1. Introduction
Chinese hamster ovary (CHO) cells are, today, the gold standard for the manufacturing of therapeutic proteins. The overall value of products derived from these cells exceeds 50 billion US $ annually, and significant research and development efforts are underway to further improve both protein quality and quantity from CHO cultures in bioreactors. Proteins like ENBREL (a TNF inhibitor) and HERCEPTIN (an anti-HER-2 breast cancer antibody) are each produced at more than one metric ton per year, and thousands of patients benefit from these protein drugs. Recent publications have expressed hope that a detailed knowledge of DNA sequences and transcription patterns of one specific CHO cell strain could provide urgently needed tools to improve the manufacture of protein pharmaceuticals [
1]. However, this research does not take into account a profoundly adverse problem with CHO cells that could very much limit “omics” approaches and/or deliver entirely irrelevant data.
As cell biologists would agree for any immortalized cell line, CHO cells are - whether being referred to as “K1”, “DG44”, “DX B11”, “CHO-Toronto”, “CHOpro3-”, or “CHO-S” - members of a widely distributed family of related, but profoundly different cell lines, as their individual behaviors/phenotypes (responsiveness to different environmental conditions) in cell culture differ quite significantly. The extent of their relatedness in genome structure, genomic sequence composition, and in transcription patterns has not been studied, and reasonable conclusions on their similarities are therefore not available. In fact, if one considers the observed variability and diversity of genomic structures in immortalized cells in general, each one of the above-mentioned CHO cell lines is a “Quasispecies”. This term was first coined by Eigen and Schuster in 1977–1978 in a series of landmark papers describing a high mutation rate environment where a large group or “cloud” of related offspring exists and where one would expect that a large fraction of the offspring carries at least one mutation [
2,
3,
4].
Understanding and appreciating the above reasoning requires a history of CHO cells prior to their use in modern biotechnology. Fortunately, most of the history of the cells has been recorded in an enormous wealth of publications resulting from fundamental research executed with CHO cells from the 1960s to the 1980s. In addition, Michael Gottesmann [
5] edited, in 1985, approximately at the time of the emergence of interest for CHO cells in the biotechnology industry, a 900-page compendium entitled
Molecular Cell Genetics that contains CO Main work exclusively dedicated to the Chinese hamster and the cells derived from this species. Unfortunately, this compendium is out of print today but was available to the author for this review.
1.2. The Early History of CHO Cells
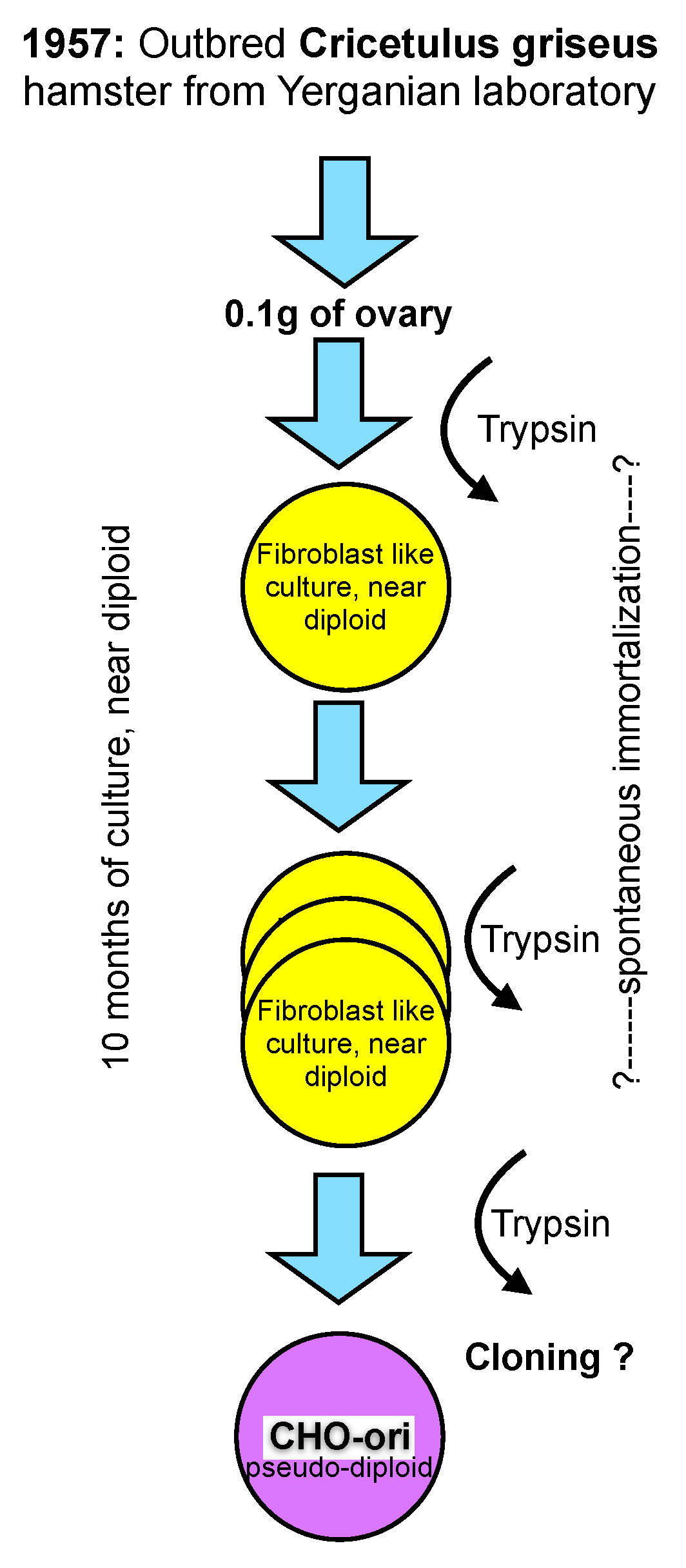
CHO cells were established in the laboratory of Dr. Theodore T. Puck in 1957 [
11], then at the Eleanor Roosevelt Institute for Cancer Research, and later at the Department of Biochemistry of the University of Colorado in Denver, from 0.1 gram of ovary tissue of a Chinese hamster. The outbred hamster was provided by Dr. George Yerganian of the Boston Children’s Cancer Research Foundation [
12]. Out-breeding tries to avoid homozygocity and, thus, maintains a vigorous and diverse genetic background of diploid animals or plants.
