

The Clinical Management of Pompe Disease: A Pediatric Perspective

Abstract
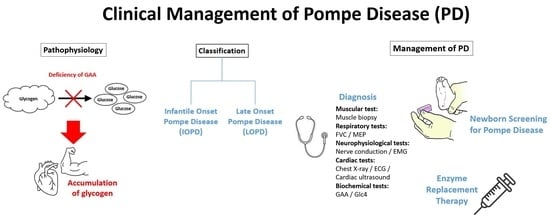
1. Introduction
2. IOPD and LOPD
3. Clinical Presentations of Pompe Disease
4. Examinations and Diagnosis of Pompe Disease
4.1. Muscular System
4.2. Respiratory System
4.3. Neurophysiological System
4.4. Cardiac System
4.5. Biochemical Tests
5. The Importance of Newborn Screening for PD
5.1. Global Practice on Pompe NBS
5.2. Next Generation Sequencing and Mutation Analysis
5.3. The Concern on NBS in Pompe Disease
5.4. Clinical Benefits of Pompe NBS-Guided Early Treatment
6. ERT for PD
Dose Optimization in ERT
7. Recommended Treatment Protocol for PD
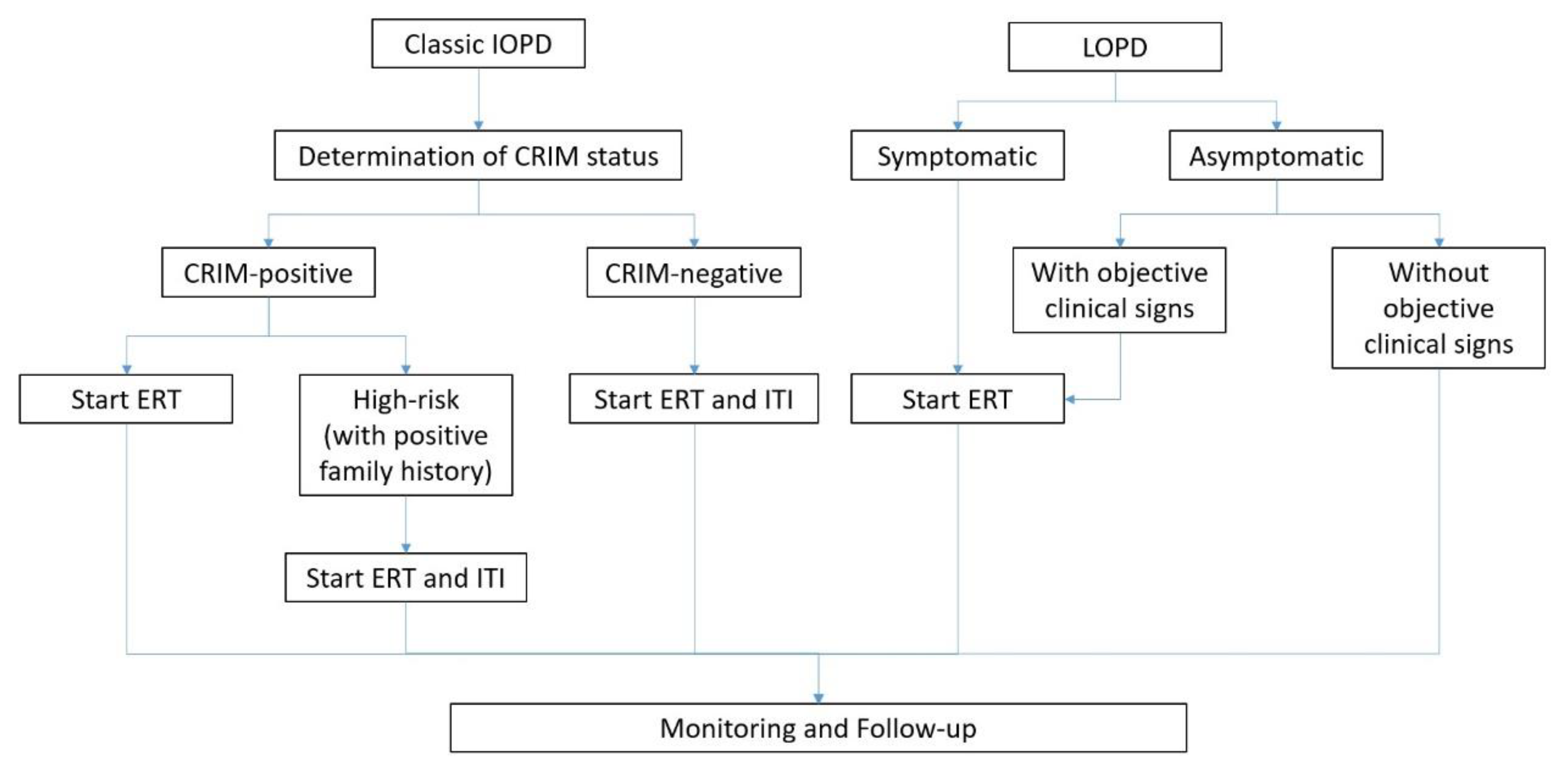
7.1. The Importance of CRIM Status and Management of CRIM-Negative Patients
7.2. ERT in Practice
7.3. Monitoring of ERT and Follow-Up
7.4. Genetic Counselling
8. Case Sharing on ERT for PD
9. Perspectives in Pompe Disease Management
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Zhao, Y.; Wang, Z.; Lu, J.; Gu, X.; Huang, Y.; Qiu, Z.; Wei, Y.; Yan, C. Characteristics of Pompe Disease in China: A Report from the Pompe Registry. Orphanet J. Rare Dis. 2019, 14, 78. [Google Scholar] [CrossRef] [PubMed]
- Hahn, A.; Schänzer, A. Long-Term Outcome and Unmet Needs in Infantile-Onset Pompe Disease. Ann. Transl. Med. 2019, 7, 283. [Google Scholar] [CrossRef] [PubMed]
- Kishnani, P.S.; Steiner, R.D.; Bali, D.; Berger, K.; Byrne, B.J.; Case, L.; Crowley, J.F.; Downs, S.; Howell, R.R.; Kravitz, R.M.; et al. Pompe Disease Diagnosis and Management Guideline. Genet. Med. 2006, 8, 267–288. [Google Scholar] [CrossRef] [PubMed]
- Kronn, D.F.; Day-Salvatore, D.; Hwu, W.-L.; Jones, S.A.; Nakamura, K.; Okuyama, T.; Swoboda, K.J.; Kishnani, P.S.; on behalf of the Pompe Disease Newborn Screening Working Group. Management of Confirmed Newborn-Screened Patients with Pompe Disease Across the Disease Spectrum. Pediatrics 2017, 140 (Suppl. 1), S24–S45. [Google Scholar] [CrossRef] [PubMed]
- Martínez, M.; Romero, M.G.; Guereta, L.G.; Cabrera, M.; Regojo, R.M.; Albajara, L.; Couce, M.L.; De Pipaon, M.S. Infantile-Onset Pompe Disease with Neonatal Debut: A Case Report and Literature Review. Medicine 2017, 96, e9186. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.; Sheng, B.; Lau, K.; Chan, H.; Kam, G.; Lee, H.; Mak, C. Clinical Manifestation of Late Onset Pompe Disease Patients in Hong Kong. Neuromuscul. Disord. 2016, 26, 873–879. [Google Scholar] [CrossRef]
