
In Silico Prediction, Characterization and Molecular Docking Studies on New Benzamide Derivatives


 ,
,  ,
,  , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Molecular Properties
2.2. Molecular Docking
2.3. Molecule Preparation for ADME-Tox Features
2.4. Assessment of Drug- and Lead-Likeness Features
2.5. Computational Pharmacokinetic and Pharmacogenomic Profiles
3. Results
3.1. Compound Selection
3.2. Ligand Preparation
3.2.1. Geometry Optimization of the Ligands
3.2.2. In Silico Exploration of Molecular Properties
3.2.3. Quantum Chemical Calculations—Graphical Models
3.2.4. Drug-Likeness, Pharmacokinetic, and Pharmacogenomic Profiles of 1a–1o Compounds
3.3. Molecular Docking Studies and Predictive Ligand–Receptor Interactions
3.3.1. Molecular Docking of the Ligands into S. aureus DNA Gyrase Active Site
3.3.2. Molecular Docking of the Ligands with E. coli DNA Gyrase
4. Discussion
4.1. Compound-Docking Outcomes Correlated with the S. aureus Co-Crystallized Interaction Group
4.2. Compound-Docking Outcomes Correlated with the E. coli Co-Crystallized Interaction Group
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Villamizar-Mogotocoro, A.-F.; Vargas-Méndez, L.Y.; Kouznetsov, V.V. Pyridine and quinoline molecules as crucial protagonists in the never-stopping discovery of new agents against tuberculosis. Eur. J. Pharm. Sci. 2020, 151, 105374. [Google Scholar] [CrossRef] [PubMed]
- Yerragunta, V.; Patil, P.; Anusha, V.; Kumaraswamy, T.; Suman, D.; Samhitha, T. Pyrimidine and its biological activity: A review. PharmaTutor 2013, 1, 39–44. [Google Scholar]
- Ran, K.; Zeng, J.; Wan, G.; He, X.; Feng, Z.; Xiang, W.; Wei, W.; Hu, X.; Wang, N.; Liu, Z.; et al. Design, Synthesis and Biological Evaluations of a Series of Pyrido[1,2-a]Pyrimidinone Derivatives as Novel Selective FGFR Inhibitors. Eur. J. Med. Chem. 2021, 220, 113499. [Google Scholar] [CrossRef] [PubMed]
- Jubete, G.; Puig de la Bellacasa, R.; Estrada-Tejedor, R.; Teixidó, J.; Borrell, J.I. Pyrido[2,3-d]pyrimidin-7(8H)-ones: Synthesis and biomedical applications. Molecules 2019, 24, 4161. [Google Scholar] [CrossRef] [PubMed]
- Neely, J.M.; Rovis, T. Pyridine Synthesis by [4 + 2] Cycloadditions of 1-Azadienes: Hetero-Diels Alder and Transition Metal-Catalysed Approaches. Org. Chem. Front. 2014, 1, 1010–1015. [Google Scholar] [CrossRef]
- Xu, D.; Sun, D.; Wang, W.; Peng, X.; Zhan, Z.; Ji, Y.; Shen, Y.; Geng, M.; Ai, J.; Duan, W. Discovery of Pyrrolo[2,3-d]Pyrimidine Derivatives as Potent Axl Inhibitors: Design, Synthesis and Biological Evaluation. Eur. J. Med. Chem. 2021, 220, 113497. [Google Scholar] [CrossRef]
- Ling, Y.; Hao, Z.Y.; Liang, D.; Zhang, C.L.; Liu, Y.F.; Wang, Y. The Expanding Role of Pyridine and Dihydropyridine Scaffolds in Drug Design. Drug. Des. Devel. Ther. 2021, 15, 4289–4338. [Google Scholar] [CrossRef]
- Lin, S.X.; Curtis, M.A.; Sperry, J. Pyridine Alkaloids with Activity in the Central Nervous System. Bioorg. Med. Chem. 2020, 28, 115820. [Google Scholar] [CrossRef]
- Comins, D.L.; Higuchi, K.; Young, D.W. Dihydropyridine Preparation and Application in the Synthesis of Pyridine Derivatives. Adv. Heterocycl. Chem. 2013, 110, 175–235. [Google Scholar] [CrossRef]
- Pollak, N.; Dölle, C.; Ziegler, M. The Power to Reduce: Pyridine Nucleotides–Small Molecules with a Multitude of Functions. Biochem. J. 2007, 402, 205–218. [Google Scholar] [CrossRef]
- Jian, Y.; Hulpia, F.; Risseeuw, M.D.P.; Forbes, H.E.; Munier-Lehmann, H.; Caljon, G.; Boshoff, H.I.M.; Van Calenbergh, S. Synthesis and Structure Activity Relationships of Cyanopyridone Based Anti-Tuberculosis Agents. Eur. J. Med. Chem. 2020, 201, 112450. [Google Scholar] [CrossRef] [PubMed]
- Marinescu, M.; Popa, C.V. Pyridine Compounds with Antimicrobial and Antiviral Activities. Int. J. M. Sci. 2022, 23, 5659. [Google Scholar] [CrossRef] [PubMed]
- Lapidot, I.; Albeck, A.; Gellerman, G.; Shatzmiller, S.; Grynszpan, F. 1,4-Dihydropyridine Cationic Peptidomimetics with Antibacterial Activity. Int. J. Pept. Res. Ther. 2015, 21, 243–247. [Google Scholar] [CrossRef]
- Klusa, V. Atypical 1,4-dihydropyridine derivatives, an approach to neuroprotection and memory enhancement. Pharmacol. Res. 2016, 113, 754–759. [Google Scholar] [CrossRef]
- Alrooqi, M.; Khan, S.; Alhumaydhi, F.A.; Asiri, S.A.; Alshamrani, M.; Mashraqi, M.M.; Alzamami, A.; Alshahrani, A.M.; Aldahish, A.A. A Therapeutic Journey of Pyridine-based Heterocyclic Compounds as Potent Anticancer Agents: A Review (From 2017 to 2021). Anticancer Agents Med. Chem. 2022, 22, 2775–2787. [Google Scholar] [CrossRef]
- Davari, A.S.; Abnous, K.; Mehri, S.; Ghandadi, M.; Hadizadeh, F. Synthesis and Biological Evaluation of Novel Pyridine Derivatives as Potential Anticancer Agents and Phosphodiesterase-3 Inhibitors. Bioorg. Chem. 2014, 57, 83–89. [Google Scholar] [CrossRef]
- Lu, T.; Goh, A.W.; Yu, M.; Adams, J.; Lam, F.; Teo, T.; Li, P.; Noll, B.; Zhong, L.; Diab, S.; et al. Discovery of (E)-3-((Styrylsulfonyl)Methyl)Pyridine and (E)-2-((Styrylsulfonyl)Methyl)Pyridine Derivatives as Anticancer Agents: Synthesis, Structure–Activity Relationships, and Biological Activities. J. Med. Chem. 2014, 57, 2275–2291. [Google Scholar] [CrossRef]
- Prachayasittikul, S.; Pingaew, R.; Worachartcheewan, A.; Sinthupoom, N.; Prachayasittikul, V.; Ruchirawat, S.; Prachayasittikul, V. Roles of Pyridine and Pyrimidine Derivatives as Privileged Scaffolds in Anticancer Agents. Mini. Rev. Med. Chem. 2017, 17, 869–901. [Google Scholar] [CrossRef]
- Fan, Z.; Shi, J.; Luo, N.; Ding, M.; Bao, X. Synthesis, Crystal Structure, and Agricultural Antimicrobial Evaluation of Novel Quinazoline Thioether Derivatives Incorporating the 1,2,4-Triazolo[4,3-a]Pyridine Moiety. J. Agric. Food Chem. 2019, 67, 11598–11606. [Google Scholar] [CrossRef]
