Diagnostic Approach to Pulmonary Hypertension in Premature Neonates


Abstract
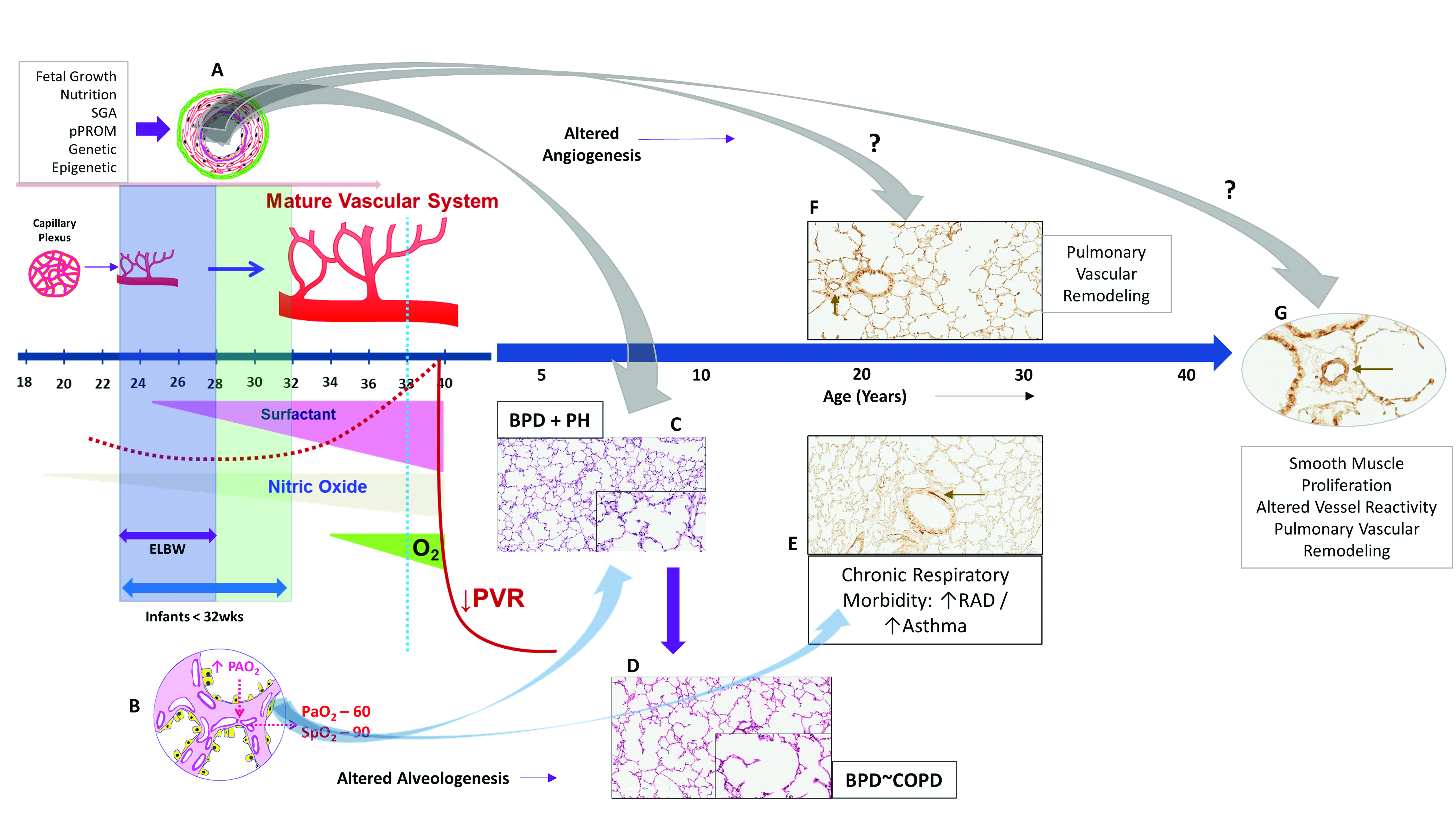
:1. Introduction
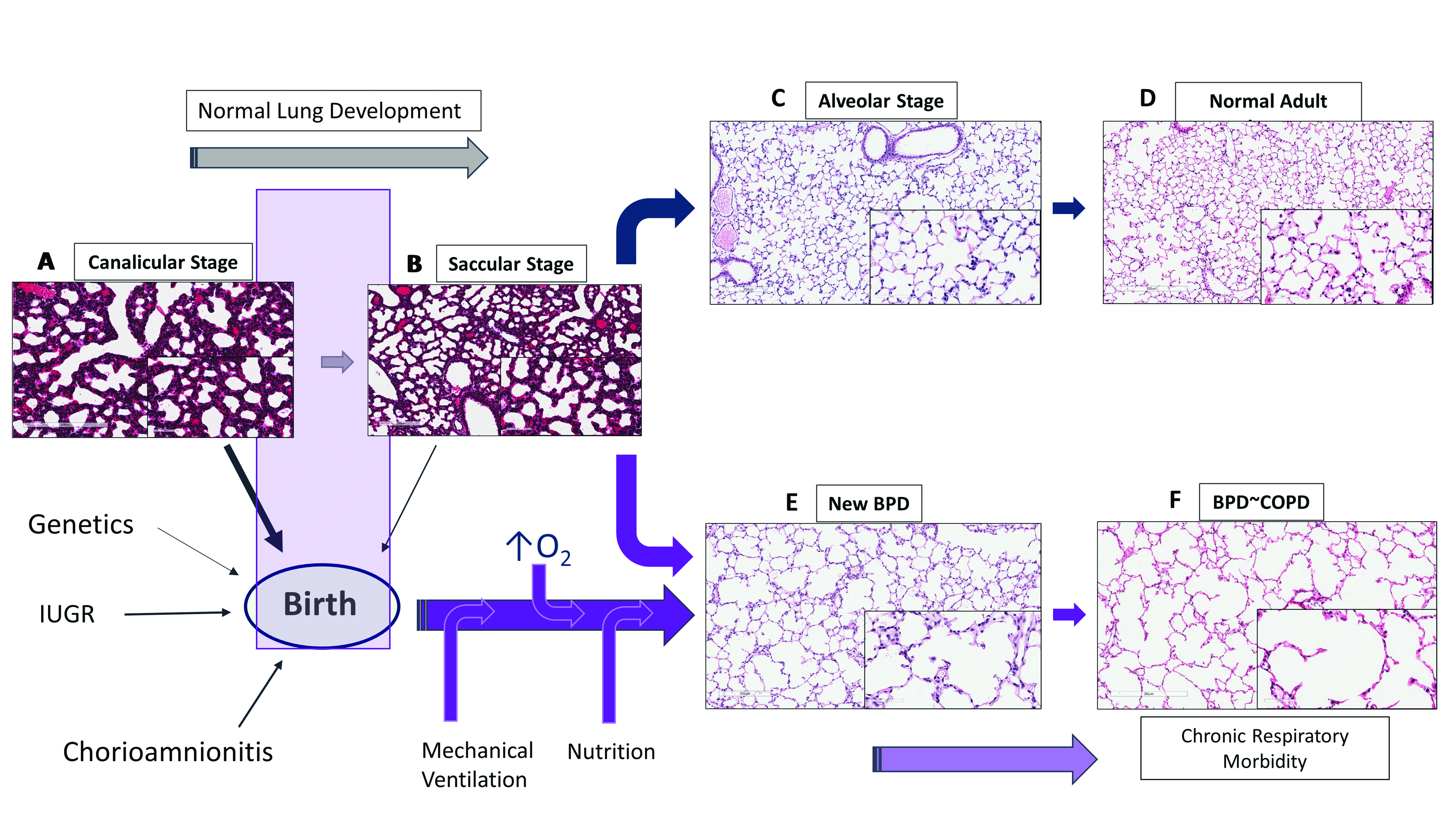
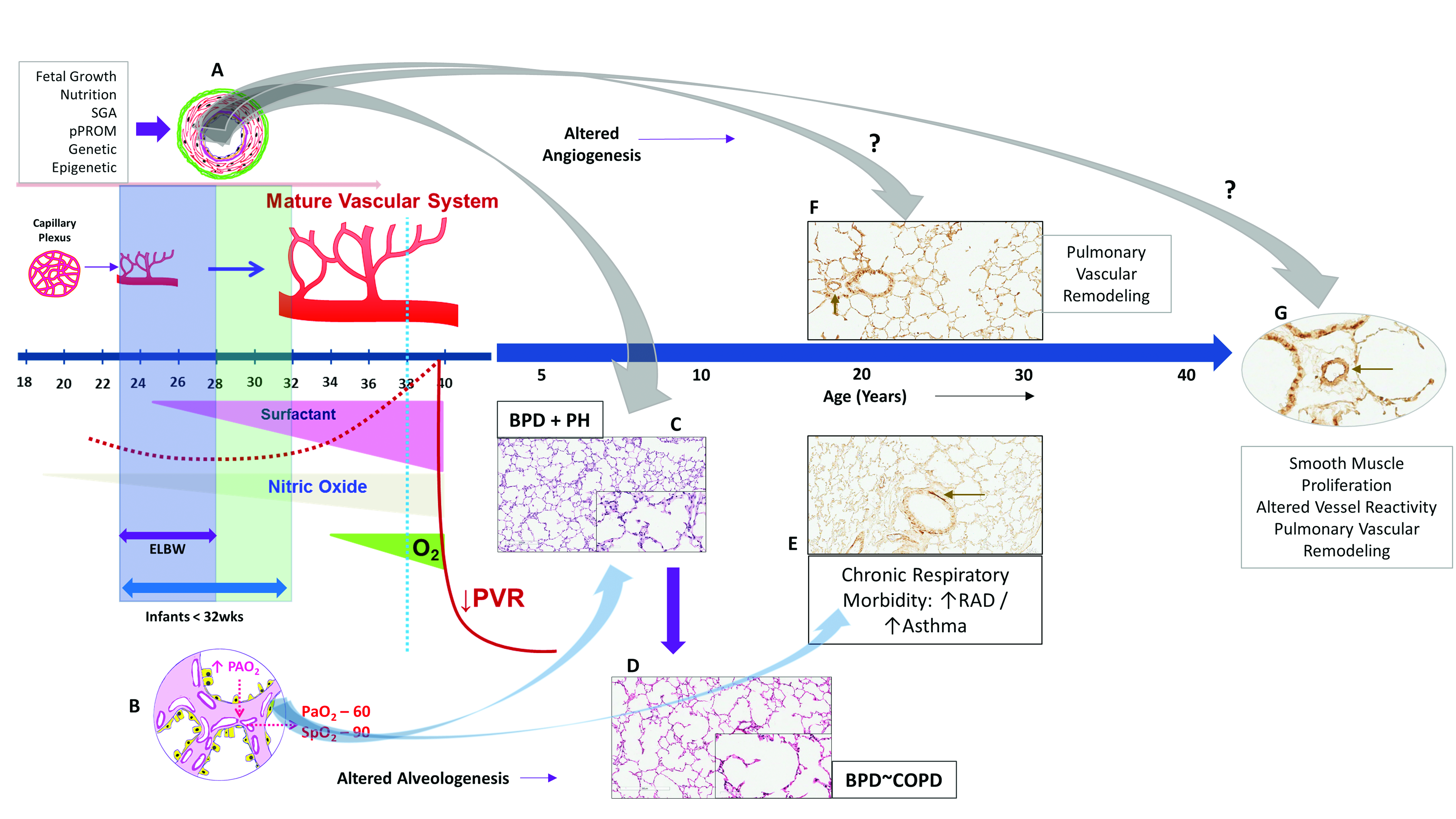
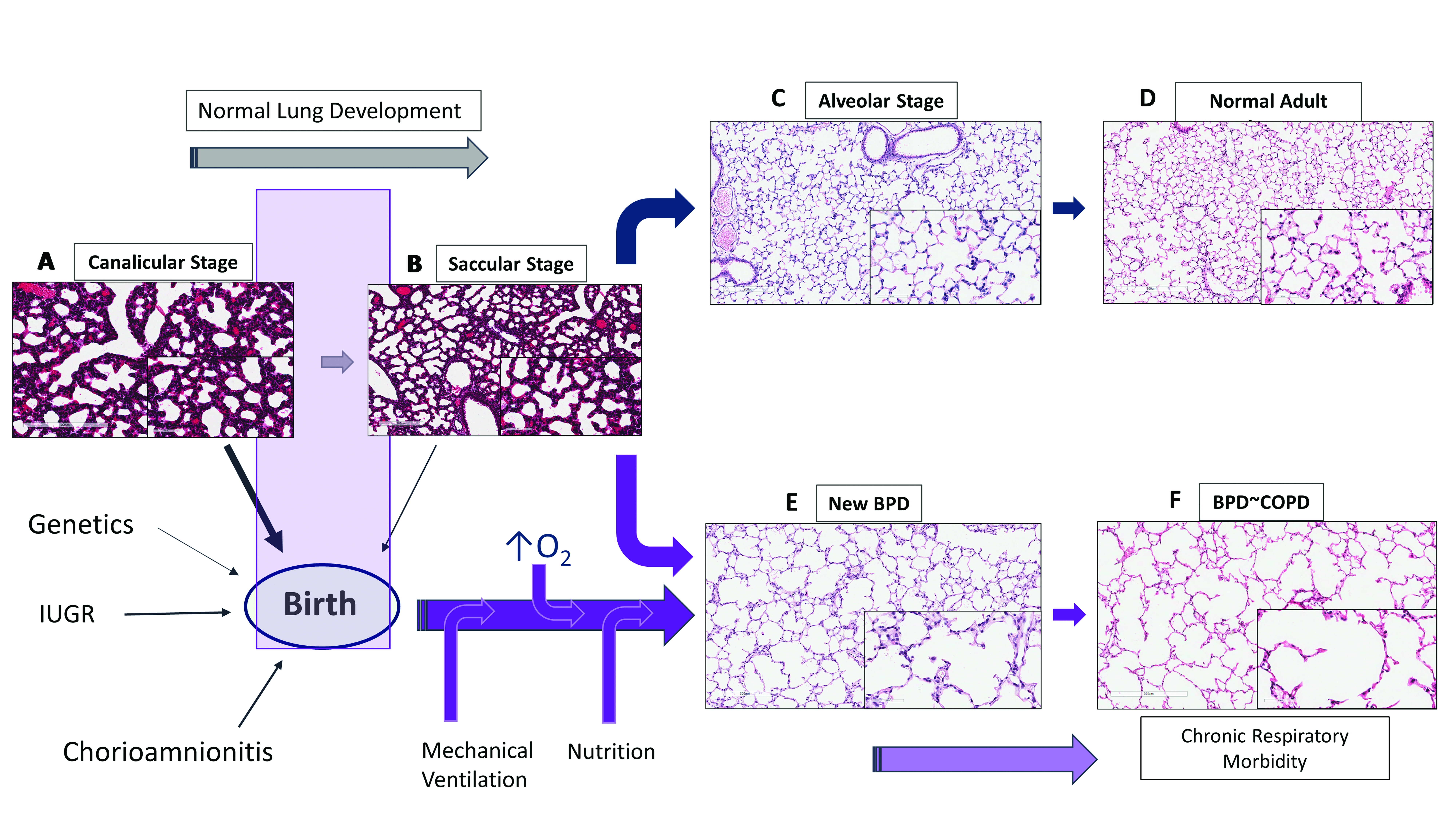
1.1. Lung Development
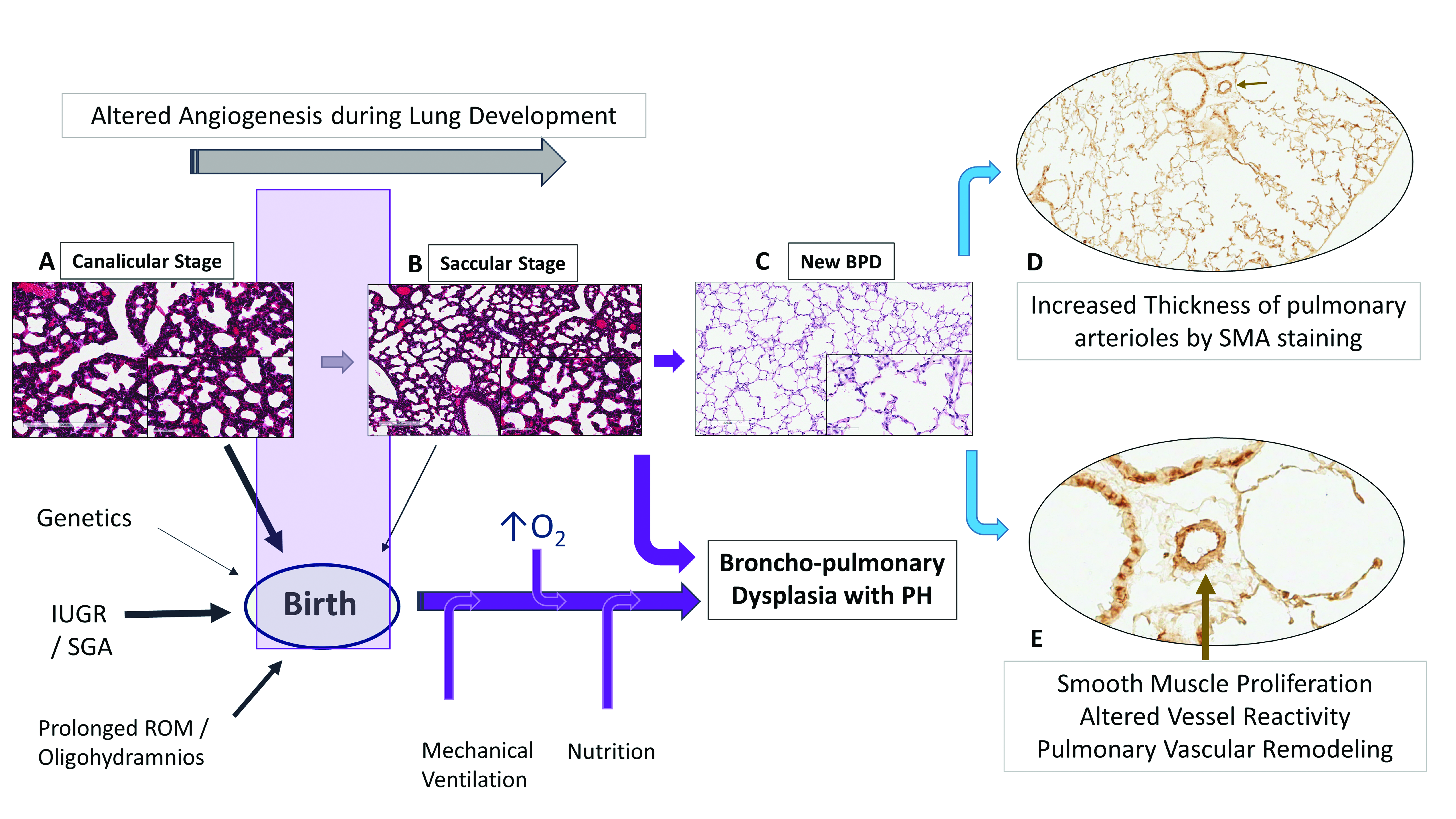
1.2. Lung Development and Angiogenesis
1.3. Vascular Dysplasia in BPD
2. Bronchopulmonary Dysplasia and Pulmonary Hypertension
2.1. Hyperoxia, Oxygen Saturations, BPD and PH
2.2. Small for Gestational Age Infant, BPD and PH
2.3. Vessel Reactivity, BPD and PH
2.4. B-Type Natriuretic Peptide (BNP)
2.5. PH and Cardiovascular Anomalies
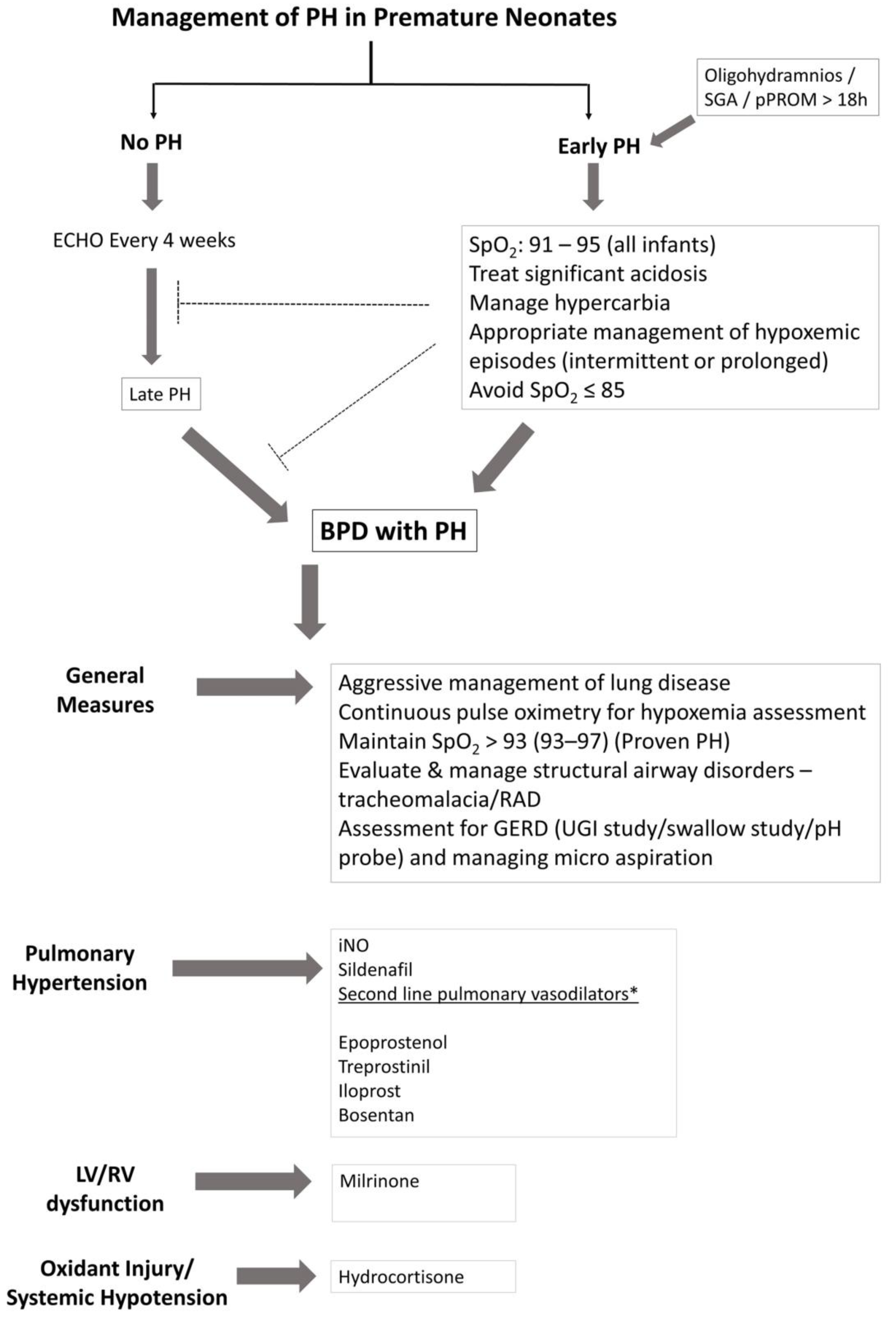
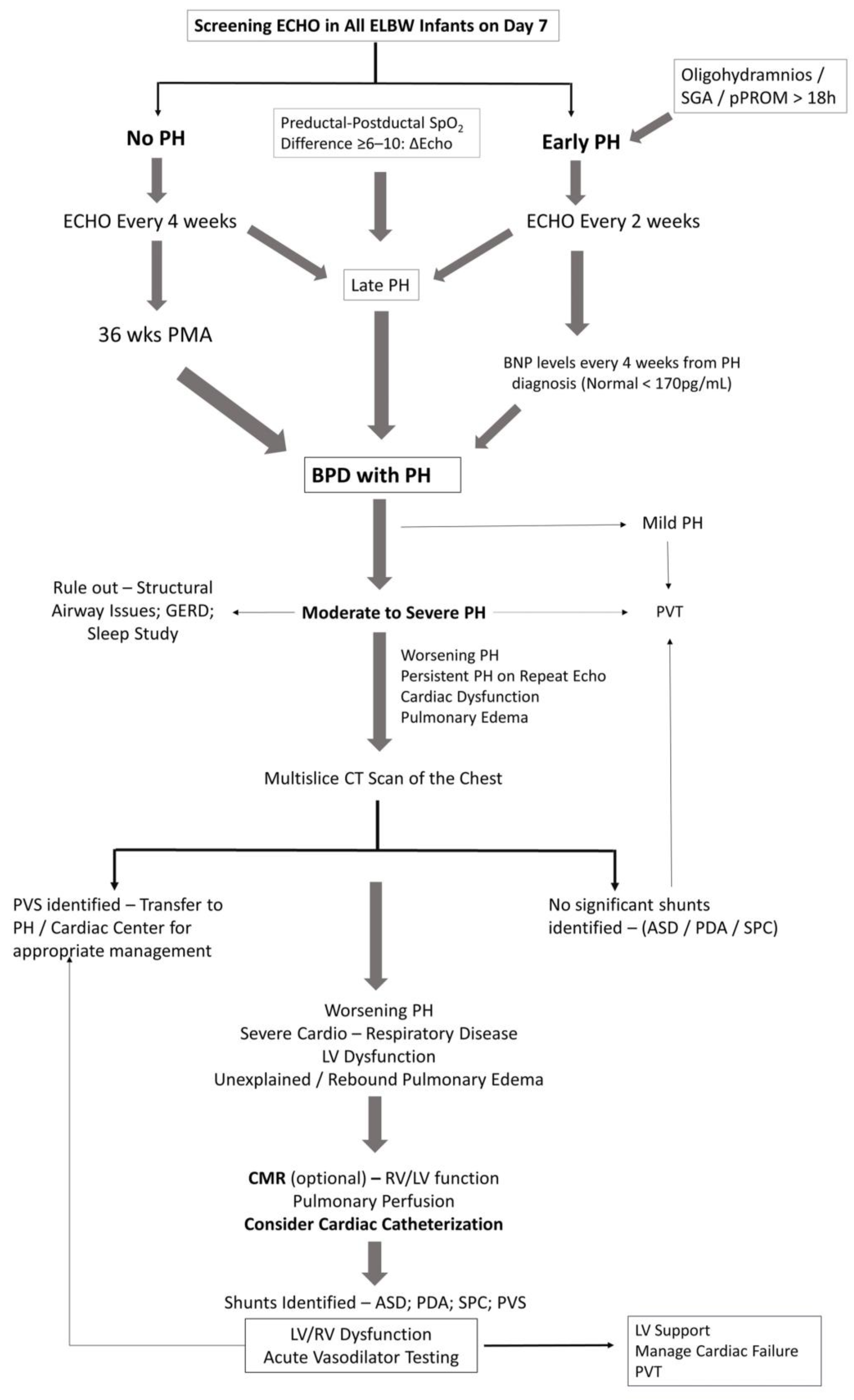
3. Diagnostic Approach to BPD with PH
3.1. General Work-Up
3.2. Echocardiography
3.3. Magnetic Resonance, CT and PH
3.4. Cardiac Catheterization
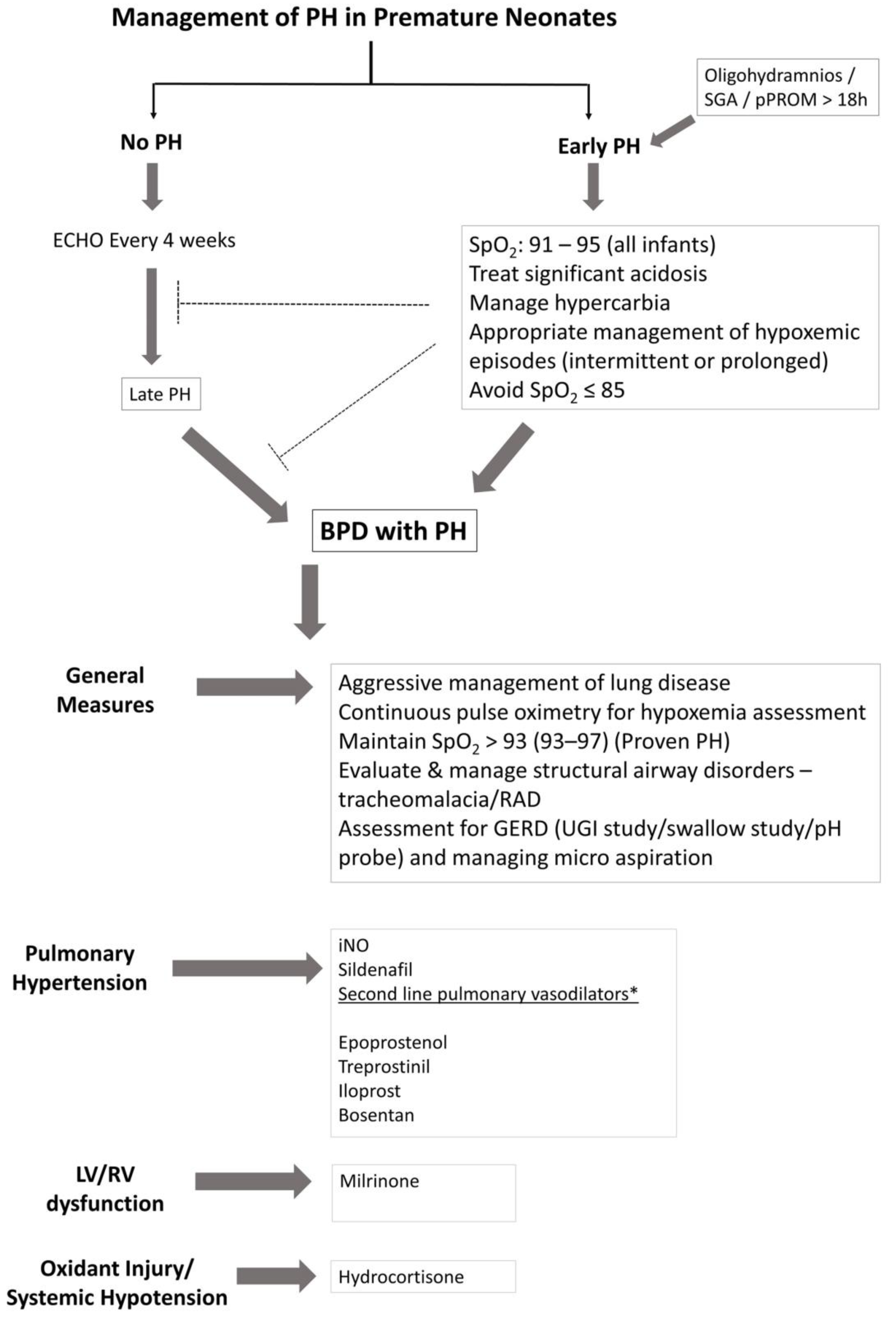
4. Management of Infants with PH
4.1. General Measures
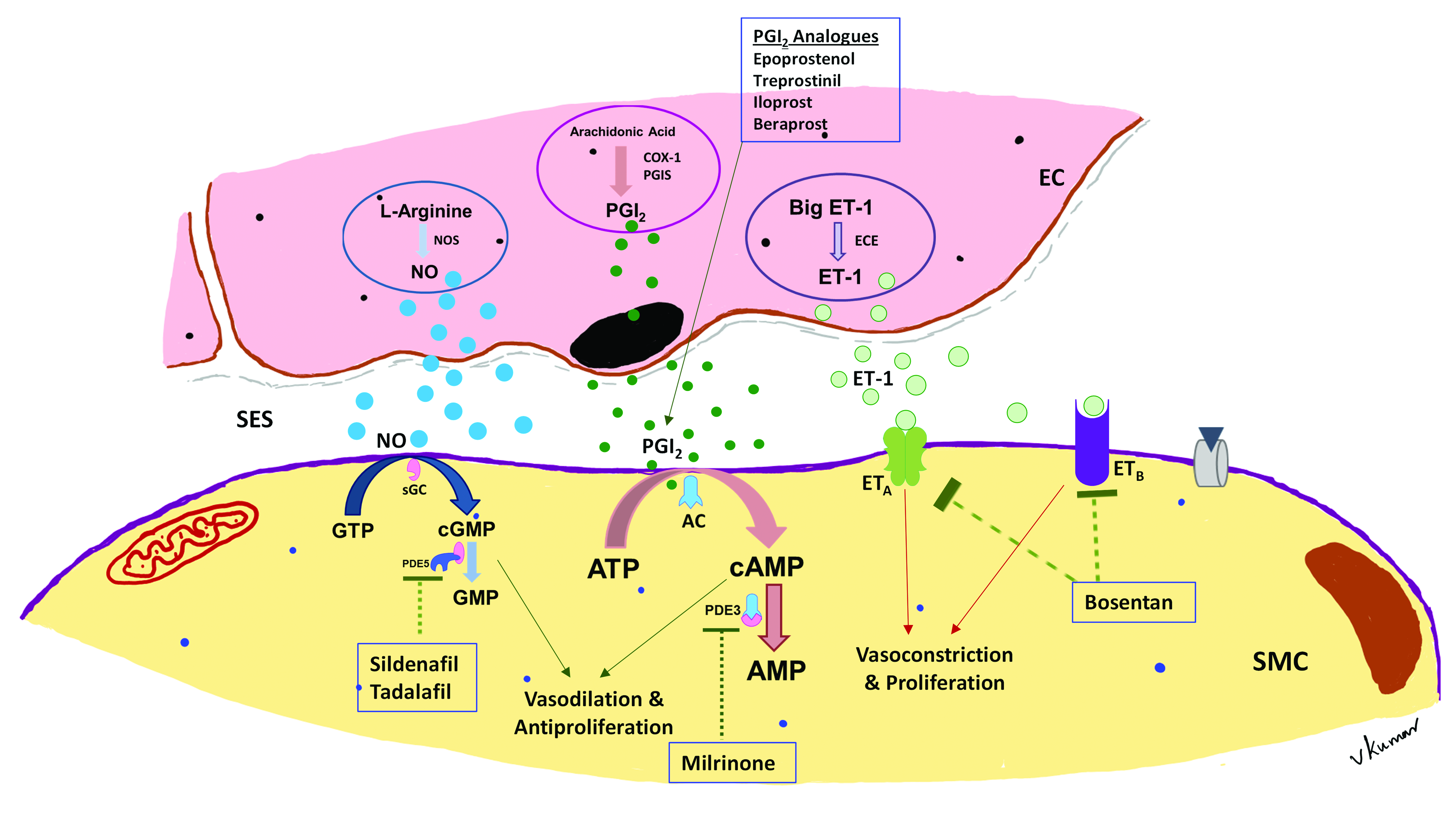
4.2. Inhaled Nitric Oxide
4.3. Sildenafil
4.4. Prostacyclin Analogues
4.5. Endothelin Antagonists
4.6. Hydrocortisone
4.7. Milrinone
4.8. Combination Therapy
4.9. Future Directions
Acknowledgments
Conflicts of Interest
References
- Blencowe, H.; Cousens, S.; Chou, D.; Oestergaard, M.; Say, L.; Moller, A.B.; Kinney, M.; Lawn, J. Born too soon: The global epidemiology of 15 million preterm births. Reprod. Health 2013, 10, S2. [Google Scholar] [CrossRef] [PubMed]
- Bokodi, G.; Treszl, A.; Kovacs, L.; Tulassay, T.; Vasarhelyi, B. Dysplasia: A review. Pediatr. Pulmonol. 2007, 42, 952–961. [Google Scholar] [CrossRef] [PubMed]
- Jobe, A.H. The new bronchopulmonary dysplasia. Curr. Opin. Pediatr. 2011, 23, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Rocha, G. Chorioamnionitis and lung injury in preterm newborns. Crit. Care Res. Pract. 2013, 2013. [Google Scholar] [CrossRef] [PubMed]
