hydrocarbons during serpentinization of olivine. Geochim. Cosmochim. Acta 2001, 65, 3769–3778. [Google Scholar] [CrossRef]
- Milesi, V.; Prinzhofer, A.; Guyot, F.; Benedetti, M.; Rodrigues, R. Contribution of siderite-water interaction for the unconventional generation of hydrocarbon gases in the Solimões Basin, North-West Brazil. Mar. Petrol. Geol. 2016, 71, 168–182. [Google Scholar] [CrossRef]
- Kishima, N. A thermodynamic study on the pyrite-pyrrhotite-magnetite-water system at 300–500 °C with relevance to the fugacity/concentration quotient of aqueous H2S. Geochim. Cosmochim. Acta 1989, 53, 2143–2155. [Google Scholar] [CrossRef]
- Etiope, G.; Samardžić, N.; Grassa, F.; Hrvatović, H.; Miošić, N.; Skopljak, F. Methane and hydrogen in hyperalkaline groundwaters of the serpentinized Dinaride ophiolite belt, Bosnia and Herzegovina. Appl. Geochem. 2017, 84, 286–296. [Google Scholar] [CrossRef]
- Klein, F.; Grozeva, N.G.; Seewald, J.S. Abiotic methane synthesis and serpentinization in olivine-hosted fluid inclusions. Proc. Natl. Acad. Sci. USA 2019, 116, 17666–17672. [Google Scholar] [CrossRef] [PubMed]
- Milkov, A.V. Molecular hydrogen in surface and subsurface natural gases: Abundance, origins and ideas for deliberate exploration. Earth Sci. Rev. 2022, 230, 104063. [Google Scholar] [CrossRef]
- Jin, Z.J.; Zhang, L.P.; Yang, L.; Hu, W.X. Primary study of geochemistry features of deep fluids and their effectiveness on oil/gas reservoir formation in sedimentary basins. Earth Sci. J. China Univ. Geosci. 2002, 27, 659–665. [Google Scholar]
- Vacquand, C.; Deville, E.; Beaumont, V.; Guyot, F.; Sissmann, O.; Pillot, D.; Arcilla, C.; Prinzhofer, A. Reduced gas seepages in ophiolitic complexes: Evidences for multiple origins of the H2–CH4–N2 gas mixtures. Geochim. Cosmochim. Acta 2018, 223, 437–461. [Google Scholar] [CrossRef]
- Burruss, R.C.; Laughrey, C.D. Carbon and hydrogen isotopic reversals in deep basin gas: Evidence for limits to the stability of hydrocarbons. Org. Geochem. 2010, 41, 1285–1296. [Google Scholar] [CrossRef]
- Zumberge, J.; Ferworn, K.; Brown, S. Isotopic reversal (‘rollover’) in shale gases produced from the Mississippian Barnett and Fayetteville formations. Mar. Pet. Geol. 2012, 31, 43–52. [Google Scholar] [CrossRef]
- Durand, B. Kerogen: Insoluble Organic Matter from Sedimentary Rocks, 1st ed.; Bayeusaine: Paris, France, 1980; pp. 35–52. [Google Scholar]
- Zhang, S.C.; Mi, J.K.; He, K. Synthesis of hydrocarbon gases from four different carbon sources and hydrogen gas using a gold-tube system by Fischer-Tropsch method. Chem. Geol. 2013, 349–350, 27–35. [Google Scholar] [CrossRef]
- Sweeney, J.J.; Burnham, A.K. Evaluation of a simple model of vitrinite reflectance based on chemical kinetics. AAPG Bull. 1990, 74, 1559–1570. [Google Scholar]
- Kissin, Y.V. Catagenesis and composition of petroleum: Origin of n-alkanes and isoalkanes in petroleum crudes. Geochim. Cosmochim. Acta 1987, 512, 445–2457. [Google Scholar] [CrossRef]
- Wei, N.; Xu, D.; Hao, B.; Guo, S.; Guo, Y.; Wang, S. Chemical reactions of organic compounds in supercritical water gasification and oxidation. Water Res. 2021, 190, 116634. [Google Scholar] [CrossRef] [PubMed]