- Bhila, V.G.; Patel, C.V.; Patel, N.H.; Brahmbhatt, D.I. One Pot Synthesis of Some Novel Coumarins Containing 5-(Substituted-2-Hydroxybenzoyl) Pyridine as a New Class of Antimicrobial and Antituberculosis Agents. Med. Chem. Res. 2013, 22, 4338–4346. [Google Scholar] [CrossRef]
- Laddha, S.S.; Bhatnagar, S.P. A New Therapeutic Approach in Parkinson’s Disease: Some Novel Quinazoline Derivatives as Dual Selective Phosphodiesterase 1 Inhibitors and Anti-Inflammatory Agents. Bioorg. Med. Chem. 2009, 17, 6796–6802. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Chauhan, A.; Drabu, S. Synthesis of Cyanopyridine and Pyrimidine Analogues as New Anti-Inflammatory and Antimicrobial Agents. Biomed. Pharmacother. 2011, 65, 375–380. [Google Scholar] [CrossRef] [PubMed]
- Thirumurugan, P.; Mahalaxmi, S.; Perumal, P.T. Synthesis and Anti-Inflammatory Activity of 3-Indolyl Pyridine Derivatives through One-Pot Multi Component Reaction. J. Chem. Sci. 2010, 122, 819–832. [Google Scholar] [CrossRef]
- Rakesh, K.P.; Gowda, D.C. Schiff’s Bases of Quinazolinone Derivatives: Synthesis and SAR Studies of a Novel Series of Potential Anti-Inflammatory and Antioxidants. Bioorg. Med. Chem. Lett. 2015, 25, 1072–1077. [Google Scholar] [CrossRef] [PubMed]
- Kamat, V.; Santosh, R.; Poojary, B.; Nayak, S.P.; Kumar, B.K.; Sankaranarayanan, M.; Faheem Khanapure, S.; Barretto, D.A.; Vootla, S.K. Pyridine- and Thiazole-Based Hydrazides with Promising Anti-inflammatory and Antimicrobial Activities along with Their In Silico Studies. ACS Omega 2020, 5, 25228–25239. [Google Scholar] [CrossRef]
- Salam Abdel, O.I.; Al-Omar, M.A.; Khalifa, N.M.; Amr Ael, G.; Abdallah, M.M. Analgesic and Anticonvulsant Activities of Some Newly Synthesized Trisubstituted Pyridine Derivatives. Z. Naturforsch. C. J. Biosci. 2013, 68, 264–268. [Google Scholar] [CrossRef]
- Helal, M.H.; El-Awdan, S.A.; Salem, M.A.; Abd-elaziz, T.A.; Moahamed, Y.A.; El-Sherif, A.A.; Mohamed, G.A.M. ynthesis, Biological Evaluation and Molecular Modeling of Novel Series of Pyridine Derivatives as Anticancer, Anti-Inflammatory and Analgesic Agents. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2015, 135, 764–773. [Google Scholar] [CrossRef]
- Carty, T.J.; Marfat, A.; Moore, P.F.; Falkner, F.C.; Twomey, T.M.; Weissman, A. Ampiroxicam, an Anti-Inflammatory Agent Which Is a Prodrug of Piroxicam. Agents Actions 1993, 39, 157–165. [Google Scholar] [CrossRef]
- Berg, J.; Fellier, H.; Christoph, T.; Grarup, J.; Stimmeder, D. The Analgesic NSAID Lornoxicam Inhibits Cyclooxygenase (COX)-1/-2, Inducible Nitric Oxide Synthase (INOS), and the Formation of Interleukin (IL)-6 in Vitro. Inflamm. Res. 1999, 48, 369–379. [Google Scholar] [CrossRef]
- El-Naggar, M.; Almahli, H.; Ibrahim, H.; Eldehna, W.; Abdel-Aziz, H. Pyridine-Ureas as Potential Anticancer Agents: Synthesis and In Vitro Biological Evaluation. Molecules 2018, 23, 1459. [Google Scholar] [CrossRef]
- Usha, C.; Santhakumari, R.; Joseph, L.; Sajan, D.; Meenakshi, R.; Sinthiya, A. Growth and Combined Experimental and Quantum Chemical Study of Glycyl-L-Valine Crystal. Heliyon 2019, 5, e01574. [Google Scholar] [CrossRef] [PubMed]
- Scrocco, E.; Tomasi, J. Electronic Molecular Structure, Reactivity and Intermolecular Forces: An Euristic Interpretation by Means of Electrostatic Molecular Potentials. Adv. Quantum Chem. 1978, 11, 115–193. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, druglikeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Dar, A.M.; Mir, S. Molecular Docking: Approaches, Types, Applications and Basic Challenges. J. Anal. Bioanal. Tech. 2017, 8, 356. [Google Scholar] [CrossRef]
- Agarwal, S.; Chadha, D.; Mehrotra, R. Molecular Modeling and Spectroscopic Studies of Semustine Binding with DNA and Its Comparison with Lomustine–DNA Adduct Formation. J. Biomol. Struct. Dyn. 2015, 33, 1653–1668. [Google Scholar] [CrossRef]
- Guedes, I.A.; de Magalhães, C.S.; Dardenne, L.E. Receptor–Ligand Molecular Docking. Biophys. Rev. 2014, 6, 75–87. [Google Scholar] [CrossRef]
- Rohs, R. Molecular Flexibility in Ab Initio Drug Docking to DNA: Binding-Site and Binding-Mode Transitions in All-Atom Monte Carlo Simulations. Nucleic Acids Res. 2005, 33, 7048–7057. [Google Scholar] [CrossRef]
- Cho, A.E.; Rinaldo, D. Extension of QM/MM Docking and Its Applications to Metalloproteins. J. Comput. Chem. 2009, 30, 2609–2616. [Google Scholar] [CrossRef]
- Limban, C.; Nuta, D.C.; Missir, A.V.; Roman, R.; Caproiu, M.T.; Dumitrascu, F.; Pintilie, L.; Stefaniu, A.; Chifiriuc, M.C.; Popa, M.; et al. Synthesis and Characterization of New Fluoro/Trifluoromethyl-Substituted Acylthiourea Derivatives with Promising Activity against Planktonic and Biofilm-Embedded Microbial Cells. Processes 2020, 8, 503. [Google Scholar] [CrossRef]
- Bordei Telehoiu, A.T.; Nuță, D.C.; Căproiu, M.T.; Dumitrascu, F.; Zarafu, I.; Ioniță, P.; Limban, C. Design, Synthesis and In Vitro Characterization of Novel Antimicrobial Agents Based on 6-Chloro-9H-carbazol Derivatives and 1,3,4-Oxadiazole Scaffolds. Molecules 2020, 25, 266. [Google Scholar] [CrossRef]
- Vlad, I.M.; Nuta, D.C.; Chirita, C.; Caproiu, M.T.; Draghici, C.; Dumitrascu, F.; Limban, C. In Silico and In Vitro Experimental Studies of New Dibenz[b,e]oxepin-11(6H)one O-(arylcarbamoyl)-oximes Designed as Potential Antimicrobial Agents. Molecules 2020, 25, 321. [Google Scholar] [CrossRef] [PubMed]
- pkCSM – Pharmacokinetics. Available online: https://biosig.lab.uq.edu.au/pkcsm/ (accessed on 11 November 2022).
