Pediatric Safety of Polysorbates in Drug Formulations

Abstract
:1. Introduction
2. Materials and Methods
Pediatric Safety Factor
3. Results and Discussion
3.1. Potential Toxicity and Hypersensitivity in Adults Due to PS20/80: A Dosing Issue?
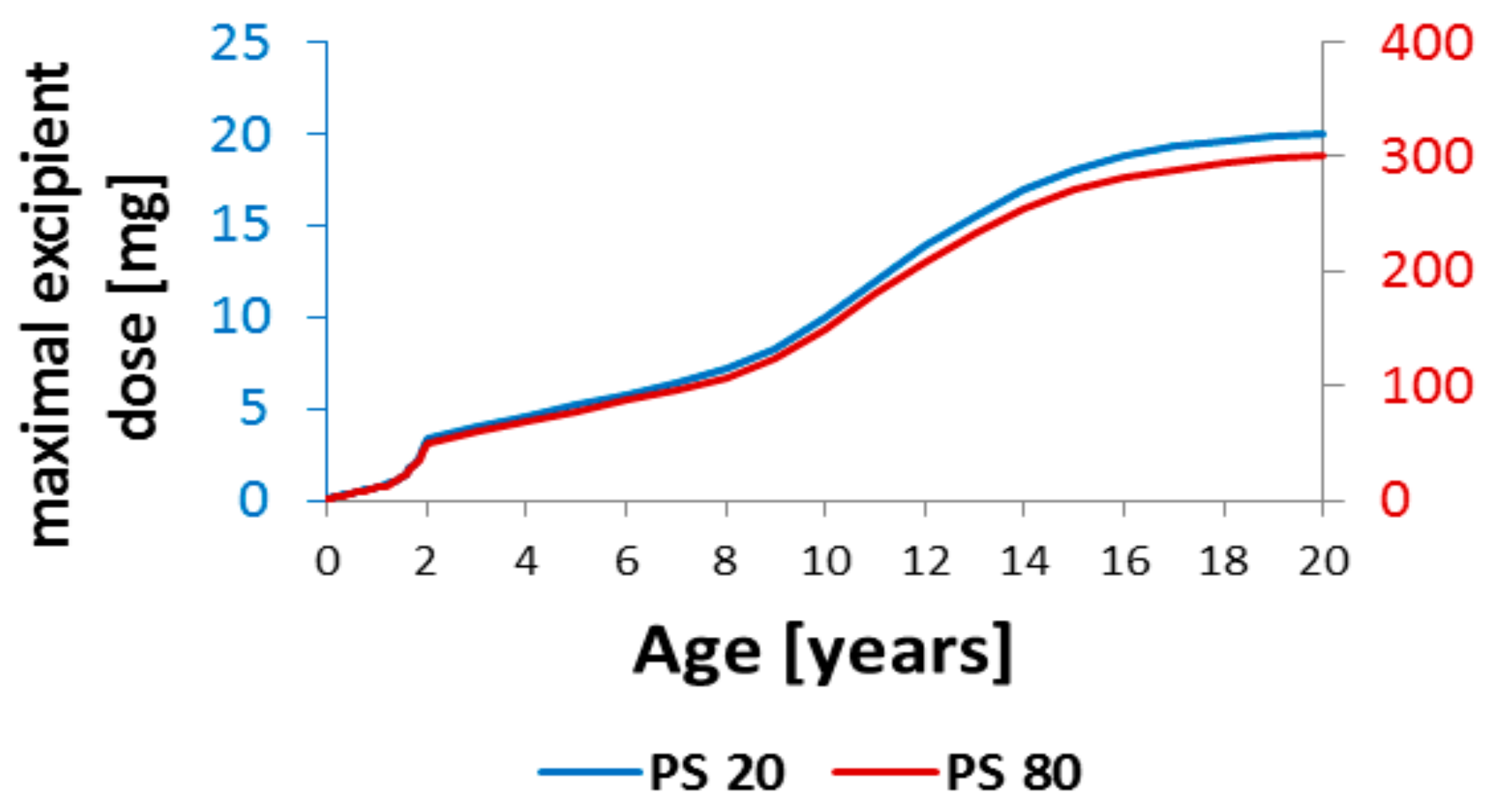
3.2. Pediatric Exposure to PS80 and PS20
3.3. Regulation of PS Use in the Pediatric Population
3.4. The Progressive Pediatric Safety Factor
3.5. Practical Considerations for the PPSF
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jones, M.T.; Mahler, H.C.; Yadav, S.; Bindra, D.; Corvari, V.; Fesinmeyer, R.M.; Gupta, K.; Harmon, A.M.; Hinds, K.D.; Koulov, A.; et al. Considerations for the Use of Polysorbates in Biopharmaceuticals. Pharm. Res. 2018, 35, 148. [Google Scholar] [CrossRef] [PubMed]
- Information for the Package Leaflet Regarding Polysorbates Used as Excipients in Medicinal Products for Human Use; European Medicines Agency, Committee for Medicinal Products for Human Use: London, UK, 2018; pp. 1–42.