- Felice, T. Pompe Disease, a Storage Cardiomyopathy. Cardiogenetics 2017, 7, 6857. [Google Scholar] [CrossRef]
- Wang, R.Y. A Newborn Screening, Presymptomatically Identified Infant with Late-Onset Pompe Disease: Case Report, Parental Experience, and Recommendations. Int. J. Neonatal Screen. 2020, 6, 22. [Google Scholar] [CrossRef]
- Herzog, A.; Hartung, R.; Reuser, A.; Hermanns, P.; Runz, H.; Karabul, N.; Gökce, S.; Pohlenz, J.; Kampmann, C.; Lampe, C.; et al. A Cross-Sectional Single-Centre Study on the Spectrum of Pompe Disease, German Patients: Molecular Analysis of the GAA Gene, Manifestation and Genotype-Phenotype Correlations. Orphanet J. Rare Dis. 2012, 7, 35. [Google Scholar] [CrossRef]
- Kishnani, P.; Hwu, W.; Mandel, H.; Nicolino, M.; Yong, F.; Corzo, D. A Retrospective, Multinational, Multicenter Study on the Natural History of Infantile-Onset Pompe Disease. J. Pediatr. 2006, 148, 671–676. [Google Scholar] [CrossRef]
- van denDorpel, J.; Poelman, E.; Harlaar, L.; vanKooten, H.; van derGiessen, L.; vanDoorn, P.; van derPloeg, A.; van denHout, J.; van derBeek, N. Distal Muscle Weakness Is a Common and Early Feature in Long-Term Enzyme-Treated Classic Infantile Pompe Patients. Orphanet J. Rare Dis. 2020, 15, 247. [Google Scholar] [CrossRef] [PubMed]
- Saich, R.; Brown, R.; Collicoat, M.; Jenner, C.; Primmer, J.; Clancy, B.; Holland, T.; Krinks, S. Is Newborn Screening the Ultimate Strategy to Reduce Diagnostic Delays in Pompe Disease? The Parent and Patient Perspective. Int. J. Neonatal Screen. 2020, 6, 1. [Google Scholar] [CrossRef] [PubMed]
- Marsden, D. Infantile Onset Pompe Disease: A Report of Physician Narratives from an Epidemiologic Study. Genet. Med. 2005, 7, 147–150. [Google Scholar] [CrossRef] [PubMed]
- Tabatabaei Shafiei, M.; Carvajal Gonczi, C.; Rahman, M.; East, A.; François, J.; Darlington, P. Detecting Glycogen in Peripheral Blood Mononuclear Cells with Periodic Acid Schiff Staining. J. Vis. Exp. 2014, 94, 52199. [Google Scholar] [CrossRef] [PubMed]
- Dolfus, C.; Simon, J.; Landemore, G.; Leroy, F.; Chapon, F. Combination of Acid Phosphatase Positivity and Rimmed Vacuoles as Useful Markers in the Diagnosis of Adult-Onset Pompe Disease Lacking Specific Clinical and Pathological Features. Folia Neuropathol. 2016, 54, 295–302. [Google Scholar] [CrossRef]
- Vissing, J.; Lukacs, Z.; Straub, V. Diagnosis of Pompe Disease: Muscle Biopsy vs. Blood-Based Assays. JAMA Neurol. 2013, 70, 923–927. [Google Scholar] [CrossRef]