- Ryan, R.M.; Ahmed, Q.; Lakshminrusimha, S. Inflammatory mediators in the immunobiology of bronchopulmonary dysplasia. Clin. Rev. Allergy Immunol. 2008, 34, 174–190. [Google Scholar] [CrossRef] [PubMed]
- Bhat, R.; Salas, A.A.; Foster, C.; Carlo, W.A.; Ambalavanan, N. Prospective analysis of pulmonary hypertension in extremely low birth weight infants. Pediatrics 2012, 129, e682–e689. [Google Scholar] [CrossRef] [PubMed]
- Check, J.; Gotteiner, N.; Liu, X.; Su, E.; Porta, N.; Steinhorn, R.; Mestan, K.K. Fetal growth restriction and pulmonary hypertension in premature infants with bronchopulmonary dysplasia. J. Perinatol. 2013, 33, 553–557. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.H.; Lakshminrusimha, S.; El Abiad, M.T.; Chess, P.R.; Ryan, R.M. Growth factors in lung development. Adv. Clin. Chem. 2005, 40, 261–316. [Google Scholar] [PubMed]
- Gortner, L.; Reiss, I.; Hilgendorff, A. Bronchopulmonary dysplasia and intrauterine growth restriction. Lancet 2006, 368, 28. [Google Scholar] [CrossRef]
- Thebaud, B.; Ladha, F.; Michelakis, E.D.; Sawicka, M.; Thurston, G.; Eaton, F.; Hashimoto, K.; Harry, G.; Haromy, A.; Korbutt, G. Vascular endothelial growth factor gene therapy increases survival, promotes lung angiogenesis, and prevents alveolar damage in hyperoxia-induced lung injury: Evidence that angiogenesis participates in alveolarization. Circulation 2005, 112, 2477–2486. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, A.J.; Pryhuber, G.S.; Huyck, H.; Watkins, R.H.; Metlay, L.A.; Maniscalco, W.M. Disrupted pulmonary vasculature and decreased vascular endothelial growth factor, Flt-1, and TIE-2 in human infants dying with bronchopulmonary dysplasia. Am. J. Respir. Crit. Care Med. 2001, 164, 1971–1980. [Google Scholar] [CrossRef] [PubMed]