From the trypsinized ovary tissue a culture emerged that appeared to be predominantly of a fibroblast type and had a near diploid karyotype with only 1% of the cell population differing in chromosome number by one less or more from the expected chromosome number of 22 (11 pairs) [
12]. However, even this small diversion from the strictly diploid character of primary cells must be considered unique and is not observed in primary cells of human origin where a fully diploid karyotype is prevalent until senescence occurs (“Hayflick limit”) after about 50 population doublings and the cells die [
13].
Figure 1.
A scheme representing the generation of the early “original” CHO cell line.
Figure 1.
A scheme representing the generation of the early “original” CHO cell line.
The culture of the primary ovary cells was maintained in culture for more than 10 months and that is longer than the time limit being established for human fetal cells by Hayflick. At an unknown time thereafter the morphology of some cells changed, and these cells overgrew the strictly fibroblast cell type seen initially. It appears that cells in culture derived from the Hamster ovary have experienced some type of spontaneous immortalization while remaining close to a diploid character. However, the mentioning of morphological changes after 10 months in culture points towards additional modifications, most likely with a genetic cause, whose origin are (still) not understood today. Subsequent recloning of these cells with a modified morphology resulted in the cell line that is now called CHO. The change of morphology from a fibroblast type of culture to a more epitheloid morphology of cells is mentioned in Puck’s early papers. Unfortunately, no further detailed information on this cloning step or the potential diversity of CHO cell lines is available. For clarity in this text, I refer to these original CHO cells as CHO-ori (see
Figure 1).
The scheme in
Figure 1 depicts my personal knowledge about the origin, history and early handling of CHO cells which, under today’s standards of scrutiny in our industry, would be unsatisfying. Clearly, in retrospect, several questions arise, particularly with respect to the immortalization of these cells, their assumed genomic constitution and their apparent phenotype change as described in the literature.
CHO-ori cells were provided to many laboratories. The cells were described as “hardy”, growing very well and fast in adherent culture, and having a high cloning efficiency, even at very low fetal bovine serum concentrations in the culture medium. 10%–20% FBS in commercial media were standard in cell culture at the time. However, Hamilton and Ham (9) reported, already, in 1977 the growth of these cells in serum-free media. The cells required the addition of proline to the culture medium, a nutritional requirement for all CHO cell lines in use today. The first reference to this is from 1963 (!). Thus, it appears that the loss or inactivation of proline synthesis is an early event in the history of these cells.
1.3. CHO-DXB11
The very first product made by CHO cells, and thus the starting point of the biotechnology era involving recombinant mammalian cells, employed a cell line called CHO-DXB11. This line was generated at Columbia University by Drs. Urlaub and Chasin, being interested in the enzyme dihydrofolate reductase (DHFR) and its genetics [
14]. The cells in Chasin’s lab were derived from CHO-K1, after a co-worker in Puck’s lab (Dr. F.T. Kao) had cloned the CHO-ori cells. According to Dr. Chasin (personal communication), the CHO-K1 subclone was established in the late 1960s. Thus as much as a decade elapsed before the well-known CHO-K1 cells were established. Similar to CHO-ori cells, CHO-K1 cells were also supplied to many laboratories around the world and experiments with these cells were described in a large number of publications. Vials of frozen CHO-K1 cells were also deposited at the American Type Culture Collection (ATCC).