- Verma, J.; Khedkar, V.M.; Coutinho, E.C. 3D-QSAR in drug design--a review. Curr. Top. Med. Chem. 2010, 10, 95–115. [Google Scholar] [CrossRef] [PubMed]
- Pal, D.; Mitra, S. A preliminary study on the in vitro antioxidant activity of the stems of O. vulgaris. J. Adv. Pharm. Tech. Res. 2010, 1, 268–272. [Google Scholar]
- Edwards, M.P.; Price, D.A. Role of Physicochemical Properties and Ligand Lipophilicity Efficiency in Addressing Drug Safety Risks. Annu. Rep. Med. Chem. 2010, 45, 380–391. [Google Scholar] [CrossRef]
- Saha, S.; Acharya, M. In silico ADME-toxicity profiling, prediction of bioactivity and CNS penetrating properties of some newer resveratrol analogues. J. PharmaSciTech 2014, 3, 98–105. [Google Scholar]
- Wang, S.; Chen, Z.; Tang, X.; Shi, L.; Zhang, L.; Yao, M. Rapid determination of partition coefficients of pharmaceuticals by phase distribution and microchip capillary electrophoresis with Octanol-Water Partition Constant 205 contactless conductivity detection. J. Sep. Sci. 2013, 36, 3615–3622. [Google Scholar] [CrossRef]
- Gałuszka, A.; Migaszewski, Z.; Namieśnik, J. The 12 Principles of Green Analytical Chemistry and the SIGNIFICANCE Mnemonic of Green Analytical Practices. TrAC-Trends Analyt. Chem 2012, 50, 78–84. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 46, 3–26. [Google Scholar] [CrossRef]
- Hefler, J.; Marfil-Garza, B.A.; Pawlick, R.L.; Freed, D.H.; Karvellas, C.J.; Bigam, D.L.; Shapiro, A.M.J. Preclinical Models of Acute Liver Failure: A Comprehensive Review. PeerJ 2021, 9, e12579. [Google Scholar] [CrossRef]
- Hitchcock, S.A.; Pennington, L.D. Structure–Brain Exposure Relationships. J. Med. Chem. 2006, 49, 7559–7583. [Google Scholar] [CrossRef]
- Kelder, J.; Grootenhuis, P.D.J.; Bayada, D.M. Polar Molecular Surface as a Dominating Determinant for Oral Absorption and Brain Penetration of Drugs. Pharm. Res. 1999, 16, 1514–1519. [Google Scholar] [CrossRef]
- Palm, K.; Stenberg, P.; Luthman, K.; Artursson, P. Polar Molecular Surface Properties Predict the Intestinal Absorption of Drugs in Humans. Pharm. Res. 1997, 14, 568–571. [Google Scholar] [CrossRef]
- Mahdavifar, Z.; Haghbayan, M. Theoretical Investigation of Pristine and Functionalized AlN and SiC Single Walled Nanotubes as an Adsorption Candidate for Methane. Appl. Surf. Sci. 2012, 263, 553–562. [Google Scholar] [CrossRef]
- Li, G.; Chen, X.; Zhou, Z.; Wang, F.; Yang, H.; Yang, J.; Liu, D. Theoretical Insights into the Structural, Relative Stable, Electronic, and Gas Sensing Properties of PbnAun (n = 2–12) Clusters: A DFT Study. RSC Adv. 2017, 7, 45432–45441. [Google Scholar] [CrossRef]
- Suganthi, S.; Balu, P.; Sathyanarayanamoorthi, V.; Kannappan, V.; Kamil, M.G.M.; Kumar, R. Structural analysis and investigation of molecular properties of Cefpodoxime acid, a third generation antibiotic. J. Mol. Struct. 2016, 1108, 1–15. [Google Scholar] [CrossRef]
- Parr, R.G.; Donnelly, R.A.; Levy, M.; Palke, W.E. Electronegativity: The density functional viewpoint. J. Chem. Phys. 1978, 68, 3801. [Google Scholar] [CrossRef]
- Parr, R.G.; Pearson, R.G. Absolute hardness: Companion parameter to absolute electronegativity. J. Am. Chem. Soc. 1983, 105, 7512–7516. [Google Scholar] [CrossRef]
- Borah, B.; Devi, T.G. Molecular property analysis of the interacting state of L-Threonine and Metformin: An experimental and computational approach. J. Mol. Struct. 2020, 1221, 128819. [Google Scholar] [CrossRef]
- Petit, J.; Meurice, N.; Kaiser, C.; Maggiora, G. Softening the Rule of Five—Where to Draw the Line? Bioorg. Med. Chem. 2012, 20, 5343–5351. [Google Scholar] [CrossRef]
- Ivanović, V.; Rančić, M.; Arsić, B.; Pavlović, A. Lipinski’s rule of five, famous extensions and famous exceptions. Chem. Naissensis 2020, 3, 171–177. [Google Scholar]
- Bax, D.B.; Chan, P.F.; Eggleston, D.S.; Fosberry, A.; Gentry, D.R.; Gorrec, F.; Giordano, I.; Hann, M.; Hennessy, A.; Hibbs, M.; et al. Type IIA Topoisomerase Inhibition by a New Class of Antibacterial Agents. Nature 2010, 466, 935–940. [Google Scholar] [CrossRef] [PubMed]
