Successful Hematopoietic Stem Cell Transplantation from a Matched Related Donor with Beta-Thalassemia Minor for Severe Aplastic Anemia


{kind=link}
Abstract
1. Introduction
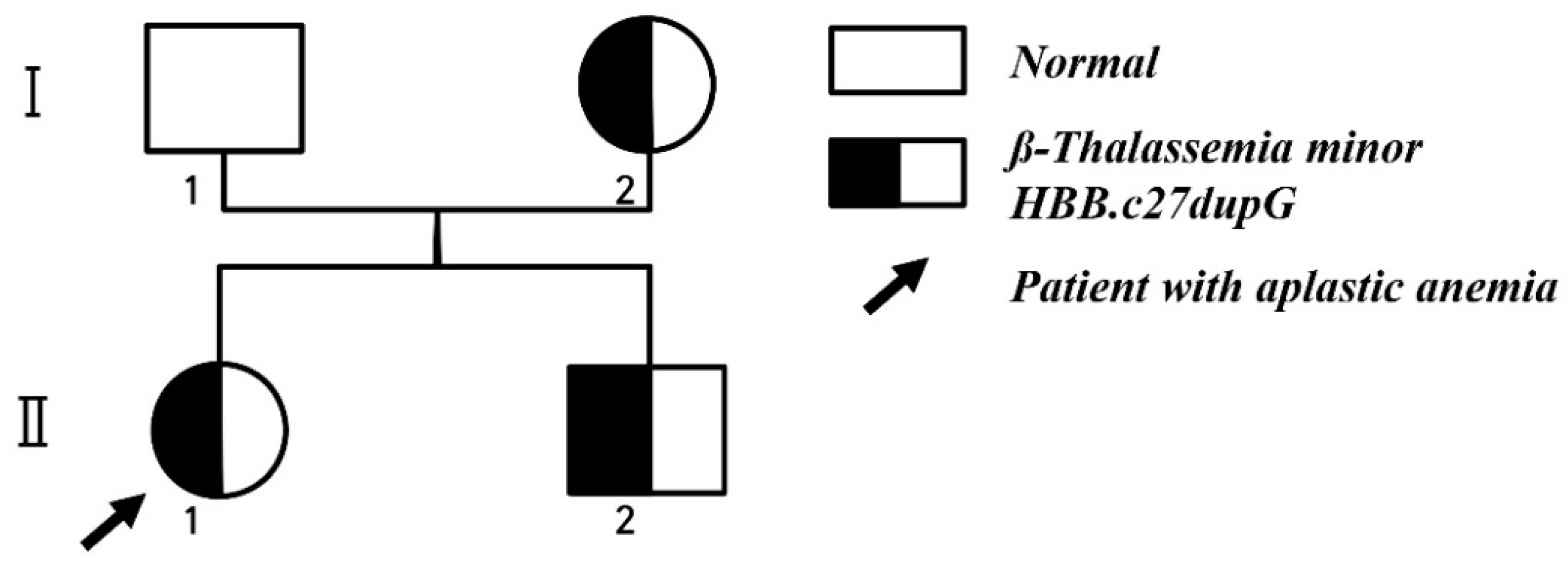
2. Case Report
3. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Young, N.S.; Bacigalupo, A.; Marsh, J.C.W. Aplastic Anemia: Pathophysiology and Treatment. Biol. Blood Marrow Transplant. 2010, 16, S119–S125. [Google Scholar] [CrossRef] [PubMed]
- Kojima, S.; Frickhofen, N.; Deeg, H.J.; Okamoto, S.; Marsh, J.; Marsh, D.J.; Bacigalupo, A.; Mizoguchi, H. Aplastic Anemia. Int. J. Hematol. 2005, 82, 408–411. [Google Scholar] [CrossRef] [PubMed]
- Kook, H.; Chung, N.G.; Kang, H.J.; Im, H.J. Acquired aplastic anemia in Korean children: Treatment guidelines from the Bone Marrow Failure Committee of the Korean Society of Pediatric Hematology Oncology. Int. J. Hematol. 2016, 103, 380–386. [Google Scholar] [CrossRef] [PubMed]
- Marsh, J.C.; Ball, S.E.; Cavenagh, J.; Darbyshire, P.; Dokal, I.; Gordon-Smith, E.C.; Keidan, J.; Laurie, A.; Martin, A.; Mercieca, J.; et al. Guidelines for the diagnosis and management of aplastic anaemia. Br. J. Haematol. 2009, 147, 43–70. [Google Scholar] [CrossRef] [PubMed]
- Adam, M.P.; Ardinger, H.H.; Pagon, R.A.; Wallace, S.E.; Bean, L.J.H.; Stephens, K.; Amemiya, A. GeneReviews® [Internet]; University of Washington: Seattle, WA, USA, 2000. [Google Scholar]
- Kramer, M.S.; Lane, D.A.; Hutchinson, T.A. The International Agranulocytosis and Aplastic Anemia Study (IAAAS). J. Clin. Epidemiol. 1988, 41, 613–616. [Google Scholar] [CrossRef]
- Locasciulli, A.; Oneto, R.; Bacigalupo, A.; Socié, G.; Korthof, E.; Bekassy, A.; Schrezenmeier, H.; Passweg, J.; Führer, M. Outcome of patients with acquired aplastic anemia given first line bone marrow transplantation or immunosuppressive treatment in the last decade: A report from the European Group for Blood and Marrow Transplantation (EBMT). Haematologica 2007, 92, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Dufour, C.; Pillon, M.; Sociè, G.; Rovò, A.; Carraro, E.; Bacigalupo, A.; Oneto, R.; Passweg, J.; Risitano, A.; Tichelli, A.; et al. Outcome of aplastic anaemia in children. A study by the severe aplastic anaemia and paediatric disease working parties of the European group blood and bone marrow transplant. Br. J. Haematol. 2015, 169, 565–573. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, N.; Kobayashi, R.; Yabe, H.; Kosaka, Y.; Yagasaki, H.; Watanabe, K.; Kudo, K.; Morimoto, A.; Ohga, S.; Muramatsu, H.; et al. First-line treatment for severe aplastic anemia in children: Bone marrow transplantation from a matched family donor versus immunosuppressive therapy. Haematologica 2014, 99, 1784–1791. [Google Scholar] [CrossRef] [PubMed]
- Schrier, S.L.; Rachmilewitz, E.; Mohandas, N. Cellular and membrane properties of alpha and beta thalassemic erythrocytes are different: Implication for differences in clinical manifestations. Blood 1989, 74, 2194–2202. [Google Scholar] [CrossRef] [PubMed]
- Moura, I.C.; Hermine, O. Erythroferrone: The missing link in β-thalassemia? Blood 2015, 126, 1974–1975. [Google Scholar] [CrossRef] [PubMed]