Dr. Chasin established the cell line CHO-DXB11 (also called DUK-XB11). The purpose of the work was to delete DHFR activity. These cells carry a deletion of one locus for DHFR and a missense mutation (T137R) of the second DFHR locus rendering the cells incapable of reducing folate, a precursor for thymidine and hypoxanthine synthesis [
14]. The cell line is not named in the paper quoted, but it is one of the gamma-ray induced mutants described. It is interesting to note that this cell line, the first to become a host system for the production of hundreds of kilograms of human tissue plasminogen activator (TPA), was the product of mutagenesis. The reasons why this and not any other CHO cell line became the pioneering cell line in biotechnology is rapidly explained: the dual inactivation of the DHFR locus rendered this cell line very useful for transgenesis with a functional DHFR gene [
15]. Transfer of a functional DHFR gene via plasmid transfection could repair the DHFR deficiency and allow easy selection of recombinant cells in well-defined media. In addition, a second, unrelated gene of interest (GOI), encoded by the same plasmid vector, could easily be transferred simultaneously and recombinant clones expressing both the functional transgenic DHFR gene and the desired co-transferred GOI could be recovered [
16]. This 1983 publication is the first to describe co-transfer of two genes into cells whereby the two corresponding DNA sequences were provided on two separate plasmids. They were simply co-transfected at different ratios. In the case of this paper quoted here, a 1:10 ratio of the DHFR plasmid to the gamma-interferon plasmid gave the highest yielding clones.
The DHFR-negative cells were grown in media containing 5%–10% fetal bovine serum (FBS). A risk factor mediated by the use of sera from cows, bovine spongiformous encephalopathy (BSE), became an important consideration for the pharmaceutical manufacturing in the 1990s. In industry, sera were generally obtained from BSE-free sources (Australia, New Zealand). Whether this practice was followed in academic labs is difficult to assess. Transfection and cloning occurred in an adherent mode, whereby cloning was done by using “cloning rings” or cotton-swaps. In both cases, an identified colony, visible to the naked eye, was targeted and many cells from such a colony were transferred into a well of a multiwell plate. Regulatory concerns, difficult to explain scientifically in view of what is discussed in this paper, requires today “single cell cloning”, frequently even twice in order to “prove” clonality.
1.4. CHO-DG44
For the researchers of metabolic studies with these cells, a low but detectable rate of reversion to DHFR activity in CHO-DXB11 cells presented a problem. In order to fully eliminate this possibility and to also provide a better DHFR-negative host system for eventual gene transfer, Dr. Chasin engaged in another round of DHFR elimination, but not with cells derived from the K1 populations. Instead, CHO-ori cells from the lab of Dr. Siminovitch lab were sourced, but in another convoluted way. Dr. Flintoff, a coworker of Siminovitch, had generated a useful mutant of CHO-ori cells, named CHO-Mtx
RIII that proved to be suitable for deletion of both DHFR alleles [
17]. In the same year (1976), Siminovitch published a highly instructive minireview on genetic diversity of cultured somatic (immortalized) cells that discusses the quasispecies concept without using the term [
18].
The elegant work by Chasin and Urlaub showing the full deletion of the two DHFR loci on chromosome 2 (actually on chromosome 2 and on a shortened marker chromosome variant Z2) resulted in the now widely-used CHO-DG44 cells [
19,
20].
1.5. Gene Amplification
The availability of these two DHFR-negative cell lines (CHO-DXB11 and CHO-DG44) allowed an approach to amplify genes with the help of an antagonist of DHFR, the chemical component methotrexate (MTX). The selection of recombinant cell lines using stepwise increases in the MTX concentration in the culture medium resulted in amplified copies of the transfected DHFR gene together with the GOI. Such induced gene amplification usually increased the productivity of the GOI [
21,
22,
23]. This approach was a key approach for enhancing protein production in clonal subpopulations of transfected CHO cell lines over a 20-year period in the biotechnology industry. During this period, most of the recombinant protein products were derived from CHO cells that had undergone MTX-induced gene amplification. Gene amplification also results in large genomic reorganizations, visible to the eye when karyotyping cells. Briefly, new chromosomal structures, known as “homogeneously staining regions” (HSRs), can be found in metaphases of MTX selected human (cancer) derived cells, as well as in CHO cells. These regions show multiple (up to thousands) repetitions of smaller chromosomal regions (amplicons), all containing DNA encoding, at least in part, sequences with DHFR activity. In fluorescence
in situ hybridizations on recombinant CHO cells, large chromosomes were found which contained entire arms and long segments within chromosomal arms, hybridizing with DHFR sequences. Copy-number analysis of such cell lines revealed hundreds and thousands of copies of DHFR in these cells [
24,
25,
26]. The genetic stability of these unusual chromosome structures within a given cell population is poorly understood [
26].