- van Tellingen, O.; Beijnen, J.H.; Verweij, J.; Scherrenburg, E.J.; Nooijen, W.J.; Sparreboom, A. Rapid esterase-sensitive breakdown of polysorbate 80 and its impact on the plasma pharmacokinetics of docetaxel and metabolites in mice. Clin. Cancer Res. 1999, 5, 2918–2924. [Google Scholar] [PubMed]
- Moore, J. Final Report on the Safety Assessment of Polysorbates 20, 21, 40, 60, 61, 65, 80, 81, and 85. J. Am. Coll. Toxicol. 1984, 3, 1–82. [Google Scholar] [CrossRef]
- Renwick, A.G.; Lazarus, N.R. Human Variability and Noncancer Risk Assessment—An Analysis of the Default Uncertainty Factor. Regul. Toxicol. Pharmacol. 1998, 27, 3–20. [Google Scholar]
- Dourson, M.; Charnley, G.; Scheuplein, R. Differential sensitivity of children and adults to chemical toxicity II. Risk and regulation. Regul. Toxicol. Pharmacol. 2002, 35, 448–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- NIH. Dailymed. Available online: https://dailymed.nlm.nih.gov/dailymed/advanced-search.cfm (accessed on 24 September 2019).
- FDA. Inactive Ingredient Search. Available online: https://www.accessdata.fda.gov (accessed on 24 September 2019).
- EFSA. Scientific Opinion on the re-evaluation of polyoxyethylene sorbitan monolaurate (E 432), polyoxyethylene sorbitan monooleate (E 433), polyoxyethylene sorbitan monopalmitate (E 434), polyoxyethylene sorbitan monostearate (E 435) and polyoxyethylene sorbitan tristearate (E 436) as food additives. EFSA J. 2015, 13, 4152. [Google Scholar] [CrossRef]
- Toxicological Evaluation of Certain Food Additives with a Review of General Principles and of Specifications: Seventeenth Report of the Joint FAO-WHO Expert Committee on Food Additives; World Health Organization Technical Report Series 539; World Health Organization & Food and Agriculture Organization of the United Nations: Geneva, Switzerland, 1974; pp. 1–40.
- Kowey, P.R.; Marinchak, R.A.; Rials, S.J.; Filart, R.A. Intravenous amiodarone. J. Am. Coll. Cardiol. 1997, 29, 1190–1198. [Google Scholar] [CrossRef] [Green Version]
- Ha, E.; Wang, W.; Wang, Y.J. Peroxide formation in polysorbate 80 and protein stability. J. Pharm. Sci. 2002, 91, 2252–2264. [Google Scholar] [CrossRef]
- Aguirre, S.A.; Gukasyan, H.J.; Younis, H.S.; Huang, W. Safety Assessment of Formulation Vehicles Following Intravitreal Administration in Rabbits. Pharm. Res. 2018, 35, 173. [Google Scholar] [CrossRef]
- Toxicology and Carcinogenesis Studies of Polysorbate 80 in F344/N Rats and B6C3F1 Mice; National Toxicology Program Technical Report Series 415; National Toxicology Program, National Institutes of Health: Research Triangle Park, NC, USA, 1992; pp. 1–225.