- Healy, A.M.; Morgenthau, L.; Zhu, X.; Farber, H.W.; Cardoso, W.V. VEGF is deposited in the subepithelial matrix at the leading edge of branching airways and stimulates neovascularization in the murine embryonic lung. Dev. Dyn. 2000, 219, 341–352. [Google Scholar] [CrossRef]
- Ng, Y.S.; Rohan, R.; Sunday, M.E.; Demello, D.E.; D’Amore, P.A. Differential expression of VEGF isoforms in mouse during development and in the adult. Dev. Dyn. 2001, 220, 112–121. [Google Scholar] [CrossRef]
- Gebb, S.A.; Shannon, J.M. Tissue interactions mediate early events in pulmonary vasculogenesis. Dev. Dyn. 2000, 217, 159–169. [Google Scholar] [CrossRef]
- Coalson, J.J.; Winter, V.T.; Siler-Khodr, T.; Yoder, B.A. Neonatal chronic lung disease in extremely immature baboons. Am. J. Respir. Crit. Care Med. 1999, 160, 1333–1346. [Google Scholar] [CrossRef] [PubMed]
- Maniscalco, W.M.; Watkins, R.H.; D’Angio, C.T.; Ryan, R.M. Hyperoxic injury decreases alveolar epithelial cell expression of vascular endothelial growth factor (VEGF) in neonatal rabbit lung. Am. J. Respir. Cell Mol. Biol. 1997, 16, 557–567. [Google Scholar] [CrossRef] [PubMed]
- Rozance, P.J.; Seedorf, G.J.; Brown, A.; Roe, G.; O’Meara, M.C.; Gien, J.; Tang, J.R.; Abman, S.H. Intrauterine growth restriction decreases pulmonary alveolar and vessel growth and causes pulmonary artery endothelial cell dysfunction in vitro in fetal sheep. Am. J. Physiol. Lung Cell. Mol. Physiol. 2011, 301, L860–L871. [Google Scholar] [CrossRef] [PubMed]
- Walsh, M.C.; Szefler, S.; Davis, J.; Allen, M.; van Marter, L.; Abman, S.; Blackmon, L.; Jobe, A. Summary proceedings from the bronchopulmonary dysplasia group. Pediatrics 2006, 117, S52–S56. [Google Scholar] [CrossRef] [PubMed]
- Al-Ghanem, G.; Shah, P.; Thomas, S.; Banfield, L.; El Helou, S.; Fusch, C.; Mukerji, A. Bronchopulmonary dysplasia and pulmonary hypertension: A meta-analysis. J. Perinatol. 2017, 37, 414–419. [Google Scholar] [CrossRef] [PubMed]