1.6. CHO-K1
In Gottesman (1987) we find the following: “One subline of the original isolate, called CHO-K1 (ATCC CCL 61) was maintained in Denver by Puck and Kao, whereas another subline was sent to Tobey at Los Alamos. This latter line was adapted to suspension growth by Thompson at the University of Toronto (CHO-S) in 1971 and has given rise to a number of Toronto subclones with similar properties including the line CHO Pro
-5 used extensively by Siminovitch and numerous colleagues in Toronto, CHO GAT of McBurney and Whitmore, subline 10001 of Gottesman at the NIH, and subline AA8 of Thompson. There are some differences in the karyotypes of the CHO-K1 and CHO-S cell lines, and CHO-S grows well in spinner and suspension culture, whereas CHO-K 1 does not. Both sublines seem to give rise readily to mutant phenotypes.” This statement shows the handling of CHO cells by many laboratories, their diversity in phenotypes (one grows the other not in suspension) and the reason for their popularity: “give rise readily to mutant phenotypes”. Today’s popularity of CHO-K1 cells is based on the successful use of these cells by a well-known contract manufacturing company that licenses them as a substrate in connection with a unique gene-transfer system based on the enzyme glutamine synthetase (GS-system). This system was originally designed for NS0 cells (a myeloma-derived cell line also used for the fusion with B cells in the generation of hybridomas) [
27] and was quickly applied to CHO cells as well. The origin of the CHO-K1 cells in the hands of the above mentioned contract manufacturer goes back to a vial of frozen cells derived in November 1989 from the European Collection of Animal Cell Cultures (ECACC). A serum-free, suspension culture was frozen in the year 2000 (11 years later) as a “development bank”. Eventually, a subline was generated that gave rise in October 2002 to a “CHO K1 SV” Master Cell Bank under “protein-free” conditions (Dr. Hilary Metcalfe, personal communication). Worldwide, at the time of writing of this review, there are five licensed pharmaceutical products that were made with the help of the GS system in combination with CHO-K1 cells.
Briefly, recombinant CHO-K1 cells can be obtained after co-transfection with a functional glutamine synthetase gene together with GOI on the same plasmid followed by selection in the absence of glutamine. In addition, the application of a GS inhibitor (methionine sulphoximine, MSX) allows either an increase of the stringency of selection or the selection for subpopulations of cells with an amplified copy number of the GS gene and the GOI. One has to assume that the principles of gene amplification with the GS system are similar to the ones discussed above for the DHFR system. Unfortunately no publications with respect to karyotypic characterization of GS/MSX amplified sequences in CHO cells have been published.
1.7. CHO-S
About 50 years ago it was recognized that some CHO cells have the capacity to grow in single-cell suspension culture [
28]. In 1973, Thompson and coworkers described suspension cultures of CHO cells [
29] and CHO-S cells were mentioned (see above quote from Gottesman 1987). Thompson’s CHO-S cells were derived from the CHO-Toronto cell line (a sister cell line of CHO-K1), also referred to as CHO pro
−5. Unfortunately, there is lack of clarity and scientific credit for the origin of CHO-S cells. CHO-S cells mentioned by Thompson and/or Gottesman, were passed on eventually through Dr. R. A. Tobey’s laboratory at the Los Alamos National Laboratory, New Mexico to a company interested in growing and eventually commercially using such cells. These cells were further cultivated by this company and have been marketed since 2002 as CHO-S. Here, I will call the CHO-S from Thompson’s lab as CHO-So (o = original) and CHO-S from that company as CHO-Sc (c = commercial). In view of their nebulous culture history it must be assumed that these two cell lines will differ with respect to optimal culture conditions and other phenotypic/genetic features. I assume that CHO-So are in freezers/liquid nitrogen tanks of labs that have worked with them, but I have not been able to locate these cells.
Neither of these CHO-S cell lines was used at Genentech in the mid-1980s for culture in single-cell suspension. Instead, recombinant clonal subpopulations derived from CHO-DUXB11, first established in adherent cultures with FBS in the medium, were individually adapted to suspension. The suspension adapted, serum-free subpopulations were not recloned prior to generation of Master Cell Banks (personal information provided by the author). This fact appears surprising, but is scientifically defendable, since “stability” and “identity” of a recombinant cell population has a higher chance of being maintained when cloning is avoided (see also discussion on stability and microevolution, below). The approach to suspension-adapt clonally derived cell lines, grown prior and during cloning in serum-containing media, without another recloning step was the basis for the first large-scale (10,000 L) stirred-tank bioreactor-based culture of CHO cells for the production of human recombinant TPA and it was also used for other products developed by Genentech in the 1990s.