- Norris, L.; Qureshi, Z.; Bookstaver, B.; Raisch, D.; Sartor, O.; Chen, H.; Chen, F.; Bennett, C. Polysorbate 80 hypersensitivity reactions: A renewed call to action. Community Oncol. 2010, 7, 425–428. [Google Scholar] [CrossRef]
- Coors, E.A.; Seybold, H.; Merk, H.F.; Mahler, V. Polysorbate 80 in medical products and nonimmunologic anaphylactoid reactions. Ann. Allergy Asthma Immunol. 2005, 95, 593–599. [Google Scholar] [CrossRef]
- Shelley, W.B.; Talanin, N.; Shelley, E.D. Polysorbate 80 hypersensitivity. Lancet 1995, 345, 1312–1313. [Google Scholar] [CrossRef]
- Steele, R.H.; Limaye, S.; Cleland, B.; Chow, J.; Suranyi, M.G. Hypersensitivity reactions to the polysorbate contained in recombinant erythropoietin and darbepoietin. Nephrology (Carlton) 2005, 10, 317–320. [Google Scholar] [CrossRef] [PubMed]
- Sanofi-Aventis. Taxotere [package Insert]. Available online: http://products.sanofi.us/Docetaxel/Docetaxel.pdf (accessed on 24 September 2019).
- Sanofi-Aventis. Jevtana [package Insert]. Available online: http://products.sanofi.us/jevtana/jevtana.pdf (accessed on 24 September 2019).
- Palacios Castano, M.I.; Venturini Diaz, M.; Lobera Labairu, T.; Gonzalez Mahave, I.; Del Pozo Gil, M.D.; Blasco Sarramian, A. Anaphylaxis Due to the Excipient Polysorbate 80. J. Investig. Allergol. Clin. Immunol. 2016, 26, 394–396. [Google Scholar] [CrossRef] [Green Version]
- Weiszhar, Z.; Czucz, J.; Revesz, C.; Rosivall, L.; Szebeni, J.; Rozsnyay, Z. Complement activation by polyethoxylated pharmaceutical surfactants: Cremophor-EL, Tween-80 and Tween-20. Eur. J. Pharm. Sci. 2012, 45, 492–498. [Google Scholar] [CrossRef]
- Bove, K.E.; Kosmetatos, N.; Wedig, K.E.; Frank, D.J.; Whitlatch, S.; Saldivar, V.; Haas, J.; Bodenstein, C.; Balistreri, W.F. Vasculopathic hepatotoxicity associated with E-Ferol syndrome in low-birth-weight infants. JAMA 1985, 254, 2422–2430. [Google Scholar] [CrossRef]
- Bodenstein, C.J. Intravenous vitamin E and deaths in the intensive care unit. Pediatrics 1984, 73, 733. [Google Scholar]
- Lorch, V.; Murphy, D.; Hoersten, L.R.; Harris, E.; Fitzgerald, J.; Sinha, S.N. Unusual syndrome among premature infants: Association with a new intravenous vitamin E product. Pediatrics 1985, 75, 598–602. [Google Scholar]
- Martone, W.J.; Williams, W.W.; Mortensen, M.L.; Gaynes, R.P.; White, J.W.; Lorch, V.; Murphy, M.D.; Sinha, S.N.; Frank, D.J.; Kosmetatos, N.; et al. Illness with fatalities in premature infants: Association with an intravenous vitamin E preparation, E-Ferol. Pediatrics 1986, 78, 591–600. [Google Scholar]
- Alade, S.L.; Brown, R.E.; Paquet, A., Jr. Polysorbate 80 and E-Ferol toxicity. Pediatrics 1986, 77, 593–597. [Google Scholar]
- Sugerman, H.J.; Hirsch, J.I.; Tatum, J.L.; Strash, A.M.; Sharp, D.E.; Greenfield, L.J. Comparative scintigraphy in oleic acid pulmonary microvascular injury. Crit. Care Med. 1982, 10, 31–33. [Google Scholar] [CrossRef] [PubMed]
- Bhat, R.; Jiang, J.X.; Walsh, J.M.; Mortensen, M.L.; Vidyasagar, D.; Evans, M.A. 616 Effect of Vitamin E and Polysorbate on Bile Acid Transport in Newborn Rabbit Hepatocytes. Pediatr. Res. 1985, 19, 213. [Google Scholar] [CrossRef] [Green Version]
- Nityanand, S.; Kapoor, N.K. Effect of chronic oral administration of Tween-80 in Charles Foster rats. Indian J. Med. Res. 1979, 69, 664–670. [Google Scholar] [PubMed]