- Bay, L.B.; Fainboim, A. Infantile-Onset Pompe Disease: Diagnosis and Management. Arch. Argent. Pediatr. 2019, 117, 271–278. [Google Scholar] [CrossRef]
- Tarnopolsky, M.; Katzberg, H.; Petrof, B.J.; Sirrs, S.; Sarnat, H.B.; Myers, K.; Dupré, N.; Dodig, D.; Genge, A.; Venance, S.L.; et al. Pompe Disease: Diagnosis and Management. Evidence-Based Guidelines from a Canadian Expert Panel. Can. J. Neurol. Sci. 2016, 43, 472–485. [Google Scholar] [CrossRef]
- Laveneziana, P.; Albuquerque, A.; Aliverti, A.; Babb, T.; Barreiro, E.; Dres, M.; Dubé, B.-P.; Fauroux, B.; Gea, J.; Guenette, J.A.; et al. ERS Statement on Respiratory Muscle Testing at Rest and during Exercise. Eur. Respir. J. 2019, 53, 1801214. [Google Scholar] [CrossRef]
- Yang, C.-F.; Niu, D.-M.; Tai, S.-K.; Wang, T.-H.; Su, H.-T.; Huang, L.-Y.; Soong, W.-J. Airway Abnormalities in Very Early Treated Infantile-Onset Pompe Disease: A Large-Scale Survey by Flexible Bronchoscopy. Am. J. Med. Genet. Part A 2020, 182, 721–729. [Google Scholar] [CrossRef]
- Müller-Felber, W.; Horvath, R.; Gempel, K.; Podskarbi, T.; Shin, Y.; Pongratz, D.; Walter, M.C.; Baethmann, M.; Schlotter-Weigel, B.; Lochmüller, H.; et al. Late Onset Pompe Disease: Clinical and Neurophysiological Spectrum of 38 Patients Including Long-Term Follow-up in 18 Patients. Neuromuscul. Disord. 2007, 17, 698–706. [Google Scholar] [CrossRef] [PubMed]
- Manganelli, F.; Ruggiero, L. Clinical Features of Pompe Disease. Acta Myol. 2013, 32, 82. [Google Scholar]
- Hobson-Webb, L.; Dearmey, S.; Kishnani, P. The Clinical and Electrodiagnostic Characteristics of Pompe Disease with Post-Enzyme Replacement Therapy Findings. Clin. Neurophysiol. 2011, 122, 2312–2317. [Google Scholar] [CrossRef] [PubMed]
- Ansong, A.; Li, J.; Nozik-Grayck, E.; Ing, R.; Kravitz, R.; Idriss, S.; Kanter, R.; Rice, H.; Chen, Y.; Kishnani, P. Electrocardiographic Response to Enzyme Replacement Therapy for Pompe Disease. Genet. Med. 2006, 8, 297–301. [Google Scholar] [CrossRef] [PubMed]
- Limongelli, G.; Fratta, F. Cardiovascular Involvement in Pompe Disease. Acta Myol. 2011, 30, 202. [Google Scholar]
- Al Jasmi, F.; Al Jumah, M.; Alqarni, F.; Al-Sanna’a, N.; Al-Sharif, F.; Bohlega, S.; Cupler, E.J.; Fathalla, W.; Hamdan, M.A.; Makhseed, N.; et al. Diagnosis and Treatment of Late-Onset Pompe Disease in the Middle East and North Africa Region: Consensus Recommendations from an Expert Group. BMC Neurol. 2015, 15, 205. [Google Scholar] [CrossRef]