- Brvar, M.; Perdih, A.; Renko, M.; Anderluh, G.; Turk, D.; Solmajer, T. Structure-Based Discovery of Substituted 4,5′-Bithiazoles as Novel DNA Gyrase Inhibitors. J. Med. Chem. 2012, 55, 6413–6426. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.Y.; Zhang, H.X.; Mezei, M.; Cui, M. Molecular docking: A powerful approach for structure-based drug discovery. Curr. Comput. Aided Drug Des. 2011, 7, 146–157. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| The Compounds 1a–1o, Chemically Derived from 2-((4-ethylphenoxy) methyl)-N-(heteroarylcarbamothioyl)benzamides | |
|---|---|
 Structure 1a Formula: C20H19N3O2S2 |  Structure 1b Formula: C22H21N3O2S |
 Structure 1c Formula: C22H21N3O2S |  Structure 1d Formula: C23H23N3O2S |
 Structure 1e Formula: C23H23N3O2S |  Structure 1f Formula: C23H23N3O2S |
 Structure 1g Formula: C22H20ClN3O2S |  Structure 1h Formula: C22H20ClN3O2S |
 Structure 1i Formula: C22H20ClN3O2S |  Structure 1j Formula: C22H20ClN3O2S |
 Structure 1k Formula: C22H20BrN3O2S |  Structure 1l Formula: C22H20BrN3O2S |
 Structure 1m Formula: C22H19Cl2N3O2S |  Structure 1n Formula: C22H19Cl2N3O2S |
 Structure 1o Formula: C22H19Br2N3O2S | |
| Compound | Molecular Property Type | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Area (Å) | Volume (Å3) | PSA (Å2) | Ovality | Log P * | Polarizability (10−30 m3) | Dipole Moment (Debye) | EHOMO (eV) | ELUMO (eV) | ∆E 1 (eV) | |
| 1a | 411.29 | 386.22 | 44.587 | 1.60 | 2.77 | 71.87 | 4.44 | −5.5964 | −2.0575 | 3.5389 |
| 1b | 423.07 | 400.53 | 46.835 | 1.61 | 2.22 | 72.98 | 4.15 | −5.5989 | −1.8515 | 3.7474 |
| 1c | 422.59 | 400.46 | 46.751 | 1.61 | 1.06 | 72.98 | 2.34 | −5.6384 | −1.9294 | 3.7090 |
| 1d | 446.42 | 422.82 | 48.360 | 1.64 | 2.39 | 75.14 | 4.93 | −4.5358 | −2.256 | 2.2798 |
| 1e | 442.95 | 418.75 | 46.912 | 1.64 | 2.39 | 74.45 | 4.48 | −5.5815 | −1.8040 | 3.7775 |
| 1f | 443.05 | 418.82 | 46.777 | 1.64 | 2.39 | 74.45 | 4.70 | −5.5835 | −1.7983 | 3.7852 |
| 1g | 438.80 | 414.21 | 46.800 | 1.63 | 2.08 | 74.12 | 2.43 | −5.6408 | −2.0175 | 3.6233 |
| 1h | 436.28 | 413.83 | 45.059 | 1.62 | 1.90 | 74.10 | 1.98 | −5.7575 | −2.0769 | 3.6806 |
| 1i | 438.01 | 414.00 | 46.732 | 1.63 | 1.90 | 74.13 | 5.11 | −5.6965 | −2.2076 | 3.4889 |
| 1j | 438.37 | 414.10 | 46.689 | 1.63 | 1.71 | 74.12 | 2.00 | −5.6844 | −2.0888 | 3.5956 |
| 1k | 443.41 | 418.70 | 46.796 | 1.64 | 2.35 | 74.48 | 2.41 | −5.6448 | −2.0313 | 3.6135 |
| 1l | 443.46 | 418.69 | 46.651 | 1.64 | 3.39 | 74.49 | 2.35 | −5.6510 | −2.0570 | 3.5940 |
| 1m | 454.83 | 427.96 | 47.476 | 1.66 | 1.95 | 75.23 | 4.17 | −5.6053 | −1.9484 | 3.6569 |
| 1n | 453.64 | 427.59 | 46.579 | 1.65 | 2.55 | 75.26 | 5.31 | −5.7401 | −2.3724 | 3.3677 |
| 1o | 464.05 | 436.95 | 47.414 | 1.67 | 2.49 | 75.95 | 4.08 | −5.6125 | −1.9484 | 3.6641 |
| Compound | Ionization Potential 2 | Electron Affinity 3 | Chemical Hardness 4 | Chemical Softness 5 | Electronegativity 6 | Chemical Potential 7 | Electrophilicity Index 8 |
|---|---|---|---|---|---|---|---|
| 1a | 5.5964 | 2.0575 | 1.7695 | 0.2826 | 3.8270 | −3.8270 | 4.1384 |
| 1b | 5.5989 | 1.8515 | 1.8737 | 0.2669 | 3.7252 | −3.7252 | 3.7031 |
| 1c | 5.6384 | 1.9294 | 1.8545 | 0.2696 | 3.7839 | −3.7839 | 3.8603 |
| 1d | 4.5358 | 2.2560 | 1.1399 | 0.4386 | 3.3959 | −3.3959 | 5.0584 |
| 1e | 5.5815 | 1.8040 | 1.8888 | 0.2647 | 3.6928 | −3.6928 | 3.6099 |
| 1f | 5.5835 | 1.7983 | 1.8926 | 0.2642 | 3.6909 | −3.6909 | 3.5989 |
| 1g | 5.6408 | 2.0175 | 1.8117 | 0.2760 | 3.8292 | −3.8292 | 4.0467 |
| 1h | 5.7575 | 2.0769 | 1.8403 | 0.2717 | 3.9172 | −3.9172 | 4.1690 |
| 1i | 5.6965 | 2.2076 | 1.7445 | 0.2866 | 3.9521 | −3.9521 | 4.4767 |
| 1j | 5.6844 | 2.0888 | 1.7978 | 0.2781 | 3.8866 | −3.8866 | 4.2012 |
| 1k | 5.6448 | 2.0313 | 1.8068 | 0.2767 | 3.8381 | −3.8381 | 4.0766 |
| 1l | 5.6510 | 2.0570 | 1.7970 | 0.2782 | 3.8540 | −3.8540 | 4.1328 |
| 1m | 5.6053 | 1.9484 | 1.8285 | 0.2735 | 3.7769 | −3.7769 | 3.9007 |
| 1n | 5.7401 | 2.3724 | 1.6839 | 0.2969 | 4.0563 | −4.0563 | 4.8856 |
| 1o | 5.6125 | 1.9484 | 1.8321 | 0.2729 | 3.7805 | −3.7805 | 3.9005 |
| Compound | Molecular Mass (<500 Da) | Log P * (<5) | Number of H-Bond Donors (<5) | Number of H-Bond Acceptors (<10) | Number of Violations of Rule of Five |