- Maron, B.A.; Abman, S.H. Translational Advances in the Field of Pulmonary Hypertension. Focusing on Developmental Origins and Disease Inception for the Prevention of Pulmonary Hypertension. Am. J. Respir. Crit. Care Med. 2017, 195, 292–301. [Google Scholar] [PubMed]
- Chandrasekharan, P.; Kozielski, R.; Kumar, V.H.; Rawat, M.; Manja, V.; Ma, C.; Lakshminrusimha, S. Early Use of Inhaled Nitric Oxide in Preterm Infants: Is there a Rationale for Selective Approach? Am. J. Perinatol. 2017, 34, 428–440. [Google Scholar] [PubMed]
- Kumar, V.H.; Hutchison, A.A.; Lakshminrusimha, S.; Morin, F.C., 3rd; Wynn, R.J.; Ryan, R.M. Characteristics of pulmonary hypertension in preterm neonates. J. Perinatol. 2007, 27, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Steinhorn, R.H. Diagnosis and treatment of pulmonary hypertension in infancy. Early Hum. Dev. 2013, 89, 865–874. [Google Scholar] [CrossRef] [PubMed]
- Woldesenbet, M.; Perlman, J.M. Histologic chorioamnionitis: An occult marker of severe pulmonary hypertension in the term newborn. J. Perinatol. 2005, 25, 189–192. [Google Scholar] [CrossRef] [PubMed]
- Kabra, N.S.; Kluckow, M.R.; Powell, J. Nitric oxide in preterm infant with pulmonary hypoplasia. Indian J. Pediatr. 2004, 71, 427–429. [Google Scholar] [CrossRef]
- Kilbride, H.W.; Thibeault, D.W. Neonatal complications of preterm premature rupture of membranes. Pathophysiology and management. Clin. Perinatol. 2001, 28, 761–785. [Google Scholar] [CrossRef]
- Tiktinsky, M.H.; Morin, F.C., 3rd. Increasing oxygen tension dilates fetal pulmonary circulation via endothelium-derived relaxing factor. Am. J. Physiol. 1993, 265, H376–H380. [Google Scholar] [PubMed]
- Kumar, V.H.; Lakshminrusimha, S.; Kishkurno, S.; Paturi, B.S.; Gugino, S.F.; Nielsen, L.; Wang, H.; Ryan, R.M. Neonatal hyperoxia increases airway reactivity and inflammation in adult mice. Pediatr. Pulmonol. 2016, 51, 1131–1141. [Google Scholar] [CrossRef] [PubMed]