- Winchester, B.; Bali, D.; Bodamer, O.; Caillaud, C.; Christensen, E.; Cooper, A.; Cupler, E.; Deschauer, M.; Fumić, K.; Jackson, M.; et al. Methods for a Prompt and Reliable Laboratory Diagnosis of Pompe Disease: Report from an International Consensus Meeting. Mol. Genet. Metab. 2008, 93, 275–281. [Google Scholar] [CrossRef]
- Umapathysivam, K.; Hopwood, J.; Meikle, P. Determination of Acid Alpha-Glucosidase Activity in Blood Spots as a Diagnostic Test for Pompe Disease. Clin. Chem. 2001, 47, 1378–1383. [Google Scholar] [CrossRef]
- Gelb, M.H.; Turecek, F.; Scott, C.R.; Chamoles, N.A. Direct Multiplex Assay of Enzymes in Dried Blood Spots by Tandem Mass Spectrometry for the Newborn Screening of Lysosomal Storage Disorders. J. Inherit. Metab. Dis. 2006, 29, 397. [Google Scholar] [CrossRef]
- An, Y.; Young, S.; Hillman, S.; VanHove, J.; Chen, Y.; Millington, D. Liquid Chromatographic Assay for a Glucose Tetrasaccharide, a Putative Biomarker for the Diagnosis of Pompe Disease. Anal. Biochem. 2000, 287, 136–143. [Google Scholar] [CrossRef]
- Tchan, M.; Henderson, R.; Kornberg, A.; Kairaitis, K.; Fuller, M.; Davis, M.; Ellaway, C.; Reardon, K.; Corbett, A.; Needham, M.; et al. Is It Pompe Disease? Australian Diagnostic Considerations. Neuromuscul. Disord. 2020, 30, 389–399. [Google Scholar] [CrossRef] [PubMed]
- Kishnani, P.; Corzo, D.; Leslie, N.; Gruskin, D.; Van derPloeg, A.; Clancy, J.; Parini, R.; Morin, G.; Beck, M.; Bauer, M.; et al. Early Treatment with Alglucosidase Alpha Prolongs Long-Term Survival of Infants with Pompe Disease. Pediatr. Res. 2009, 66, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Molster, C.; Urwin, D.; DiPietro, L.; Fookes, M.; Petrie, D.; van derLaan, S.; Dawkins, H. Survey of Healthcare Experiences of Australian Adults Living with Rare Diseases. Orphanet J. Rare Dis. 2016, 11, 30. [Google Scholar] [CrossRef] [PubMed]
- Chiang, S.; Chen, P.; Hwu, W.; Lee, A.; Chen, L.; Lee, N.; Chiou, L.; Chien, Y. Performance of the Four-Plex Tandem Mass Spectrometry Lysosomal Storage Disease Newborn Screening Test: The Necessity of Adding a 2nd Tier Test for Pompe Disease. Int. J. Neonatal Screen. 2018, 4, 41. [Google Scholar] [CrossRef] [PubMed]
- Bodamer, O.A.; Scott, C.R.; Giugliani, R.; on behalf of the Pompe Disease Newborn Screening Working Group. Newborn Screening for Pompe Disease. Pediatrics 2017, 140 (Suppl. 1), S4–S13. [Google Scholar] [CrossRef] [PubMed]
- Sawada, T.; Kido, J.; Nakamura, K. Newborn Screening for Pompe Disease. Int. J. Neonatal Screen. 2020, 6, 31. [Google Scholar] [CrossRef]