|---|---|---|---|---|---|
| 1a | 397.51 | 4.79 | 2 | 5 | 0 |
| 1b | 391.49 | 4.49 | 2 | 5 | 0 |
| 1c | 391.49 | 4.15 | 2 | 5 | 0 |
| 1d | 405.51 | 6.03 | 2 | 5 | 1 |
| 1e | 405.51 | 4.85 | 2 | 5 | 0 |
| 1f | 405.51 | 4.85 | 2 | 5 | 0 |
| 1g | 425.93 | 5.11 | 2 | 5 | 1 |
| 1h | 425.93 | 5.11 | 2 | 5 | 1 |
| 1i | 425.93 | 5.11 | 2 | 5 | 1 |
| 1j | 425.93 | 5.11 | 2 | 5 | 1 |
| 1k | 470.38 | 5.18 | 2 | 5 | 1 |
| 1l | 470.38 | 5.51 | 2 | 5 | 1 |
| 1m | 460.38 | 6.92 | 2 | 5 | 1 |
| 1n | 460.38 | 6.07 | 2 | 5 | 1 |
| 1o | 549.28 | 7.04 | 2 | 5 | 2 |
| Compound | Lipinski | Ghose | Veber | Egan | Bioavailability Score |
|---|---|---|---|---|---|
| 1a | Yes | Yes | Yes | Yes | 0.55 |
| 1b | Yes | Yes | Yes | Yes | 0.55 |
| 1c | Yes | Yes | Yes | Yes | 0.55 |
| 1d | Yes | Yes | Yes | Yes | 0.55 |
| 1e | Yes | Yes | Yes | Yes | 0.55 |
| 1f | Yes | Yes | Yes | Yes | 0.55 |
| 1g | Yes | Yes | Yes | Yes | 0.55 |
| 1h | Yes | Yes | Yes | Yes | 0.55 |
| 1i | Yes | Yes | Yes | Yes | 0.55 |
| 1j | Yes | Yes | Yes | Yes | 0.55 |
| 1k | Yes | Yes | Yes | Yes | 0.55 |
| 1l | Yes | Yes | Yes | Yes | 0.55 |
| 1m | Yes | Yes | Yes | Yes | 0.55 |
| 1n | Yes | Yes | Yes | Yes | 0.55 |
| 1o | No; 2 violations: MW > 500, MLOGP > 4.15 | No; 2 violations: MW > 480, MR > 130 | Yes | Yes | 0.17 |
| Compounds | Intestinal Absorption % | BBB Permeability | AMES | hERG I Inhibitor | Hepatotoxicity | Skin Sensitization |
|---|---|---|---|---|---|---|
| 1a | 90.166 | −0.05 | no | no | no | no |
| 1b | 91.808 | −0.124 | no | no | yes | no |
| 1c | 91.829 | −0.005 | no | no | yes | no |
| 1d | 91.373 | −0.089 | no | no | yes | no |
| 1e | 91.447 | −0.068 | no | no | yes | no |
| 1f | 91.484 | −0.073 | no | no | yes | no |
| 1g | 90.375 | −0.083 | no | no | no | no |
| 1h | 90.478 | 0.033 | no | no | yes | no |
| 1i | 90.377 | 0.014 | no | no | no | no |
| 1j | 90.282 | −0.008 | no | no | no | no |
| 1k | 90.308 | −0.084 | no | no | no | no |
| 1l | 90.29 | −0.016 | no | no | yes | no |
| 1m | 88.364 | 0.004 | no | no | no | no |
| 1n | 88.753 | −0.013 | no | no | no | no |
| Compounds | CYP3A4 Substrate | CYP1A2 Inhibitor | CYP2C19 Inhibitor | CYP2C9 Inhibitor | CYP2D6 Substrate/Inhibitor | CYP3A4 Inhibitor |
|---|---|---|---|---|---|---|
| 1a | yes | yes | yes | yes | no/no | yes |
| 1b | yes | yes | yes | yes | no/no | yes |
| 1c | yes | yes | yes | yes | no/no | yes |
| 1d | yes | yes | yes | yes | no/no | yes |
| 1e | yes | yes | yes | yes | no/no | yes |
| 1f | yes | yes | yes | yes | no/no | yes |
| 1g | yes | no | yes | yes | no/no | yes |
| 1h | yes | no | yes | yes | no/no | yes |
| 1i | yes | no | yes | yes | no/no | yes |
| 1j | yes | no | yes | yes | no/no | yes |
| 1k | yes | no | yes | yes | no/no | yes |
| 1l | yes | no | yes | yes | no/no | yes |
| 1m | yes | no | yes | yes | no/no | yes |
| 1n | yes | no | yes | yes | no/no | yes |
| Compound | Score | RMSD (Å) | Interacting Group | Hydrogen Bond * | Bond Length (Å) |
|---|---|---|---|---|---|
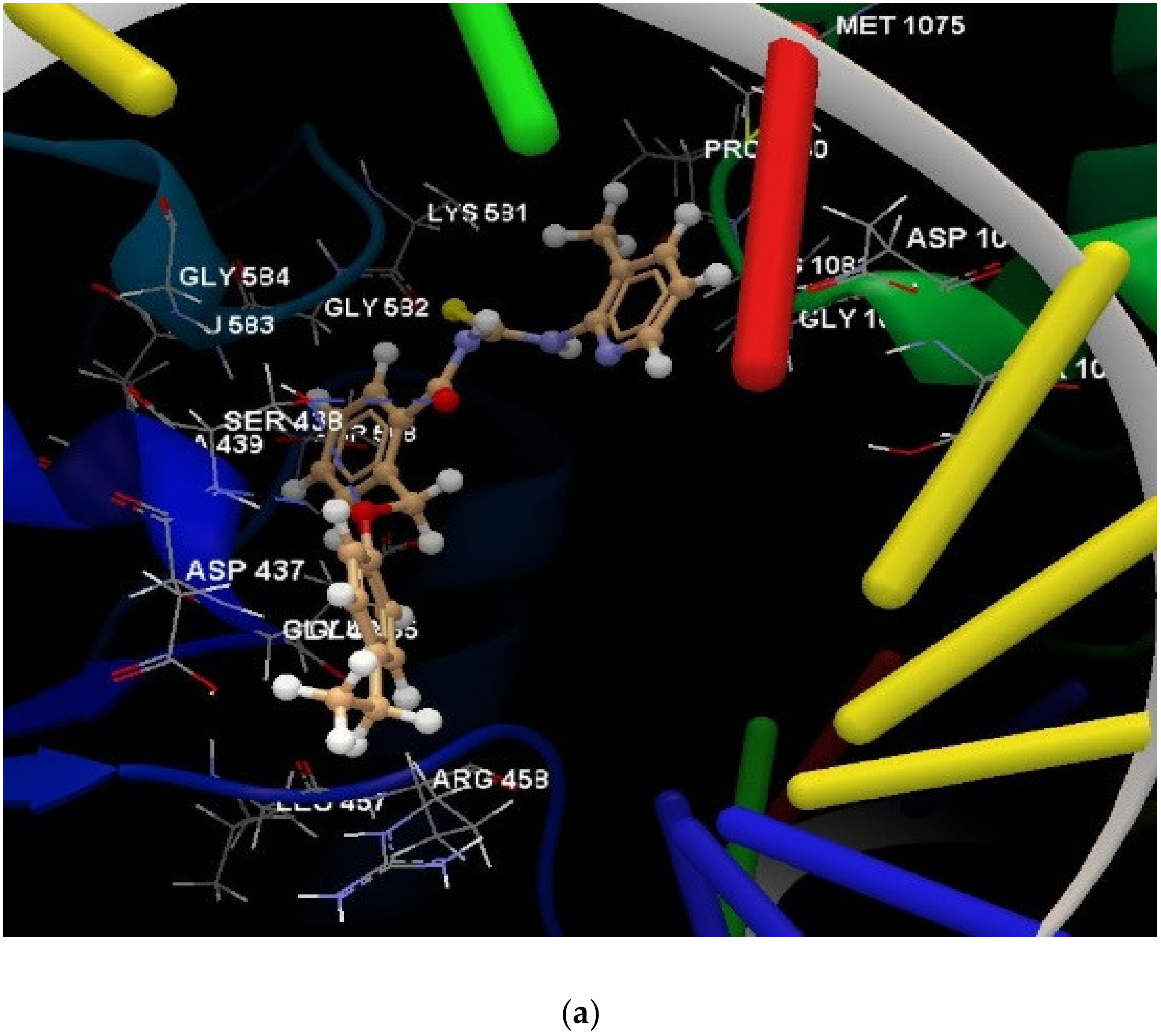
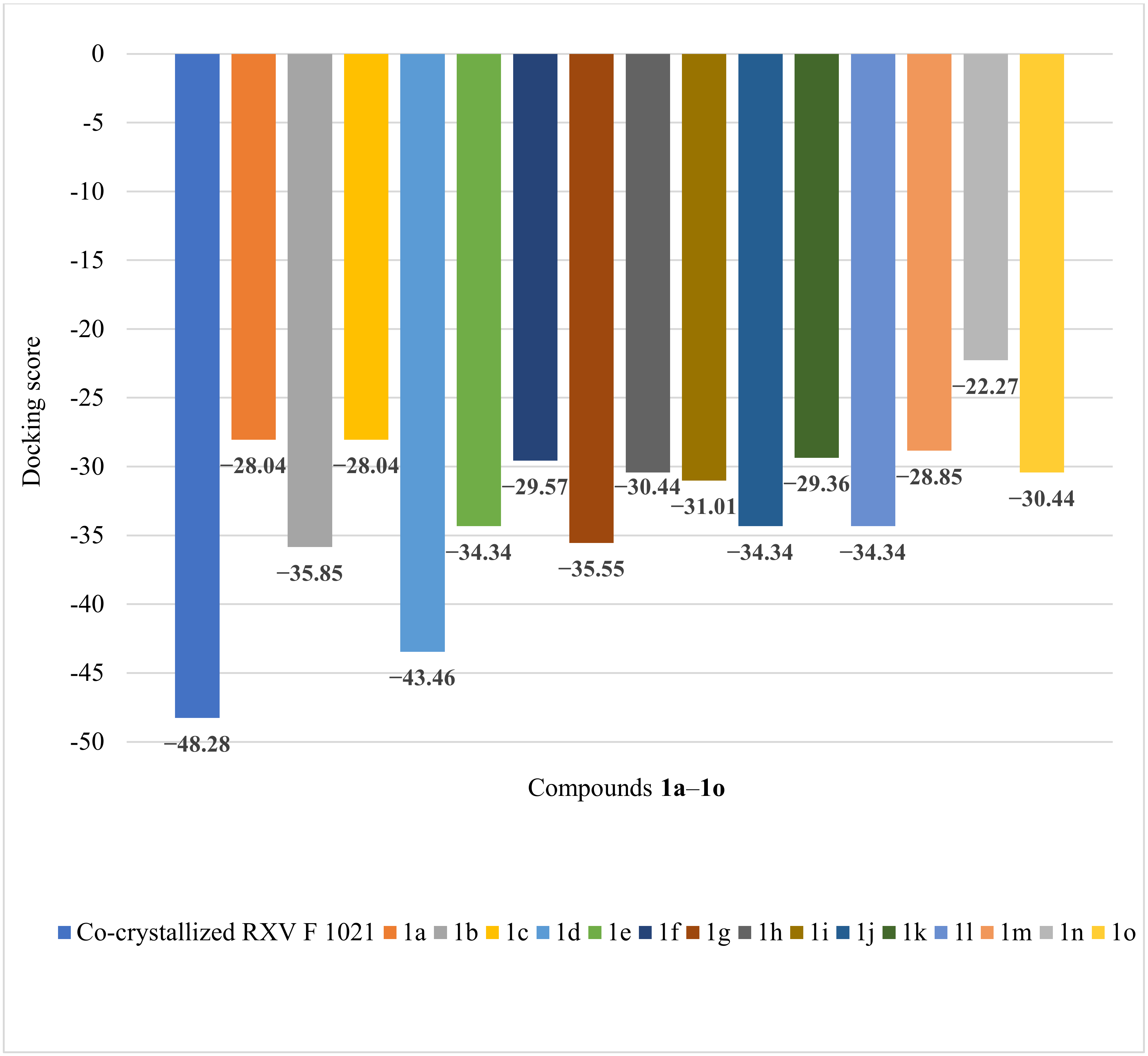
| Co-crystallized RXV F 1021 | −48.28 | 1.29 | SER1084(B), ASP1083(B), MET1075(B), VAL1071(B), MET1121(B), GLY1072(B), ALA1068(B), ARG1069(B), ARG1122(D), MET1121(D), ALA1068(D), GLY1072(D), MET1075(D), VAL1071(D), ASP1083(D) | N sp2 (N21)–O sp2 from ASP1083(B) | 2.902 |
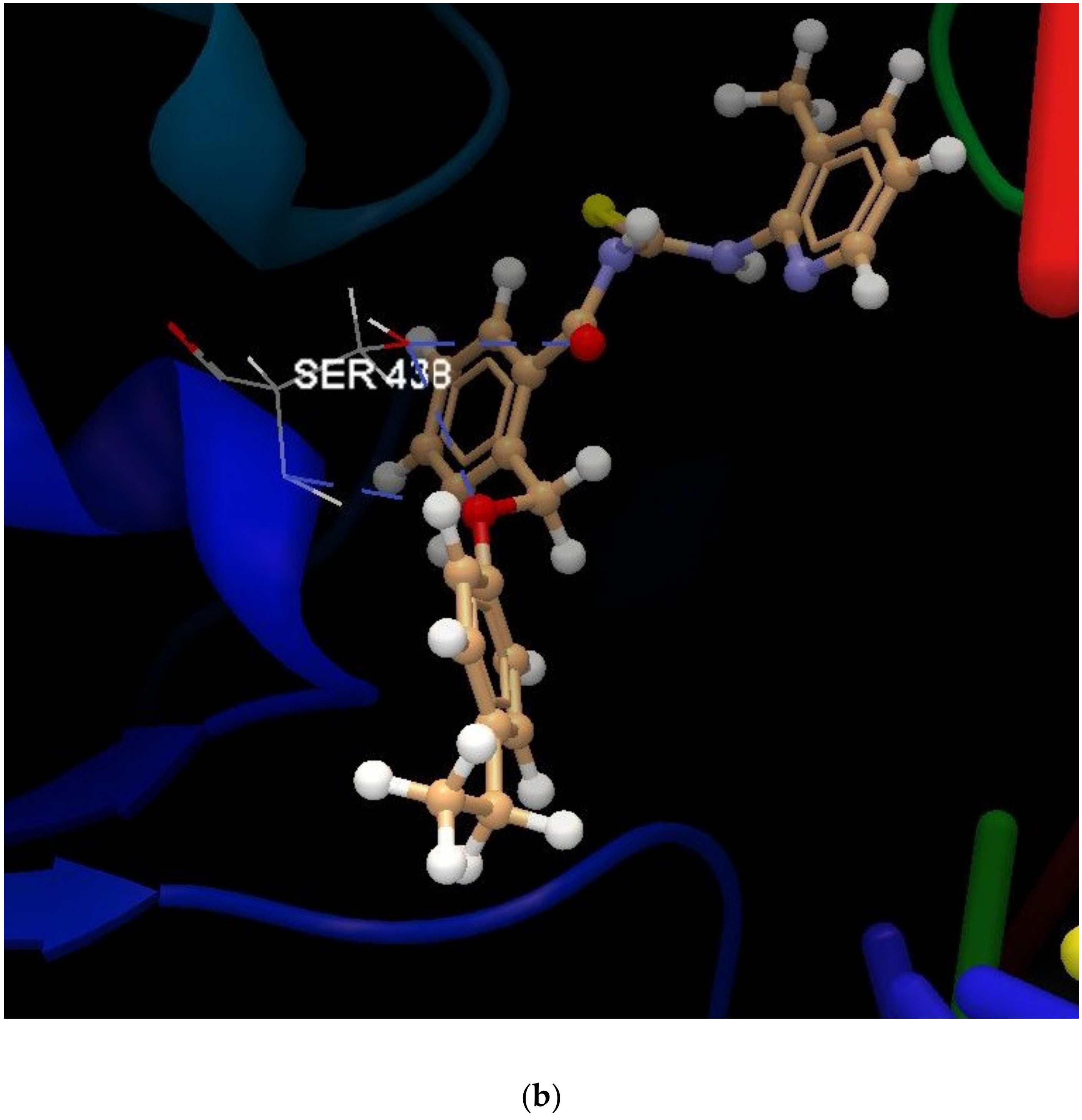
| 1a | −28.04 | 0.98 | MET 586, GLU 585, GLY 584, ALA 439, SER 438, ASP 437, LEU 583, TIR 580, GLY 582, GLY 436, LYS 581, ASP 508, GLU 435, LEU 457, ARG 458, GLY 459, LYS 460, ILE 516 (see Figure S6 in Supplementary Materials) | O sp2 (O21)–O sp3 from SER 438 O sp2 (O21)–N sp2 from SER 438 O sp2 (O21)–N sp2 from ASP 437 N sp2 (N19)–O sp3 from ASP 437 N sp2 (N23)–O sp3 from ASP 437 (see Figure S7 in Supplementary Materials) | 3.202 2.673 3.183 3.259 2.950 |
| 1b | −35.85 | 0.54 | GLY 440, ASP 437, SER 438, GLY 584, ALA 439, LEU 583, GLY 436, GLY 582, ASP 508, GLU 435, LEU 457, ARG 458, GLY 459, LYS 460, ASP 512, ILE 516 (see Figure S8 in Supplementary Materials) | O sp3 (O7)–N sp2 from ASP 437 N sp3 (N17)–O sp2 from GLU 435 (see Figure S9 in Supplementary Materials) | 3.136 3.086 |