- Veness-Meehan, K.A.; Bottone, F.G., Jr.; Stiles, A.D. Effects of retinoic acid on airspace development and lung collagen in hyperoxia-exposed newborn rats. Pediatr. Res. 2000, 48, 434–444. [Google Scholar] [CrossRef] [PubMed]
- Warner, B.B.; Stuart, L.A.; Papes, R.A.; Wispe, J.R. Functional and pathological effects of prolonged hyperoxia in neonatal mice. Am. J. Physiol. 1998, 275, L110–L117. [Google Scholar] [PubMed]
- Supplemental Therapeutic Oxygen for Prethreshold Retinopathy Of Prematurity (STOP-ROP), a randomized, controlled trial. I: primary outcomes. Pediatrics 2000, 105, 295–310.
- Northway, W.H., Jr.; Rosan, R.C.; Porter, D.Y. Pulmonary disease following respirator therapy of hyaline-membrane disease. Bronchopulmonary dysplasia. N. Engl. J. Med. 1967, 276, 357–368. [Google Scholar] [CrossRef] [PubMed]
- Shaffer, S.G.; O’Neill, D.; Bradt, S.K.; Thibeault, D.W. Chronic vascular pulmonary dysplasia associated with neonatal hyperoxia exposure in the rat. Pediatr. Res. 1987, 21, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Goss, K.N.; Kumari, S.; Tetri, L.H.; Barton, G.; Braun, R.K.; Hacker, T.A.; Eldridge, M.W. Postnatal Hyperoxia Exposure Durably Impairs Right Ventricular Function and Mitochondrial Biogenesis. Am. J. Respir. Cell Mol. Biol. 2017, 56, 609–619. [Google Scholar] [CrossRef] [PubMed]
- BOOST-II Australia and United Kingdom Collaborative Groups; Tarnow-Mordi, W.; Stenson, B.; Kirby, A.; Juszczak, E.; Donoghoe, M.; Deshpande, S.; Morley, C.; King, A.; et al. Outcomes of Two Trials of Oxygen-Saturation Targets in Preterm Infants. N. Engl. J. Med. 2016, 374, 749–760. [Google Scholar]
- Carlo, W.A.; Finer, N.N.; Walsh, M.C.; Rich, W.; Gantz, M.G.; Laptook, A.R.; Yoder, B.A.; Faix, R.G.; Das, A.; Poole, W.K.; et al. Target ranges of oxygen saturation in extremely preterm infants. N. Engl. J. Med. 2010, 362, 1959–1969. [Google Scholar] [PubMed]