- Sawada, T.; Kido, J.; Sugawara, K.; Momosaki, K.; Yoshida, S.; Kojima-Ishii, K.; Inoue, T.; Matsumoto, S.; Endo, F.; Ohga, S.; et al. Current Status of Newborn Screening for Pompe Disease in Japan. Orphanet J. Rare Dis. 2021, 16, 516. [Google Scholar] [CrossRef]
- Ames, E.G.; Fisher, R.; Kleyn, M.; Ahmad, A. Current Practices for U.S. Newborn Screening of Pompe Disease and MPSI. Int. J. Neonatal Screen. 2020, 6, 72. [Google Scholar] [CrossRef]
- Becker, J.A.; Vlach, J.; Raben, N.; Nagaraju, K.; Adams, E.M.; Hermans, M.M.; Reuser, A.J.; Brooks, S.S.; Tifft, C.J.; Hirschhorn, R.; et al. The African Origin of the Common Mutation in African American Patients with Glycogen-Storage Disease Type II. Am. J. Hum. Genet. 1998, 62, 991. [Google Scholar] [CrossRef]
- Laforêt, P.; Nicolino, M.; Eymard, B.; Puech, J.P.; Caillaud, C.; Poenaru, L.; Fardeau, M. Juvenile and Adult-Onset Acid Maltase Deficiency in France. Neurology 2000, 55, 1122–1128. [Google Scholar] [CrossRef]
- Montalvo, A.L.E.; Bembi, B.; Donnarumma, M.; Filocamo, M.; Parenti, G.; Rossi, M.; Merlini, L.; Buratti, E.; De Filippi, P.; Dardis, A.; et al. Mutation Profile of the GAA Gene in 40 Italian Patients with Late Onset Glycogen Storage Disease Type II. Hum. Mutat. 2006, 27, 999–1006. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, Z.; Jin, W.; Lv, H.; Zhang, W.; Que, C.; Huang, Y.; Yuan, Y. Clinical and GAA Gene Mutation Analysis in Mainland Chinese Patients with Late-Onset Pompe Disease: Identifying c.2238G > C as the Most Common Mutation. BMC Med. Genet. 2014, 15, 141. [Google Scholar] [CrossRef] [PubMed]
- Momosaki, K.; Kido, J.; Yoshida, S.; Sugawara, K.; Miyamoto, T.; Inoue, T.; Okumiya, T.; Matsumoto, S.; Endo, F.; Hirose, S.; et al. Newborn Screening for Pompe Disease in Japan: Report and Literature Review of Mutations in the GAA Gene in Japanese and Asian Patients. J. Hum. Genet. 2019, 64, 741–755. [Google Scholar] [CrossRef]
- Chien, Y.-H.; Lee, N.-C.; Thurberg, B.L.; Chiang, S.-C.; Zhang, X.K.; Keutzer, J.; Huang, A.-C.; Wu, M.-H.; Huang, P.-H.; Tsai, F.-J.; et al. Pompe Disease in Infants: Improving the Prognosis by Newborn Screening and Early Treatment. Pediatrics 2009, 124, e1116–e1125. [Google Scholar] [CrossRef] [PubMed]
- Chien, Y.-H.; Lee, N.-C.; Chen, C.-A.; Tsai, F.-J.; Tsai, W.-H.; Shieh, J.-Y.; Huang, H.-J.; Hsu, W.-C.; Tsai, T.-H.; Hwu, W.-L. Long-Term Prognosis of Patients with Infantile-Onset Pompe Disease Diagnosed by Newborn Screening and Treated since Birth. J. Pediatr. 2015, 166, 985–991.e2. [Google Scholar] [CrossRef] [PubMed]