1.8. Diversity of Culture Conditions and the Cytogenetics of CHO Cells
CHO cells have been maintained by hundreds of different laboratories under highly diverse conditions. Therefore, the fluidity of genomic structures in immortalized cells will have to be considered. Decades of research into culture of immortalized cells have taught one important lesson: any culture of clonal or non-clonal cell lines will have a dramatic and lasting effect on the diversity of genotypes exhibited by the cell population. Insights into the persistent and continuing fluidity of genomes of immortalized cells go back to the 1960s. The “father of mammalian cytogenetics”, Dr. T.C. Hsu, published, in 1961 a landmark paper entitled “Chromosomal Evolution in Cell Populations” [
30] that summarized more than a decade of work after the visual analysis of chromosome structures and their identification had become a standard technique. Chromosomes could be counted and identified and, thus, provided a suitable means to begin to understand the genomic organization of plants and animals. However, in contrast to the clearly recognizable and stable (in structure) chromosomes of diploid animals and plants, chromosomes of animal-derived immortalized cells showed a strong tendency to be non-identical in number (from cell to cell) and apparently were able to change their organizational structures. Chromosomes of cell lines were not stable and unchanged for the long periods of time that are assured in (wild-type) biological species. The extremely rapid genome modifying impact of immortalization is strikingly visible in the unique and highly unusual chromosomal structures of such cells. For CHO cell lines, nothing different was seen. An example of this is given in
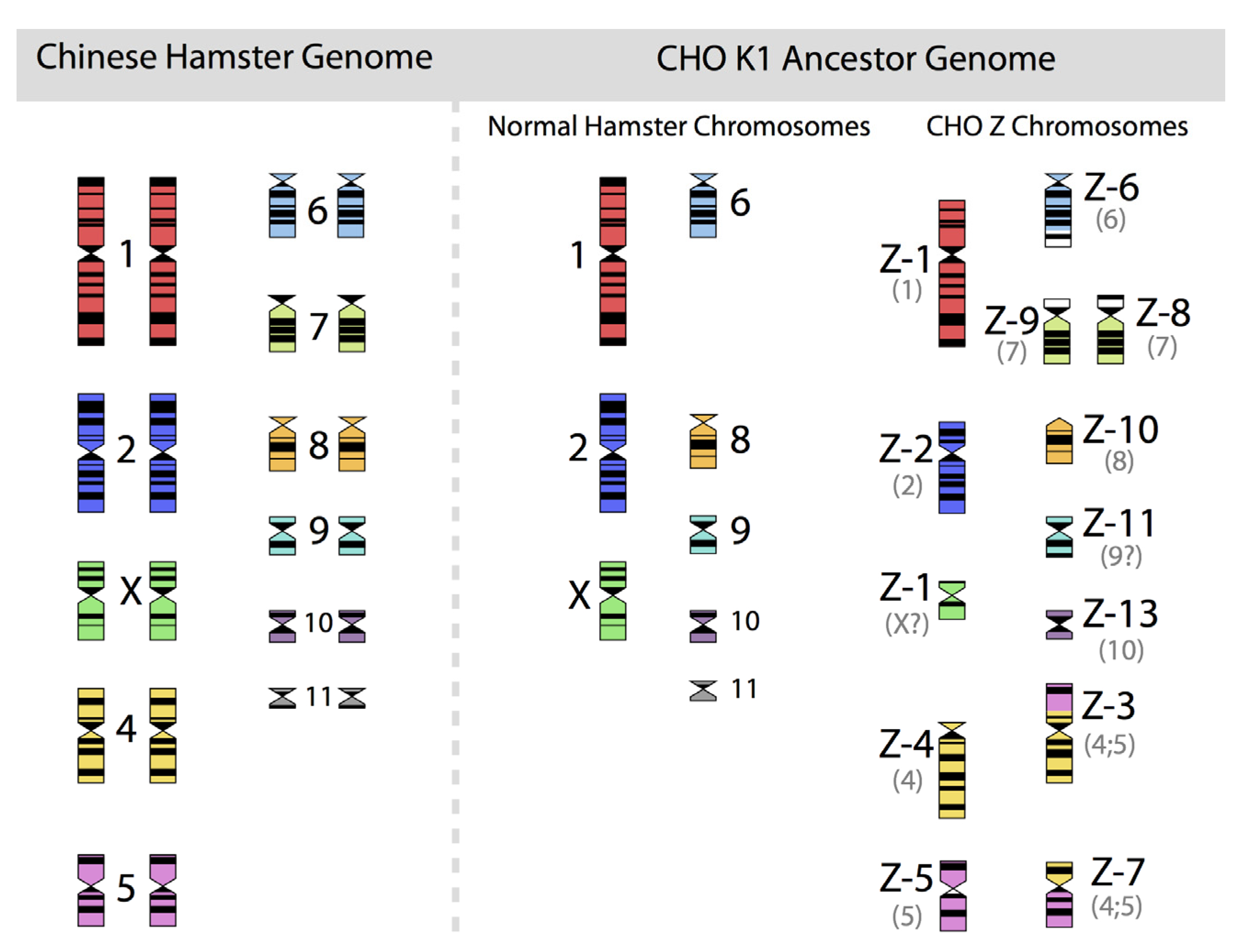
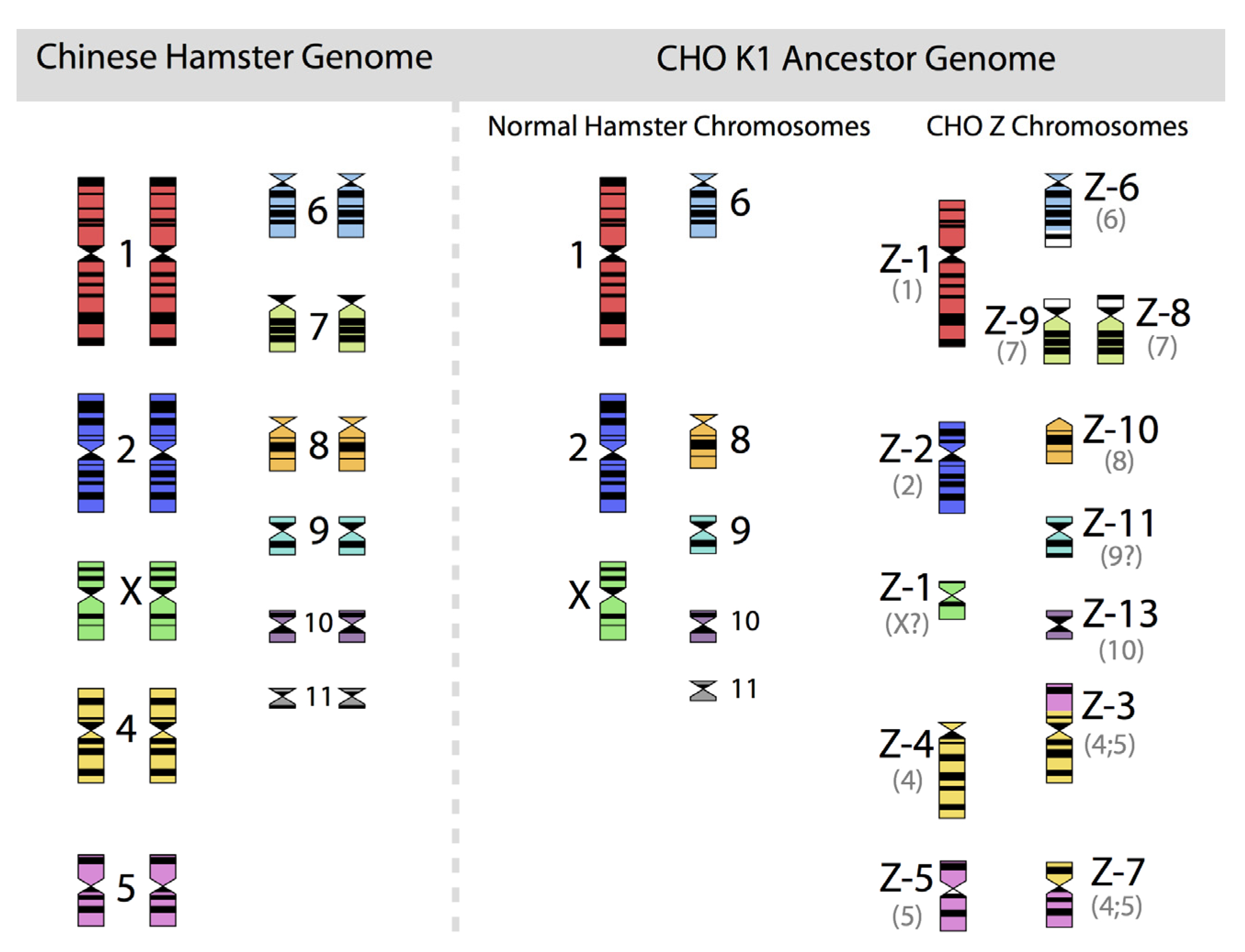
Figure 2, in which the diploid chromosomes of the Chinese hamster are shown together with those of an ancestral CHO-K1 cell line, based on the karyotyping work of Deaven and Peterson [
31].
Figure 2.
The 22 chromosomes of the Chinese hamster and of the 21 chromosomes of CHO-K1 as identified by G-banding techniques. Redrawn by C.P. Wurm after the publication of Deaven and Peterson (1973) [
31]. Part of this figure was first published in Nature Biotechnology, 2011 [
32].
Figure 2.
The 22 chromosomes of the Chinese hamster and of the 21 chromosomes of CHO-K1 as identified by G-banding techniques. Redrawn by C.P. Wurm after the publication of Deaven and Peterson (1973) [
31]. Part of this figure was first published in Nature Biotechnology, 2011 [
32].