- Group BIUKC; Group BIAC; Group BINZC; Stenson, B.J.; Tarnow-Mordi, W.O.; Darlow, B.A.; Simes, J.; Juszczak, E.; Askie, L.; Battin, M.; et al. Oxygen saturation and outcomes in preterm infants. N. Engl. J. Med. 2013, 368, 2094–2104. [Google Scholar] [PubMed]
- Schmidt, B.; Whyte, R.K.; Asztalos, E.V.; Moddemann, D.; Poets, C.; Rabi, Y.; Solimano, A.; Roberts, R.S.; Canadian Oxygen Trial Group. Effects of targeting higher vs. lower arterial oxygen saturations on death or disability in extremely preterm infants: A randomized clinical trial. JAMA 2013, 309, 2111–2120. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.J.; Polin, R.A.; Committee on Fetus and Newborn. Oxygen Targeting in Extremely Low Birth Weight Infants. Pediatrics 2016, 138, e20161576. [Google Scholar] [CrossRef] [PubMed]
- Reiss, I.; Landmann, E.; Heckmann, M.; Misselwitz, B.; Gortner, L. Increased risk of bronchopulmonary dysplasia and increased mortality in very preterm infants being small for gestational age. Arch. Gynecol. Obstet. 2003, 269, 40–44. [Google Scholar] [CrossRef] [PubMed]
- Sartori, C.; Allemann, Y.; Trueb, L.; Delabays, A.; Nicod, P.; Scherrer, U. Augmented vasoreactivity in adult life associated with perinatal vascular insult. Lancet 1999, 353, 2205–2207. [Google Scholar] [CrossRef]
- Xu, X.F.; Lv, Y.; Gu, W.Z.; Tang, L.L.; Wei, J.K.; Zhang, L.Y.; Du, L.Z. Epigenetics of hypoxic pulmonary arterial hypertension following intrauterine growth retardation rat: Epigenetics in PAH following IUGR. Respir. Res. 2013, 14, 20. [Google Scholar] [CrossRef] [PubMed]
- Walsh, M.C.; Di Fiore, J.M.; Martin, R.J.; Gantz, M.; Carlo, W.A.; Finer, N. Association of Oxygen Target and Growth Status With Increased Mortality in Small for Gestational Age Infants: Further Analysis of the Surfactant, Positive Pressure and Pulse Oximetry Randomized Trial. JAMA Pediatr. 2016, 170, 292–294. [Google Scholar] [CrossRef] [PubMed]