- Milverton, J.; Newton, S.; Merlin, T. The Effectiveness of Enzyme Replacement Therapy for Juvenile-Onset Pompe Disease: A Systematic Review. J. Inherit. Metab. Dis. 2019, 42, 57–65. [Google Scholar] [CrossRef]
- Suzuki, Y.; Tsuji, A.; Omura, K.; Nakamura, G.; Awa, S.; Kroos, M.; Reuser, A.J.J. Km Mutant of Acid α-Glucosidase in a Case of Cardiomyopathy without Signs of Skeletal Muscle Involvement. Clin. Genet. 1988, 33, 376–385. [Google Scholar] [CrossRef]
- El-Gharbawy, A.H.; Mackey, J.; DeArmey, S.; Westby, G.; Grinnell, S.G.; Malovrh, P.; Conway, R.; Kishnani, P.S. An Individually, Modified Approach to Desensitize Infants and Young Children with Pompe Disease, and Significant Reactions to Alglucosidase Alfa Infusions. Mol. Genet. Metab. 2011, 104, 118. [Google Scholar] [CrossRef]
- Landis, J.L.; Hyland, H.; Kindel, S.J.; Punnoose, A.; Geddes, G.C. Pompe Disease Treatment with Twice a Week High Dose Alglucoside Alfa in a Patient with Severe Dilated Cardiomyopathy. Mol. Genet. Metab. Reports 2018, 16, 1–4. [Google Scholar] [CrossRef]
- van Gelder, C.M.; Poelman, E.; Plug, I.; Hoogeveen-Westerveld, M.; van der Beek, N.A.M.E.; Reuser, A.J.J.; van der Ploeg, A.T. Effects of a Higher Dose of Alglucosidase Alfa on Ventilator-Free Survival and Motor Outcome in Classic Infantile Pompe Disease: An Open-Label Single-Center Study. J. Inherit. Metab. Dis. 2016, 39, 383–390. [Google Scholar] [CrossRef]
- Chien, Y.-H.; Tsai, W.-H.; Chang, C.-L.; Chiu, P.-C.; Chou, Y.-Y.; Tsai, F.-J.; Wong, S.-L.; Lee, N.-C.; Hwu, W.-L. Earlier and Higher Dosing of Alglucosidase Alfa Improve Outcomes in Patients with Infantile-Onset Pompe Disease: Evidence from Real-World Experiences. Mol. Genet. Metab. Reports 2020, 23, 100591. [Google Scholar] [CrossRef] [PubMed]
- Jones, H.N.; Crisp, K.D.; Moss, T.; Strollo, K.; Robey, R.; Sank, J.; Canfield, M.; Case, L.E.; Mahler, L.; Kravitz, R.M.; et al. Effects of Respiratory Muscle Training (RMT) in Children with Infantile-Onset Pompe Disease and Respiratory Muscle Weakness. J. Pediatr. Rehabil. Med. 2014, 7, 255–265. [Google Scholar] [CrossRef] [PubMed]
- van Capelle, C.I.; Goedegebure, A.; Homans, N.C.; Hoeve, H.L.J.; Reuser, A.J.; van der Ploeg, A.T. Hearing Loss in Pompe Disease Revisited: Results from a Study of 24 Children. J. Inherit. Metab. Dis. 2010, 33, 597. [Google Scholar] [CrossRef]
- Roberts, M.; Kishnani, P.S.; van derPloeg, A.T.; Müller-Felber, W.; Merlini, L.; Prasad, S.; Case, L.E. The Prevalence and Impact of Scoliosis in Pompe Disease: Lessons Learned from the Pompe Registry. Mol. Genet. Metab. 2011, 104, 574–582. [Google Scholar] [CrossRef]