- Worel, N.; Buser, A.; Greinix, H.T.; Hägglund, H.; Navarro, W.; Pulsipher, M.A.; Nicoloso De Faveri, G.; Bengtsson, M.; Billen, A.; Espino, G.; et al. Suitability Criteria for Adult Related Donors: A Consensus Statement from the Worldwide Network for Blood and Marrow Transplantation Standing Committee on Donor Issues. Biol. Blood Marrow Transplant. 2015, 21, 2052–2060. [Google Scholar] [CrossRef] [PubMed]
- Galanello, R.; Origa, R. Beta-thalassemia. Orphanet J. Rare Dis. 2010, 5, 11. [Google Scholar] [CrossRef] [PubMed]
- Taher, A.; Isma’eel, H.; Cappellini, M.D. Thalassemia intermedia: Revisited. Blood Cells Mol. Dis. 2006, 37, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Eapen, M.; Le Rademacher, J.; Antin, J.H.; Champlin, R.E.; Carreras, J.; Fay, J.; Passweg, J.R.; Tolar, J.; Horowitz, M.M.; Marsh, J.C.W.; et al. Effect of stem cell source on outcomes after unrelated donor transplantation in severe aplastic anemia. Blood 2011, 118, 2618–2621. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Lee, J.H.; Joo, Y.D.; Bae, S.H.; Hyun, M.S.; Lee, J.H.; Kim, D.Y.; Lee, W.S.; Ryoo, H.M.; Kim, M.K.; et al. A randomized comparison of cyclophosphamide vs. reduced dose cyclophosphamide plus fludarabine for allogeneic hematopoietic cell transplantation in patients with aplastic anemia and hypoplastic myelodysplastic syndrome. Ann. Hematol. 2012, 91, 1459–1469. [Google Scholar] [CrossRef] [PubMed]
- Chung, N.-G.; Lee, J.W.; Jang, P.-S.; Jeong, D.-C.; Cho, B.; Kim, H.-K. Reduced dose cyclophosphamide, fludarabine and antithymocyte globulin for sibling and unrelated transplant of children with severe and very severe aplastic anemia. Pediatric Transpl. 2013, 17, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Im, H.J.; Koh, K.N.; Kang, S.H.; Yoo, J.W.; Choi, E.S.; Cho, Y.U.; Jang, S.; Park, C.J.; Seo, J.J. Comparable Outcome with a Faster Engraftment of Optimized Haploidentical Hematopoietic Stem Cell Transplantation Compared with Transplantations from Other Donor Types in Pediatric Acquired Aplastic Anemia. Biol. Blood Marrow Transplant. 2019, 25, 965–974. [Google Scholar] [CrossRef] [PubMed]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jung, M.Y.; Lim, Y.T.; Lim, H.; Hah, J.O.; Lee, J.M. Successful Hematopoietic Stem Cell Transplantation from a Matched Related Donor with Beta-Thalassemia Minor for Severe Aplastic Anemia. Children 2020, 7, 162. https://doi.org/10.3390/children7100162
Jung MY, Lim YT, Lim H, Hah JO, Lee JM. Successful Hematopoietic Stem Cell Transplantation from a Matched Related Donor with Beta-Thalassemia Minor for Severe Aplastic Anemia. Children. 2020; 7(10):162. https://doi.org/10.3390/children7100162
Chicago/Turabian StyleJung, Mi Young, Young Tae Lim, Hyunji Lim, Jeong Ok Hah, and Jae Min Lee. 2020. "Successful Hematopoietic Stem Cell Transplantation from a Matched Related Donor with Beta-Thalassemia Minor for Severe Aplastic Anemia" Children 7, no. 10: 162. https://doi.org/10.3390/children7100162
APA StyleJung, M. Y., Lim, Y. T., Lim, H., Hah, J. O., & Lee, J. M. (2020). Successful Hematopoietic Stem Cell Transplantation from a Matched Related Donor with Beta-Thalassemia Minor for Severe Aplastic Anemia. Children, 7(10), 162. https://doi.org/10.3390/children7100162