| 1c | −28.04 | 1.11 | ASP 437, SER 438, ALA 439, GLY 436, LEU 457, GLU 435, ASP 508, ARG 458, GLY 459, LYS 460 (see Figure S10 in Supplementary Materials) | N sp2 (N19)–O sp3 from SER 438 N sp3 (N17)–N sp2 from ASP 437 N sp3 (N17)–N sp2 from SER 438 (see Figure S11 in Supplementary Materials) | 3.027 3.114 2.987 |
| 1d | −43.46 | 0.74 | ASP 437, SER 438, GLY 436, LEU 457, ALA 439, GLY 584, LEU 583, GLU 435, GLY 582, LYS 581, ASP 508, PRO 1080, HYS 1081, MET 1075, GLY 1082, ASP 1083, SER 1084, ARG 458 (see Figure S12 in Supplementary Materials) | O sp3 (O7)–N sp2 from SER 438 O sp3 (O7)–O sp3 from SER 438 O sp2 (O21)–O sp3 from SER 438 (see Figure S13 in Supplementary Materials) | 3.229 3.022 3.026 |
| 1e | −34.34 | 0.64 | GLY 441, ASP 437, SER 438, ALA 439, GLY 436, LEU 457, GLU 435, ASP 508, ARG 458, GLY 459, LYS 460 (see Figure S14 in Supplementary Materials) | N sp2 (N19)–O sp3 from SER 438 N sp3 (N17)–O sp3 from SER 438 N sp3 (N17)–N sp2 from SER 438 (see Figure S15 in Supplementary Materials) | 2.817 3.071 3.165 |
| 1f | −29.57 | 0.52 | ASP 437, SER 438, ALA 439, GLY 436, LEU 457, GLU 435, ASP 508, ARG 458, GLY 459, LYS 460 (see Figure S16 in Supplementary Materials) | N sp2 (N23)–O sp3 from SER 438 N sp2 (N19)–O sp3 from SER 438 N sp3 (N17)–O sp3 from SER 438 N sp3 (N17)–N sp2 from SER 438 N sp3 (N17)–N sp2 from ASP 437 O sp2 (O21)–N sp2 from ASP 437 (see Figure S17 in Supplementary Materials) | 3.023 2.599 3.108 2.786 3.196 3.150 |
| 1g | −35.55 | 0.34 | ASP 437, SER 438, ALA 439, GLY 436, LEU 457, GLU 435, ASP 508, ARG 458, GLY 459, LYS 460 (see Figure S18 in Supplementary Materials) | O sp3 (O7)–N sp2 from SER 438 O sp3 (O7)–N sp2 from ASP 437 (see Figure S19 in Supplementary Materials) | 3.091 3.140 |
| 1h | −30.44 | 0.62 | GLY 440, ASP 437, SER 438, ALA 439, GLY 582, GLY 436, GLU 435, LEU 457, ARG 458, ASP 1083, GLY 1082, MET 1075, HYS 1081, PRO 1080, ASP 508 (see Figure S20 in Supplementary Materials) | O sp3 (O7)–N sp2 from ASP 437 (see Figure S21 in Supplementary Materials) | 3.100 |
| 1i | −31.01 | 1.31 | GLY 440, ASP 437, SER 438, GLY 584, ALA 439, LEU 583, ASP 508, GLY 582, GLY 436, GLU 435, LEU 457, ARG 458, GLY 459, LYS 460, ILE 461 (see Figure S22 in Supplementary Materials) | N sp2 (N19)–O sp3 from SER 438 N sp3 (N17)–O sp3 from SER 438 N sp3 (N17)–N sp2 from SER 438 N sp3 (N17)–N sp2 from ASP 437 (see Figure S23 in Supplementary Materials) | 2.875 3.326 2.895 3.095 |
| 1j | −34.34 | 0.24 | ASP 437, SER 438, ALA 439, GLY 584, LEU 583, GLY 436, LEU 457, GLU 435, ASP 508, ARG 458, GLY 459, LYS 460, ILE 461 (see Figure S24 in Supplementary Materials) | N sp2 (N19)–O sp3 from SER 438 N sp3 (N17)–O sp3 from SER 438 N sp3 (N17)–N sp2 from SER 438 (see Figure S25 in Supplementary Materials) | 2.941 3.092 3.176 |
| 1k | −29.36 | 1.31 | ASP 437, SER 438, GLY 584, ALA 439, GLU 585, LEU 583, GLY 582, GLY 436, LEU 457, GLU 435, LYS 581, ASP 508, ARG 458, GLY 459, LYS 460, ILE 516 (see Figure S26 in Supplementary Materials) | O sp2 (O21)–O sp3 from SER 438 O sp2 (O21)–N sp2 from SER 438 (see Figure S27 in Supplementary Materials) | 2.824 3.359 |
| 1l | −34.34 | 0.20 | ASP 437, SER 438, ALA 439, GLY 436, LUE 457, GLU 435, ASP 508, ARG 458, GLY 459, LYS 460 (see Figure S28 in Supplementary Materials) | N sp2 (N19)–O sp3 from SER 438 N sp3 (N17)–O sp3 from SER 438 O sp2 (O21)–N sp2 from ASP 437 O sp3 (O7)–N sp2 from GLY 459 (see Figure S29 in Supplementary Materials) | 2.714 3.132 3.089 3.123 |
| 1m | −28.85 | 1.67 | GLY 440, GLY 584, ALA 439, LEU 583, DER 438, ASP 437, GLY 582, GLY 436, LEU 457, GLU 435, ASP 508, ARG 458, GLY 459, LYS 460 (see Figure S30 in Supplementary Materials) | O sp3 (O7)–O sp3 from SER 438 O sp3 (O7)–N sp2 from SER 438 O sp3 (O7)–N sp2 from ASP 437 (see Figure S31 in Supplementary Materials) | 3.329 2.996 3.204 |
| 1n | −22.27 | 0.50 | GLY 440, ASP 437, SER 438, ALA 439, GLY 584, GLU 585, LEU 583, GLY 436, GLY 582, LYS 582, ASP 508, GLU 435, ARG 458, GLY 459, ILE 516 (see Figure S32 in Supplementary Materials) | O sp2 (O21)–O sp3 from SER 438 O sp2 (O21)–N sp2 from SER 438 O sp2 (O21)–N sp2 from ASP 437 (see Figure S33 in Supplementary Materials) | 3.176 2.762 3.156 |