- Elberson, V.D.; Nielsen, L.C.; Wang, H.; Kumar, H.S. Effects of intermittent hypoxia and hyperoxia on angiogenesis and lung development in newborn mice. J. Neonatal Perinat. Med. 2015, 8, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Dumas de la Roque, E.; Smeralda, G.; Quignard, J.F.; Freund-Michel, V.; Courtois, A.; Marthan, R.; Muller, B.; Guibert, C.; Dubois, M. Altered vasoreactivity in neonatal rats with pulmonary hypertension associated with bronchopulmonary dysplasia: Implication of both eNOS phosphorylation and calcium signaling. PLoS ONE 2017, 12, e0173044. [Google Scholar] [CrossRef] [PubMed]
- Kuhr, F.K.; Smith, K.A.; Song, M.Y.; Levitan, I.; Yuan, J.X. New mechanisms of pulmonary arterial hypertension: Role of Ca2+ signaling. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H1546–H1562. [Google Scholar] [CrossRef] [PubMed]
- Vasantha, H.S.; Kumar, H.W.; Kishkurno, S.; Paturi, B.S.; Nielsen, L.; Ryan, R.M. Long-term effects of neonatal hyperoxia in adult mice the anatomical records. Unpublished.
- Thibeault, D.W.; Truog, W.E.; Ekekezie, II. Acinar arterial changes with chronic lung disease of prematurity in the surfactant era. Pediatr. Pulmonol. 2003, 36, 482–489. [Google Scholar] [CrossRef] [PubMed]
- Czernik, C.; Rhode, S.; Metze, B.; Schmalisch, G.; Buhrer, C. Persistently elevated right ventricular index of myocardial performance in preterm infants with incipient bronchopulmonary dysplasia. PLoS ONE 2012, 7, e38352. [Google Scholar] [CrossRef] [PubMed]
- Skinner, J.R.; Boys, R.J.; Hunter, S.; Hey, E.N. Pulmonary and systemic arterial pressure in hyaline membrane disease. Arch. Dis. Child. 1992, 67, 366–373. [Google Scholar] [CrossRef] [PubMed]
- Collaco, J.M.; Dadlani, G.H.; Nies, M.K.; Leshko, J.; Everett, A.D.; McGrath-Morrow, S.A. Risk Factors and Clinical Outcomes in Preterm Infants with Pulmonary Hypertension. PLoS ONE 2016, 11, e0163904. [Google Scholar] [CrossRef] [PubMed]