- Wang, Z.; Okamoto, P.; Keutzer, J. A New Assay for Fast, Reliable CRIM Status Determination in Infantile-Onset Pompe Disease. Mol. Genet. Metab. 2014, 111, 92–100. [Google Scholar] [CrossRef]
- Mendelsohn, N.J.; Messinger, Y.H.; Rosenberg, A.S.; Kishnani, P.S. Elimination of Antibodies to Recombinant Enzyme in Pompe’s Disease. N. Engl. J. Med. 2009, 360, 194–195. [Google Scholar] [CrossRef]
- van der Ploeg, A.T.; Kruijshaar, M.E.; Toscano, A.; Laforêt, P.; Angelini, C.; Lachmann, R.H.; Pascual, S.I.P.; Roberts, M.; Rösler, K.; Stulnig, T.; et al. European Consensus for Starting and Stopping Enzyme Replacement Therapy in Adult Patients with Pompe Disease: A 10-Year Experience. Eur. J. Neurol. 2017, 24, 768.e31. [Google Scholar] [CrossRef]
- Raben, N.; Ralston, E.; Chien, Y.-H.; Baum, R.; Schreiner, C.; Hwu, W.-L.; Zaal, K.J.M.; Plotz, P.H. Differences in the Predominance of Lysosomal and Autophagic Pathologies between Infants and Adults with Pompe Disease: Implications for Therapy. Mol. Genet. Metab. 2010, 101, 324. [Google Scholar] [CrossRef] [PubMed]
- Llerena, J.C.; Nascimento, O.J.; Oliveira, A.S.B.; Dourado, M.E.T.; Marrone, C.D.; Siqueira, H.H.; Sobreira, C.F.R.; Dias-Tosta, E.; Werneck, L.C. Guidelines for the Diagnosis, Treatment and Clinical Monitoring of Patients with Juvenile and Adult Pompe Disease. Arq. Neuropsiquiatr. 2015, 74, 166–176. [Google Scholar] [CrossRef]
- Ambrosino, N.; Confalonieri, M.; Crescimanno, G.; Vianello, A.; Vitacca, M. The Role of Respiratory Management of Pompe Disease. Respir. Med. 2013, 107, 1124–1132. [Google Scholar] [CrossRef]
- Pena, L.D.M.; Barohn, R.J.; Byrne, B.J.; Desnuelle, C.; Goker-Alpan, O.; Ladha, S.; Laforêt, P.; Mengel, K.E.; Pestronk, A.; Pouget, J.; et al. Safety, Tolerability, Pharmacokinetics, Pharmacodynamics, and Exploratory Efficacy of the Novel Enzyme Replacement Therapy Avalglucosidase Alfa (NeoGAA) in Treatment-Naïve and Alglucosidase Alfa-Treated Patients with Late-Onset Pompe Disease: A Phase 1, Open-Label, Multicenter, Multinational, Ascending Dose Study. Neuromuscul. Disord. 2019, 29, 167–186. [Google Scholar] [CrossRef] [PubMed]
- Koeberl, D.D.; Luo, X.; Sun, B.; McVie-Wylie, A.; Dai, J.; Li, S.; Banugaria, S.G.; Chen, Y.-T.; Bali, D.S. Enhanced Efficacy of Enzyme Replacement Therapy in Pompe Disease Through Mannose-6-Phosphate Receptor Expression in Skeletal Muscle. Mol. Genet. Metab. 2011, 103, 107. [Google Scholar] [CrossRef] [PubMed]