| 1o | −30.44 | 1.87 | LEU 583, GLY 584, ALA 439, SER 438, ASP 437, GLY 436, ASP 508, GLU 435, LEU 457, ARG 459, GLY 459, LYS 460 (see Figure S34 in Supplementary Materials) | O sp3 (O7)–O sp3 from SER 438 O sp3 (O7)–N sp2 from SER 438 O sp3 (O7)–N sp2 from ASP 437 (see Figure S35 in Supplementary Materials) | 3.229 2.950 3.252 |
| Compound | Score | RMSD (Å) | Interacting Group | Hydrogen Bond | Bond Length (Å) |
|---|---|---|---|---|---|
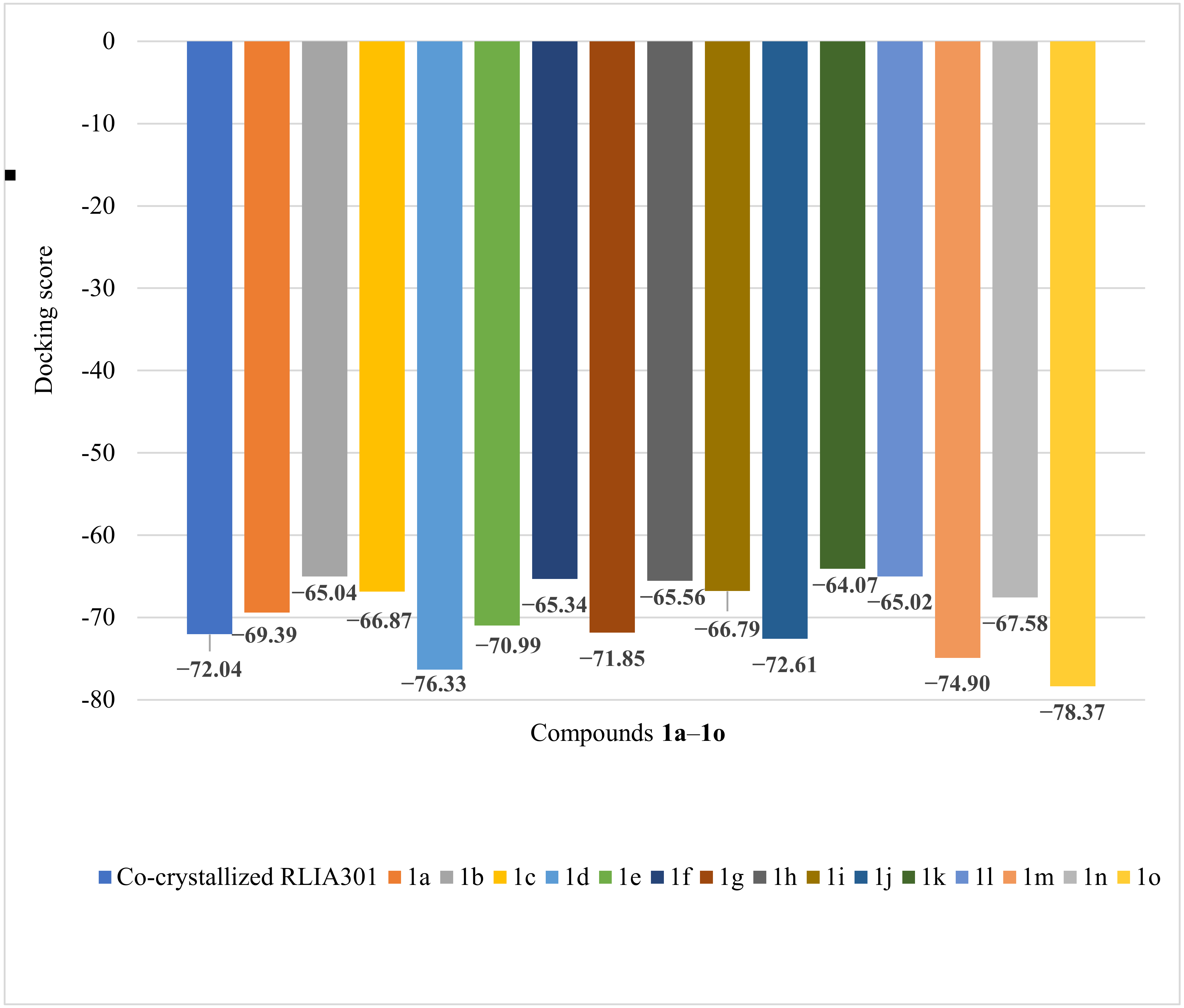
| Co-crystallized RLIA301 | −72.04 | 0.08 | LYS 103:A, ALA 100:A, GLY 102:A, GLY 101:A, ILE 94:A, PRO 79:A, ILE 78:A, ARG 76:A, GLY 75:A, THR 165:A, ASP 73:A, GLN 72:A, VAL 71:A, VAL 167:A, GLU 50:A, ASN 46:A, VAL 120:A, VAL 43:A, ALA 47:A | O sp2(O12)–N sp2 from ARG 136:A O sp2 (O12)–N sp2 from ARG 136:A O sp2 (O12)–N sp2 from ARG 76:A N sp3 (N13)–O sp2 from GLY 101:A H–O sp2 from ASP 73:A | 3.069 2.700 3.138 3.146 1.822 |
| 1a | −69.39 | 0.32 | VAL 118, GLY 117, HIS 99, PHE 104, ALA 100, GLY 119, VAL 120, ILE 94, LYS 103, GLY 101, GLY 102, VAL 43, ASN 46, VAL 167, VAL 71, ALA 47, THR 165, ASP 73, ILE 78, PRO 79, GLU 50, GLY 77, GLY 75, ARG 76, ARG 136 (see Figure S36 in Supplementary Materials) | N sp2 (N24)–N sp2 from LYS 103 (see Figure S37 in Supplementary Materials) | 3.159 |
| 1b | −65.04 | 0.19 | HIS 99, PHE 104, VAL 118, GLY 117, GLY 119, LIS 103, ALA 100, GLY 102, GLY 101, ILE 94, MET 95, VAL 120, GLU 42, ASN 46, VAL 43, ALA 47, GLU 50, VAL 167, ASP 73, GLY 75, THR 165, ARG 136, ARG 76, GY77, ILE 78, PRO 79 (see Figure S38 in Supplementary Materials) | N sp3 (N18)–N sp2 from ASN 46 (see Figure S39 in Supplementary Materials) | 2.727 |
| 1c | −66.87 | 0.58 | HYS 99, MET 95, GLY 119, VAL 120, PHE 104, ALA 100, ILE 94, GLY 101, LYS 103, GLY 102, ILE 78, PRO 79, GLY 77, ARG 76, ARG 136, VAL 43, VAL 44, VAL 71, VAL 167, LEU 132, ASN 46, MET 166, ALA 47 GLN 72, ASP 73, THR 165, GLU 50 (see Figure S40 in Supplementary Materials) | O sp3 (O8)–N sp2 from ASN 46 N sp3 (N18)–O sp2 from GLY 101 N sp3 (N22)–O sp2 from GLY 101 N sp2 (N26)–N sp2 from ARG 76 (see Figure S41 in Supplementary Materials) | 3.058 3.182 2.830 2.878 |
| 1d | −76.33 | 1.51 | GLY117, VAL 118, ASP105, PHE104, HIS 99, SER 121, VAL 120, LYS 103, ALA 100, GLY 102, GLY 101, ILE 94, GLU 42, VAL 44, VAL 43, ASN 46, ALA 47, VAL 71, GLY 50, GLN 72, ASP 73, VAL 167, MET 166, THR 165, ILE 78, GLY 77, ARG 76, ARG 136, PRO 79, GLY 119 (see Figure S42 in Supplementary Materials) | O sp2 (O21)–N sp2 from ASN 46 N sp3 (N19)–N sp2 from ASN 46 N sp2 (N23)–N sp2 from ASN 46 (see Figure S43 in Supplementary Materials) | 2.642 2.686 3.178 |
| 1e | −70.99 | 1.60 | VAL 97, SER 121, ILE 94, LEU 98, ALA 100, HIS 99, GLY 119, GLY 101, VAL 118, GLY 117, GLY 102, PHE 104, LIS 103, ASP 105, VAL 120, PRO 79, ILE 78, VAL 43, GLY 77, VAL 44, ASN 46, ALA 47, ARG 76, GLU 50, ASP 73, GLN 72, ARG 136, THR 165, VAL 71, MET 166, VAL 167 (see Figure S44 in Supplementary Materials) | N sp3 (N20)–O sp2 from ALA 100 (see Figure S45 in Supplementary Materials) | 3.163 |
| 1f | −65.34 | 0.10 | ASP 73, ALA 47, VAL 43, GLU 42, ASN 46, GLU 50, GLY 75, THR 165, ARG 76, GLY 77, ILE 78, ARG 136, PRO 79, GLY 117, VAL 118, GLY 119, VAL 120, LYS 103, PHE 104, ASP 105, HYS 99, ILE 94, GLY 101, GLY 102, ALA 100 (see Figure S46 in Supplementary Materials) | N sp3 (N18)–N sp2 from ASN 46 N sp2 (N24)–N sp2 from ASN 46 (see Figure S47 in Supplementary Materials) | 3.305 3.205 |
| 1g | −71.85 | 0.72 | HIS 99, PHE 104, VAL 118, GLY 117, GLY 119, ALA 100, VAL 120, GLU 42, LIS 103, GLY 102, GLY 101, ILE 94, VAL 43, ASN 46, VAL 167, ALA 47, GLU 50, ASP 73, GLY 75, THR 165, GLY 77, ARG 76, ARG 136, ILE 78, PRO 79 (see Figure S48 in Supplementary Materials) | N sp3 (N18)–N sp2 from ASN 46 (see Figure S49 in Supplementary Materials) | 2.914 |