- Cramer, B.; Krooss, B.M.; Littke, R. Modelling isotope fractionation during primary cracking of natural gas: A reaction kinetic approach. Chem. Geol. 1998, 149, 235–250. [Google Scholar] [CrossRef]
- Tang, Y.C.; Perry, J.K.; Jenden, P.D.; Schoell, M. Mathematical modeling of stable carbon isotope ratios in natural gases. Geochim. Cosmochim. Acta 2000, 64, 2673–2687. [Google Scholar] [CrossRef]
- He, K.; Zhang, S.C.; Mi, J.K.; Zhang, W.L. The evolution of chemical groups and isotopic fractionation at different maturation stages during lignite pyrolysis. Fuel 2008, 211, 492–506. [Google Scholar] [CrossRef]
- Berner, U.; Faber, E.; Scheeder, G.; Panten, D. Primary cracking of algal and landplant kerogens: Kinetic models of isotope variations in methane, ethane and propane. Chem. Geol. 1995, 126, 233–245. [Google Scholar] [CrossRef]
- Galimov, E.M. Isotope organic geochemistry. Org. Geochem. 2006, 37, 1200–1262. [Google Scholar] [CrossRef]
- Suda, K.; Ueno, Y.; Yoshizaki, M.; Nakamura, H.; Kurokawa, K.; Nishiyama, E.; Yoshino, K.; Hongoh, Y.; Kawachi, K.; Omori, S.; et al. Origin of methane in serpentinite-hosted hydrothermal systems: The CH4–H2–H2O hydrogen isotope systematics of the Hakuba Happo hot spring. Earth Planet. Sc. Lett. 2014, 386, 112–125. [Google Scholar] [CrossRef]
- Turner, A.C.; Pester, N.J.; Bill, M.; Conrad, M.E.; Knauss, K.G.; Stolper, D.A. Experimental determination of hydrogen isotope exchange rates between methane and water under hydrothermal conditions. Geochim. Cosmochim. Acta 2022, 329, 231–255. [Google Scholar] [CrossRef]
- Pester, N.J.; Conrad, M.E.; Knauss, K.G.; De Paolo, D.J. Kinetics of D/H isotope fractionation between molecular hydrogen and water. Geochim. Cosmochim. Acta 2018, 242, 191–212. [Google Scholar] [CrossRef]
- Kiyosu, Y. Hydrogen isotopic compositions of hydrogen and methane from some volcanic areas in northeastern Japan. Earth Planet. Sci. Lett. 1983, 62, 41–52. [Google Scholar] [CrossRef]
- Proskurowski, G.; Lilley, M.D.; Kelly, D.S.; Olson, E.J. Low temperature volatile production at the Lost City Hydrothermal Field, evidence from a hydrogen stable isotope geothermometer. Chem. Geol. 2006, 229, 331–343. [Google Scholar] [CrossRef]
- Hao, Y.; Pang, Z.; Tian, J.; Wang, Y.; Li, Z.; Li, L.; Xing, L. Origin and evolution of hydrogen-rich gas discharges from a hot spring in the eastern coastal area of China. Chem. Geol. 2020, 538, 119477. [Google Scholar] [CrossRef]
- Tilley, B.; Muehlenbachs, K. Isotope reversals and universal stages and trends of gas maturation in sealed, self-contained petroleum systems. Chem. Geol. 2013, 339, 194–204. [Google Scholar] [CrossRef]
- Jenden, P.D.; Drazan, D.J.; Kaplan, I.R. Mixing of thermogenic natural gases in northern Appalachian Basin. AAPG Bull. 1993, 77, 980–998. [Google Scholar]
- Mangenot, X.; Xie, H.; Crémière, A.; Giunta, T.; Lilley, M.; Sissmann, O.; Orphan, V.; Schimmelmann, A.; Gaucher, E.C.; Girard, J.P.; et al. 2H–2H clumping in molecular hydrogen method and preliminary results. Chem. Geol. 2023, 621, 121278. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).