- McVie-Wylie, A.J.; Lee, K.L.; Qiu, H.; Jin, X.; Do, H.; Gotschall, R.; Thurberg, B.L.; Rogers, C.; Raben, N.; O’Callaghan, M.; et al. Biochemical and pharmacological characterization of different recombinant acid α-glucosidase preparations evaluated for the treatment of pompe disease. Mol. Genet. Metab. 2008, 94, 448. [Google Scholar] [CrossRef] [PubMed]
- Kishnani, P.S.; Attarian, S.; Borges, J.L.; Bouhour, F.; Chien, Y.-H.; Choi, Y.-C.; Clemens, P.; Day, J.; Diaz-Manera, J.; Er-dem-Ozdamar, S.; et al. Efficacy and Safety Results of the Avalglucosidase Alfa Phase 3 COMET Trial in Late-Onset Pompe Disease Patients. Mol. Genet. Metab. 2021, 132, S57. [Google Scholar] [CrossRef]
- Zhang, P.; Sun, B.; Osada, T.; Rodriguiz, R.; Yang, X.Y.; Luo, X.; Kemper, A.R.; Clay, T.M.; Koeberl, D.D. Immunodominant Liver-Specific Expression Suppresses Transgene-Directed Immune Responses in Murine Pompe Disease. Hum. Gene Ther. 2012, 23, 460. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.K.; Collins, S.W.; Conlon, T.J.; Mah, C.S.; Lawson, L.A.; Martin, A.D.; Fuller, D.D.; Cleaver, B.D.; Clément, N.; Phillips, D.; et al. Phase I/II Trial of Adeno-Associated Virus–Mediated Alpha-Glucosidase Gene Therapy to the Diaphragm for Chronic Respiratory Failure in Pompe Disease: Initial Safety and Ventilatory Outcomes. Hum. Gene Ther. 2013, 24, 630. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| IOPD | LOPD |
|---|---|
Musculoskeletal
| Musculoskeletal
|
| Infantile Form | Late-Onset Form |
|---|---|
Potential patient group(s)
| Potential patient group(s)
|
| Outcomes | Assessments |
|---|---|
| Skeletal muscle strength |
|
| Vital capacity |
|
| Functional exercise capacity |
|
| Quality of life |
|
| Hearing |
|
| Laboratory tests |
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marques, J.S. The Clinical Management of Pompe Disease: A Pediatric Perspective. Children 2022, 9, 1404. https://doi.org/10.3390/children9091404
Marques JS. The Clinical Management of Pompe Disease: A Pediatric Perspective. Children. 2022; 9(9):1404. https://doi.org/10.3390/children9091404
Chicago/Turabian StyleMarques, Jorge Sales. 2022. "The Clinical Management of Pompe Disease: A Pediatric Perspective" Children 9, no. 9: 1404. https://doi.org/10.3390/children9091404
APA StyleMarques, J. S. (2022). The Clinical Management of Pompe Disease: A Pediatric Perspective. Children, 9(9), 1404. https://doi.org/10.3390/children9091404