More recently, Omasa and his group constructed genomic BAC libraries of lines of available CHO-K1 and CHO-DG44 cells in order to establish a map of the hamster chromosomes as fragments of them are distributed in the chromosomes of those cell lines. The BAC based maps solidify the earlier made karyotyping based findings by Deaven and Peterson: Dramatic rearrangement of chromosomal fragments as compared to the diploid (Hamster) genome in both cases. Also, only few structures appear “stable” when comparing DG44 and K1 cells [
33]. All immortalized cells present similar restructuring of their genomic DNA. What is not shown is the fact that the genomic structure of CHO-K1 shown in
Figure 2 is only one of many different genomic organizations present in a population of CHO-K1 cells. Other cells may show certain similarities to this pattern, but they will rarely (or never?) exhibit one that is identical when studying 100 karyotypes of individual cells. Deaven and Peterson observed a distribution of chromosome numbers per cell, ranging from 19–23. 60%–70% of the cells had 21 chromosomes. Although CHO-K1 cells do not have the 11 pairs of chromosomes of the hamster genome, the majority of the chromosome structures of the hamster genome are present albeit rearranged, with only a few elements (G-banding pattern fragments) not clearly accounted for. Much more recently, in PhD work (2006) done under the guidance of Prof. Alan Dickson (University of Manchester) by E. Hazelwood, a similarly complex genomic situation of K1 cells as they are/were used by the above mentioned contract manufacturer was revealed: A CHO-K1 cell line showed metaphase spreads over a broad chromosome number range with 16 to 30 chromosomes (100 cells studied), with 18%, 23%, and 18% of cells showing 19, 20, or 21 chromosomes, respectively. A CHO-K1 SV cell line, grown under protein free conditions (mentioned earlier), showed also a very broad chromosome number distribution of 10–30 chromosomes. In this instance, 10%, 13%, 17%, 7%, and 12% (total 59%) of cells showed 16, 17, 18, 19, or 20 chromosomes, respectively. Studies with clonal subpopulations of these cells revealed similar complexities of the karyotypes – none of them matching even approximately the statistics shown for the “parental” cells [
34]. The American Type Culture Collection describes the CHO-K1 cell line’s genotype as “Chromosome frequency distribution 50 cells: 2
n = 22. Stemline number is hypodiploid.” Thus, the same name “CHO-K1” refers to very different cell populations, most clearly represented by their karyotype.
This is not surprising: In cultivated, immortalized animal cells, single-cell cloning, with and without prior gene transfer, but also just the modification of cell culture conditions for a given cell population, leads to new and genetically diverse cells as pointed out by T.S. Hsu. Each of these populations of cells represents a new quasispecies family in the terminology of Eigen and Schuster. If cloning is performed, as is now essential for a manufacturing cell lines, we never know the genomic composition of the one cell that gives rise to the resulting population of cells. For example, we don’t even know whether cloning efficiencies of cells with 19, or 20, or 21 chromosomes are different. However, it is not unreasonable to assume that they are different. Whenever we have a chance to do karyotype analysis on a clonal population, we find diversity in chromosome structures. Clonal cell populations analyzed post-transfection and subjected to stringent selective forces show a bewildering genomic restructuring, as judged by simple karyotyping or chromosome counting. Each clonal population analyzed is different. The modal chromosome numbers vary and individual, recognizable chromosomes show rearrangements [
35].
A recent paper [
36] on the genomic landscape of one particular strain of HeLa cells [
37], theimmortalized cell line that was the foundation of animal cell culture technologies used today, sheds a revealing light on the dynamics of genome remodeling under continuous cultivation. A remarkably high level of aneuploidy and numerous large structural variants were found at unprecedented resolution. Almost a quarter of this HeLa cell line genome showed “Loss of Heterzygosity”. The original genome of Henrietta Lacks, the unfortunate woman who developed cervical cancer and whose cells are the source of the many HeLa cell lines being studied over the last 50 years, would be close to 100% heterozygote and thus would show significant sequence variations between the allelic DNAs representing the two sets of the 23 chromosomes. The HeLa cell line studied shows an average chromosome number of 64 and many segments of the original chromosome have a ploidy status ranging from triploid to octoploid. One large homozygote fragment of chromosome 5 with a size of about 40 million basepairs is apparently present with eight copies in HeLa cells. Another fragment, about 90 million base pairs, essentially the entire q-arm of chromosome 3 is present as three copies. These karyotype features result obviously from losses of fragments or arms of chromosomes while the corresponding allelic fragments are duplicated or multiplied. The authors state: “The extensive genomic rearrangements are indicative of catastrophic chromosome shattering”. Up to 2000 genes in HeLa cells are expressed at higher ranges than those seen in human tissues. More than 700 large deletions and almost 15,000 small deletions (as compared to the human genome) were detected. Most interesting in view of the major chromosomal rearrangements in CHO are the results of multiplex fluorescent
in situ hybridizations (MFISH), a chromosome-painting method. Unfortunately, only 12 metaphase spreads were analyzed in this way. As with CHO, these 12 metaphase spreads show common structural rearrangements of the karyotype, but also a number of “single cell events”. The latter show unique translocations of chromosomal fragments, not seen in any of the other cells, indicative of a continuing dynamic of restructuring of the HeLa genome. Since HeLa cells usually do not undergo single cell cloning as CHO cells do, the fate of cells with unique rearrangements is difficult to predict. They may be passed on as a more or less constant and small part of the entire population, if they do not negatively affect the duplication of a cell. Unfortunately, the term “unique” and “single cell observations” are not to be taken at face value. An analysis of the karyotype of 12 cells does not provide a sufficient basis to make conclusions about populations of hundreds of millions of dividing cells.
Only very recently a few papers [
36,
38,
39] have been published that shed a light on mechanisms for these catastrophic events in human cancer cells. It remains to be seen how similar such events are between spontaneously (
in vitro) immortalized (animal) cells and human cancer cells.