- Isaksson, M.; Jansson, L. Contact allergy to Tween 80 in an inhalation suspension. Contact Dermat. 2002, 47, 312–313. [Google Scholar] [CrossRef]
- Kicker, J.S.; Haizlip, J.A.; Buck, M.L. Hepatotoxicity after continuous amiodarone infusion in a postoperative cardiac infant. J. Pediatr. Pharmacol. Ther. 2012, 17, 189–195. [Google Scholar] [CrossRef]
- Lucente, P.; Iorizzo, M.; Pazzaglia, M. Contact sensitivity to Tween 80 in a child. Contact Dermat. 2000, 43, 172. [Google Scholar]
- Masi, S.; de Clety, S.C.; Anslot, C.; Detaille, T. Acute amiodarone toxicity due to an administration error: Could excipient be responsible? Br. J. Clin. Pharmacol. 2009, 67, 691–693. [Google Scholar] [CrossRef] [Green Version]
- de Zwart, L.L.; Haenen, H.E.; Versantvoort, C.H.; Wolterink, G.; van Engelen, J.G.; Sips, A.J. Role of biokinetics in risk assessment of drugs and chemicals in children. Regul. Toxicol. Pharmacol. 2004, 39, 282–309. [Google Scholar] [CrossRef]
- Zoetis, T.; Hurtt, M.E. Species comparison of anatomical and functional renal development. Birth defects research. Birth Defects Res. B Dev. Reprod. Toxicol. 2003, 68, 111–120. [Google Scholar] [CrossRef]
- EC. Communication from the Commission on the Precautionary Principle. Available online: http://ec.europa.eu/dgs/health_consumer/library/pub/pub07_en.pdf (accessed on 24 September 2019).
- Souza, A., Jr.; Santos, D.; Fonseca, S.; Medeiros, M.; Batista, L.; Turner, M.; Coelho, H. Toxic excipients in medications for neonates in Brazil. Eur. J. Pediatr. 2014, 173, 935–945. [Google Scholar] [CrossRef]
- Whittaker, A.; Currie, A.E.; Turner, M.A.; Field, D.J.; Mulla, H.; Pandya, H.C. Toxic additives in medication for preterm infants. Arch. Dis. Child. Fetal Neonatal Ed. 2009, 94, F236–F240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lass, J.; Naelapaa, K.; Shah, U.; Kaar, R.; Varendi, H.; Turner, M.A.; Lutsar, I. Hospitalised neonates in Estonia commonly receive potentially harmful excipients. BMC Pediatr. 2012, 12, 136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nellis, G.; Metsvaht, T.; Varendi, H.; Toompere, K.; Lass, J.; Mesek, I.; Nunn, A.J.; Turner, M.A.; Lutsar, I.; Consortium, E. Potentially harmful excipients in neonatal medicines: A pan-European observational study. Arch. Dis. Child. 2015, 100, 694–699. [Google Scholar] [CrossRef] [PubMed]
- Better Medicines for Children from Concept to Reality; Progress Report on the Paediatric Regulation (EC) No. 1901/2006; European Commission: Brussels, Belgium, 2013; pp. 1–36.
- Report on the Survey of All Paediatric Uses of Medicinal Products in Europe; European Medicines Agency: London, UK, 2010; pp. 1–37.
- Regulation (EC) No 1901/2006 of the European Parliament and of the Council of 12 December 2006 on Medicinal Products for Paediatric use and Amending Regulation (EEC) No 1768/92, Directive 2001/20/EC, Directive 2001/83/EC and Regulation (EC) No 726/2004; European Commission: Brussels, Belgium, 2006; pp. 1–31.
- Thabet, Y.; Klingmann, V.; Breitkreutz, J. Drug Formulations: Standards and Novel Strategies for Drug Administration in Pediatrics. J. Clin. Pharmacol. 2018, 58 (Suppl. 10), S26–S35. [Google Scholar] [CrossRef] [Green Version]
- FDA. Food and Drug Administration Safety and Innovation Act (FDASIA). Available online: http://www.fda.gov/RegulatoryInformation/LawsEnforcedbyFDA/SignificantAmendmentstotheFDCAct/FDASIA/default.htm (accessed on 24 September 2019).