| 1h | −65.56 | 0.44 | HIS 99, ALA 100, GLY 102, GLY 119, LYS 103, ILE 91, GLY 101, MET 95, VAL 120, VAL 43, ASN 46, VAL 44, ALA 47, GLU 50, VAL 71, VAL 167, MET 166, GLN 72, ASP 73, THR 165, GLY 77, ARG 76, ILE 78, PRO 79, ARG 136 (see Figure S50 in Supplementary Materials) | O sp3 (O8)–N sp2 from ASN 46 (see Figure S51 in Supplementary Materials) | 3.072 |
| 1i | −66.79 | 0.65 | ARG 136, PRO 79, ILE 78, ILE 94, MET 95, VAL 97, ALA 100, GLY 101, GLY 102, HIS 99, LYS 103, SER 121, GLY 77, THR 165, VAL 167, MET 166, GLY 164, GLY 119, GLY 75, VAL 118, GLU 50, ASN 46, ALA 47, GLU 42, VAL 71, VAL 43, ASP 73 (see Figure S52 in Supplementary Materials) | O sp2 (O22)–N sp3 from LYS 103 N sp3 (N18)–O sp2 from GLY 77 (see Figure S53 in Supplementary Materials) | 3.298 3.396 |
| 1j | −72.61 | 0.11 | VAL 118, GLY 117, HIS 99, PHE 104, GLY 119, GLU 42, VAL 120, ALA 100, ILE 94, GLY 101, GLY 102, LIS 103, VAL 167, VAL 43, ASN 46, GLU 50, ASP 73, THR 165, ILE 78, PRO 79, GLY 77, ARG 76, GLY 75, ARG 136, ALA 47 (see Figure S54 in Supplementary Materials) | N sp3 (N18)–N sp2 from ASN 46 (see Figure S55 in Supplementary Materials) | 3.091 |
| 1k | −64.07 | 0.70 | GLY 117, VAL 118, ASP 45, ASP 49, GLU 42, VAL 43, ASN 46, ALA 47, GLY 119, GLU 50, LYS 103, GLY 102, VAL 71, ASP 73, VAL 120, ALA 100, PRO 79, ILE 94, ILE 78, GLY 77, THR 165, MET 95, MET 166, VAL 167 (see Figure S56 in Supplementary Materials) | N sp3 (N18)–N sp2 from ASN 46 O sp2 (O22)–N sp2 from ASN 46 O sp3 (O8)–N sp2 from ASN 46 (see Figure S57 in Supplementary Materials) | 3.171 3.195 3.073 |
| 1l | −65.02 | 0.21 | HIS 99, VAL 118, GLY 117, PHE 104, ALA 100, GLY 119, VAL 120, GLU 42, ILE 94, GLY 101, GLY 102, LYS 103, VAL 43, VAL 44, ASN 46, ALA 47, PRO 79, ILE 78, GLU 50, VAL 167, VAL 71, MET 166, THR 165, ASP 73, GLY 77, ARG 76, ARG 136 (see Figure S58 in Supplementary Materials) | N sp3 (N20)–N sp2 from ALA 100 (see Figure S59 in Supplementary Materials) | 3.353 |
| 1m | −74.90 | 1.49 | GLY 117, VAL 118, GLY 119, VAL 120, SER 121, PHE 104, HIS 99, LEU 98, VAL 97, ALA 100, ILE 94, GLY 101, GLY 102, LIS 103, VAL 43, ASN 46, GLU 50, ASP 73, THR 165, GLY 75, ILE 78, GLY 77, ARG 76, ARG 136, PRO 79, ALA 47 (see Figure S60 in Supplementary Materials) | N sp3 (N20)–N sp2 from ASN 46 (see Figure S61 in Supplementary Materials) | 3.082 |
| 1n | −67.58 | 0.78 | HIS 99, PHE 104, ALA 100, GLY 119, SER 121, VAL 120, LYS 103, MET 95, ILE 94, GLY 101, GLY102, VAL 43, ASN 45, VAL 44, VAL 167, ALA 47, VAL 71, GLN 72, ASP 73, GLU 30, MET 166, THR 165, ILE 78, PRO 79, GLY 77, ARG 76, ARG 136 (see Figure S62 in Supplementary Materials) | N sp2 (N18)–O sp2 from GLY 101 N sp2 (N20)–O sp2 from GLY 101 (see Figure S63 in Supplementary Materials) | 3.178 2.755 |
| 1o | −78.37 | 1.10 | ASP 105, GLY 117, VAL 118, PHE 104, HIS 99, GLY 119, GLU 42, LYS 103, ALA 100, GLY 102, GLY 101, ILE 94, VAL 120, ASN 46, VAL 43, ALA 47, GLU 50, VAL 167, ASP 73, GLY 75, THR 165, ARG 76, GLY 77, ILE 78, PRO 79, ARG 136 (see Figure S64 in Supplementary Materials) | N sp3 (N17)–Nsp2 from ASN 46 N sp3 (N19)–N sp2 from ASN 46 N sp2 (N23)–N sp2 from ASN 46 (see Figure S65 in Supplementary Materials) | 3.113 2.900 3.135 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roman, R.; Pintilie, L.; Nuță, D.; Avram, S.; Buiu, C.; Sogor, C.; Limban, C. In Silico Prediction, Characterization and Molecular Docking Studies on New Benzamide Derivatives. Processes 2023, 11, 479. https://doi.org/10.3390/pr11020479
Roman R, Pintilie L, Nuță D, Avram S, Buiu C, Sogor C, Limban C. In Silico Prediction, Characterization and Molecular Docking Studies on New Benzamide Derivatives. Processes. 2023; 11(2):479. https://doi.org/10.3390/pr11020479
Chicago/Turabian StyleRoman, Roxana, Lucia Pintilie, Diana Nuță, Speranța Avram, Catalin Buiu, Catalina Sogor, and Carmen Limban. 2023. "In Silico Prediction, Characterization and Molecular Docking Studies on New Benzamide Derivatives" Processes 11, no. 2: 479. https://doi.org/10.3390/pr11020479
APA StyleRoman, R., Pintilie, L., Nuță, D., Avram, S., Buiu, C., Sogor, C., & Limban, C. (2023). In Silico Prediction, Characterization and Molecular Docking Studies on New Benzamide Derivatives. Processes, 11(2), 479. https://doi.org/10.3390/pr11020479