1.9. Stability, Gene Pools and Microevolution
In the context of pharmaceutical production, stability is defined as the reproducible protein yield and quality from a given cell line over extended periods of time, from thawing of the cell line from a master cell bank until a given time point that is considered the longest period allowed for production. The minimal accepted period for requested proof of stability is about three months, based on the fact that manufacturing processes at large scale take significant time and will involve many cell population doublings. The expansion of cells towards the large-scale production vessel from a frozen vial can take up to four weeks. Subsequently, the production phase in the large-scale reactor can take up to three weeks for a fed-batch process and even longer for a perfusion-based manufacturing approach. Since several batches of product are typically being produced sequentially from one thawed vial of cells, a three-month time window for this work is calculated very tightly. For approved protein products, stability studies that cover at least six months of culture are standard.
It is difficult to imagine the huge number of cells that can be generated within a three-month time window that are needed for scale up and manufacturing. I need to elaborate, in order to highlight the occurring genetic “bottlenecking”: CHO cells double their number about once a day (and shorter times have been reported), thus within three months about 90 doublings of the initial cell population will occur. Cell banks, the starting population of cells in vials for pharmaceutical manufacturing, are typically made with 1–2 × 106 cells per vial, corresponding to about 30 µL of cell biomass (compacted cells). If unrestricted for subsequent growth after thawing, this biomass could multiply within the three-month time window to a biomass volume of approximately 10192 L or 10180 km3! However, a single 10,000 Liter bioreactor will contain “only” about 5 × 1012 cells (corresponding to a biomass of about 300 L). Thus, any large-scale production run will only use a minute fraction of the progeny of cells derived from the starting culture after thawing the cells. Thus, scale-up is, in biological terms, equivalent to the expansion of a single invading species into an unexploited environment (where most progeny die/are selected against). In scale-up and maintenance of cells, many restrictions on the growth of these cells occur and thus a new population of quasispecies will evolve. Due to the genetic diversity of the invading population of quasispecies, the final bioreactor will certainly contain a quasispecies different from the starting one deposited in the master cell bank.
Independent of the diversity in CHO populations, the stability of the transgene(s) within these populations represents another problem that is not sufficiently studied and understood. Due to the lack of control over the site(s) of integration of the GOI within a single CHO cell, the issue of its stability within the genome is another unresolved problem. In this context it is noteworthy that regulators and some companies are insisting more and more on “true” clonality and that a single cell cloning exercise is not satisfactory. In view of the discussion above, this level of scrutiny is difficult to justify scientifically. In spite of decades of research in this field, no controllable and reproducible gene transfer system has been developed for CHO cells so far. For this reason, manufacturers screen thousands of clonal cell populations (all of which are to be considered quasispecies populations) and study them in extended subcultivations in order to predict with a reasonable probability that the productivity is maintained a) over time at small scale and b) after transfer to large scale for manufacturing. Essentially, what we do is to generate, each time we clone cells, a founder population that undergoes micro-evolution while we optimize and scale up our cells into large bioreactors. The diversity of these founder populations must be significant, since cloning efficiencies in CHO cells are high (>80%), yet cells differ dramatically in their individual genomic composition. The hardiness of these cells however allows rapid expansion of the number of cells derived from the unique genome composition of the founder cell while rapidly restructuring it, as had already been shown by T.S. Hsu in 1961 [
30].
Since true clonality cannot be preserved and thus does not solve the perceived stability problem, the best approach for maintaining a balanced gene pool in a given quasispecies population is to minimize growth-restricting (selective) conditions. Clearly, for commercial pharmaceutical manufacturing, the maintenance of the gene pool composition of the cells in a Master Cell Bank must be assured by all means. By keeping cell populations in environments with little environmental changes one can hope that a trend towards a modified gene pool would be minimized. Unfortunately, many standard cell culture techniques are possibly favoring or selecting for modifications of a given gene pool in a quasispecies population of CHO cells. For example, the shift of cells from adherent culture to suspension cultures represents a major environmental modification and thus will lead to the selection of subpopulations. In addition, the composition of media that either prevent or allow cells to grow to high density can be considered a selective condition. Finally, even work with controlled bioreactors may be a cause for a population bottleneck. For example, certain reactors have poor gas exchange capacities and thus need to be stirred or otherwise mixed vigorously and, frequently in addition, need to be sparged with pure oxygen gas, just to maintain basic metabolic activities for the few million cells cultivated. Such conditions can kill sensitive cells and will select for populations of cells that are adapted to these harsher conditions. Other bioreactor systems are known to have higher gas transfer rates than stirred-tank bioreactors and, thus, require less energy (correlated to shear stress and liquid turbulence) in order to distribute oxygen to cells. The milder conditions of such bioreactors would be expected to maintain sensitive cells and thus would not be that restrictive/selective on a population of cells that is scaled from milliliter cultures to hundreds and thousands of liters.
Awareness of the importance of environment conditions in cell culture for maintaining stability have only recently been discussed in groups of scientists who deal with manufacturing issues. In this context, however, the complexity of genomic compositions and the diversity of genomes in cell populations has not been a topic.

{kind=link}
{kind=link}