- Buckley, L.A.; Salunke, S.; Thompson, K.; Baer, G.; Fegley, D.; Turner, M.A. Challenges and strategies to facilitate formulation development of pediatric drug products: Safety qualification of excipients. Int. J. Pharm. 2018, 536, 563–569. [Google Scholar] [CrossRef] [Green Version]
- Salunke, S.; Tuleu, C. The STEP database through the end-users eyes—USABILITY STUDY. Int. J. Pharm. 2015, 492, 316–331. [Google Scholar] [CrossRef]
- Valeur, K.S.; Hertel, S.A.; Lundstrom, K.E.; Holst, H. Safe excipient exposure in neonates and small children-protocol for the SEEN project. Dan. Med. J. 2017, 64, A5324. [Google Scholar]
- Schmitt, G. Safety of Excipients in Pediatric Formulations-A Call for Toxicity Studies in Juvenile Animals? Children (Basel) 2015, 2, 191–197. [Google Scholar] [CrossRef] [Green Version]
- Guidance for Industry-Estimating the Maximum Safe Starting Dose in Initial Clinical Trials for Therapeutics in Adult Healthy Volunteers; Food and Drug Administration: Rockville, MD, USA, 2005; pp. 1–27.
- Determination of the Appropriate FQPA Safety Factor(s) in Tolerance Assessment; Environmental Protection Agency: Washington, DC, USA, 2002; pp. 1–74.
- EFSA. Guidance on Default Assumptions Used by the EFSA Scientific Panels and Committee, and EFSA Units in the Absence of Actual Measured Data; European Food Safety Authority: Parma, Italy, 2011; pp. 1–30. [Google Scholar]
- Ivanovska, V.; Rademaker, C.M.; van Dijk, L.; Mantel-Teeuwisse, A.K. Pediatric drug formulations: A review of challenges and progress. Pediatrics 2014, 134, 361–372. [Google Scholar] [CrossRef] [Green Version]
- Nair, A.B.; Jacob, S. A simple practice guide for dose conversion between animals and human. J. Basic Clin. Pharm. 2016, 7, 27–31. [Google Scholar] [CrossRef] [Green Version]
- FDA. E11 Clinical Investigation of Medicinal Products in the Pediatric Population. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/e11-clinical-investigation-medicinal-products-pediatric-population (accessed on 24 September 2019).
- Lu, H.; Rosenbaum, S. Developmental pharmacokinetics in pediatric populations. J. Pediatr. Pharmacol. Ther. 2014, 19, 262–276. [Google Scholar] [CrossRef] [PubMed]
- Čukuranović, R.; Vlajković, S. Age related anatomical and functional characteristics of human kidney. Ser. Med. Biol. 2005, 12, 61–69. [Google Scholar]
- Beath, S.V. Hepatic function and physiology in the newborn. Semin. Neonatol. 2003, 8, 337–346. [Google Scholar] [CrossRef]
- Smolinske, S.C. Handbook of Food, Drug, and Cosmetic Excipients; CRC Press: Denver, CO, USA, 1992. [Google Scholar]
- List of Criteria for Screening PIPs with Regard to Paediatric Specific Quality Issues and Referring Them to the PDCO FWG for Discussion; European Medicines Agency: London, UK, 2013; pp. 1–8.

{kind=link}
| Age Category | Week | Year | Month | Height | Weight | PPSF |
|---|---|---|---|---|---|---|
| newborn infants (0 to 27 days) | 0 | 0.00 | 0.0 months | 49.5 cm | 3.3 kg | 10.0 |
| 1 | 0.02 | 0.2 months | 50.7 cm | 3.4 kg | 9.9 | |
| 2 | 0.04 | 0.5 months | 51.9 cm | 3.7 kg | 9.6 | |
| 3 | 0.06 | 0.7 months | 52.9 cm | 3.9 kg | 9.4 | |
| 4 | 0.08 | 0.9 months | 53.9 cm | 4.2 kg | 9.0 | |
| infants and toddlers (28 days to 23 months) | 5 | 0.10 | 1.2 months | 54.8 cm | 4.5 kg | 8.7 |
| 6 | 0.12 | 1.4 months | 55.6 cm | 4.8 kg | 8.4 | |
| 7 | 0.13 | 1.6 months | 56.5 cm | 5.0 kg | 8.2 | |
| 8 | 0.15 | 1.8 months | 57.2 cm | 5.2 kg | 8.0 | |
| 9 | 0.17 | 2.1 months | 58.0 cm | 5.4 kg | 7.8 | |
| 10 | 0.19 | 2.3 months | 58.7 cm | 5.6 kg | 7.6 | |
| 11 | 0.21 | 2.5 months | 59.3 cm | 5.8 kg | 7.4 | |
| 12 | 0.23 | 2.8 months | 60.0 cm | 5.9 kg | 7.2 | |
| 13 | 0.25 | 3.0 months | 60.6 cm | 6.1 kg | 7.0 | |
| 0.33 | 4 months | 63.0 cm | 6.7 kg | 6.4 | ||
| 0.42 | 5 months | 65.0 cm | 7.2 kg | 5.9 | ||
| 0.50 | 6 months | 66.7 cm | 7.6 kg | 5.4 | ||
| 0.58 | 7 months | 68.2 cm | 8.0 kg | 5.0 | ||
| 0.67 | 8 months | 69.7 cm | 8.3 kg | 4.7 | ||
| 0.75 | 9 months | 71.1 cm | 8.6 kg | 4.4 | ||
| 0.83 | 10 months | 72.4 cm | 8.8 kg | 4.2 | ||
| 0.92 | 11 months | 73.7 cm | 9.1 kg | 3.9 | ||
| 1.00 | 12 months | 74.9 cm | 9.3 kg | 3.6 | ||
| 1.08 | 13 months | 76.1 cm | 9.5 kg | 3.4 | ||
| 1.17 | 14 months | 77.2 cm | 9.7 kg | 3.2 | ||
| 1.25 | 15 months | 78.3 cm | 10.0 kg | 2.9 | ||
| 1.33 | 16 months | 79.4 cm | 10.2 kg | 2.7 | ||
| 1.42 | 17 months | 80.5 cm | 10.4 kg | 2.5 | ||
| 1.50 | 18 months | 81.5 cm | 10.6 kg | 2.3 | ||
| 1.58 | 19 months | 82.5 cm | 10.8 kg | 2.1 | ||
| 1.67 | 20 months | 83.5 cm | 11.0 kg | 1.8 | ||
| 1.75 | 21 months | 84.4 cm | 11.2 kg | 1.6 | ||
| 1.83 | 22 months | 85.3 cm | 11.4 kg | 1.4 | ||
| 1.92 | 23 months | 86.2 cm | 11.6 kg | 1.2 | ||
| children | 2 | 24 months | 86.4 cm | 11.8 kg | 1.0 |
| Age Category | Week | Year | PPSF | Polysorbate 20 | Polysorbate 80 |
|---|---|---|---|---|---|
| newborn infants (0 to 27 days) | 0 | 0.00 | 10.0 | 0.09 mg | 1.4 mg |
| 1 | 0.02 | 9.9 | 0.10 mg | 1.5 mg | |
| 2 | 0.04 | 9.6 | 0.11 mg | 1.7 mg | |
| 3 | 0.06 | 9.4 | 0.12 mg | 1.8 mg | |
| 4 | 0.08 | 9.0 | 0.13 mg | 2.0 mg | |
| infants and toddlers (28 days to 23 months) | 5 | 0.10 | 8.7 | 0.15 mg | 2.2 mg |
| 6 | 0.12 | 8.4 | 0.16 mg | 2.4 mg | |
| 7 | 0.13 | 8.2 | 0.17 mg | 2.6 mg | |
| 8 | 0.15 | 8.0 | 0.19 mg | 2.8 mg | |
| 9 | 0.17 | 7.8 | 0.20 mg | 3.0 mg | |
| 10 | 0.19 | 7.6 | 0.21 mg | 3.2 mg | |
| 11 | 0.21 | 7.4 | 0.22 mg | 3.4 mg | |
| 12 | 0.23 | 7.2 | 0.23 mg | 3.5 mg | |
| 13 | 0.25 | 7.0 | 0.25 mg | 3.7 mg | |
| 0.33 | 6.4 | 0.30 mg | 4.5 mg | ||
| 0.42 | 5.9 | 0.35 mg | 5.2 mg | ||
| 0.50 | 5.4 | 0.40 mg | 6.0 mg | ||
| 0.58 | 5.0 | 0.46 mg | 6.9 mg | ||
| 0.67 | 4.7 | 0.50 mg | 7.6 mg | ||
| 0.75 | 4.4 | 0.56 mg | 8.4 mg | ||
| 0.83 | 4.2 | 0.60 mg | 9.0 mg | ||
| 0.92 | 3.9 | 0.67 mg | 10.0 mg | ||
| 1.00 | 3.6 | 0.74 mg | 11.1 mg | ||
| 1.08 | 3.4 | 0.80 mg | 12.0 mg | ||
| 1.17 | 3.2 | 0.87 mg | 13.0 mg | ||
| 1.25 | 2.9 | 0.99 mg | 14.8 mg | ||
| 1.33 | 2.7 | 1.08 mg | 16.2 mg | ||
| 1.42 | 2.5 | 1.19 mg | 17.8 mg | ||
| 1.50 | 2.3 | 1.32 mg | 19.8 mg | ||
| 1.58 | 2.1 | 1.47 mg | 22.0 mg | ||
| 1.67 | 1.8 | 1.75 mg | 26.2 mg | ||
| 1.75 | 1.6 | 2.00 mg | 30.0 mg | ||
| 1.83 | 1.4 | 2.33 mg | 34.9 mg | ||
| 1.92 | 1.2 | 2.76 mg | 41.4 mg | ||
| children | 2 | 1.0 | 3.37 mg | 50.6 mg | |
| (2–11 years) | 3 | 1.0 | 4.03 mg | 60.4 mg | |
| 4 | 1.0 | 4.63 mg | 69.4 mg | ||
| 5 | 1.0 | 5.23 mg | 78.4 mg | ||
| 6 | 1.0 | 5.80 mg | 87.0 mg | ||
| 7 | 1.0 | 6.46 mg | 96.9 mg | ||
| 8 | 1.0 | 7.20 mg | 108.0 mg | ||
| 9 | 1.0 | 8.29 mg | 124.3 mg | ||
| 10 | 1.0 | 10.00 mg | 150.0 mg | ||
| 11 | 1.0 | 12.00 mg | 180.0 mg | ||
| adolescents | 12 | 1.0 | 13.94 mg | 209.1 mg | |
| (12–18 years) | 13 | 1.0 | 15.51 mg | 232.7 mg | |
| 14 | 1.0 | 17.00 mg | 255.0 mg | ||
| 15 | 1.0 | 18.06 mg | 270.9 mg | ||
| 16 | 1.0 | 18.83 mg | 282.4 mg | ||
| 17 | 1.0 | 19.29 mg | 289.3 mg | ||
| 18 | 1.0 | 19.57 mg | 293.6 mg | ||
| adults | 19 | 1.0 | 19.86 mg | 297.9 mg | |
| 20 | 1.0 | 20 mg | 300 mg |
| PS20 | PS80 | |
|---|---|---|
| NOAEL | 8 mg | 72 mg |
| PDCO | 9.9 mg | 9.9 mg |
| PPSF * | 0.09 mg | 1.4 mg |
| Age | PS20 | PS80 |
|---|---|---|
| 28 days | 0.13 mg | 2.0 mg |
| 6 months | 0.4 mg | 6.0 mg |
| 12 months | 0.74 mg | 11.1 mg |
| 24 months | 3.37 mg | 50.6 mg |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kriegel, C.; Festag, M.; Kishore, R.S.K.; Roethlisberger, D.; Schmitt, G. Pediatric Safety of Polysorbates in Drug Formulations. Children 2020, 7, 1. https://doi.org/10.3390/children7010001
Kriegel C, Festag M, Kishore RSK, Roethlisberger D, Schmitt G. Pediatric Safety of Polysorbates in Drug Formulations. Children. 2020; 7(1):1. https://doi.org/10.3390/children7010001
Chicago/Turabian StyleKriegel, Christina, Matthias Festag, Ravuri S.K. Kishore, Dieter Roethlisberger, and Georg Schmitt. 2020. "Pediatric Safety of Polysorbates in Drug Formulations" Children 7, no. 1: 1. https://doi.org/10.3390/children7010001
APA StyleKriegel, C., Festag, M., Kishore, R. S. K., Roethlisberger, D., & Schmitt, G. (2020). Pediatric Safety of Polysorbates in Drug Formulations. Children, 7(1), 1. https://doi.org/10.3390/children7010001