- Mourani, P.M.; Sontag, M.K.; Younoszai, A.; Miller, J.I.; Kinsella, J.P.; Baker, C.D.; Poindexter, B.B.; Ingram, D.A.; Abman, S.H. Early pulmonary vascular disease in preterm infants at risk for bronchopulmonary dysplasia. Am. J. Respir. Crit. Care Med. 2015, 191, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Mirza, H.; Ziegler, J.; Ford, S.; Padbury, J.; Tucker, R.; Laptook, A. Pulmonary hypertension in preterm infants: Prevalence and association with bronchopulmonary dysplasia. J. Pediatr. 2014, 165, 909–914. [Google Scholar] [CrossRef] [PubMed]
- Ruskoaho, H. Cardiac hormones as diagnostic tools in heart failure. Endocr. Rev. 2003, 24, 341–356. [Google Scholar] [CrossRef] [PubMed]
- Casserly, B.; Klinger, J.R. Brain natriuretic peptide in pulmonary arterial hypertension: Biomarker and potential therapeutic agent. Drug Des. Dev. Ther. 2009, 3, 269–287. [Google Scholar]
- Cantinotti, M.; Storti, S.; Parri, M.S.; Murzi, M.; Clerico, A. Reference values for plasma B-type natriuretic peptide in the first days of life. Clin. Chem. 2009, 55, 1438–1440. [Google Scholar] [CrossRef] [PubMed]
- Law, Y.M.; Hoyer, A.W.; Reller, M.D.; Silberbach, M. Accuracy of plasma B-type natriuretic peptide to diagnose significant cardiovascular disease in children: The Better Not Pout Children! Study. J. Am. Coll. Cardiol. 2009, 54, 1467–1475. [Google Scholar] [CrossRef] [PubMed]
- Bernus, A.; Wagner, B.D.; Accurso, F.; Doran, A.; Kaess, H.; Ivy, D.D. Brain natriuretic peptide levels in managing pediatric patients with pulmonary arterial hypertension. Chest 2009, 135, 745–751. [Google Scholar] [CrossRef] [PubMed]
- Cuna, A.; Kandasamy, J.; Sims, B. B-type natriuretic peptide and mortality in extremely low birth weight infants with pulmonary hypertension: A retrospective cohort analysis. BMC Pediatr. 2014, 14, 68. [Google Scholar] [CrossRef] [PubMed]
- Del Cerro, M.J.; Sabate Rotes, A.; Carton, A.; Deiros, L.; Bret, M.; Cordeiro, M.; Verdu, C.; Barrios, M.I.; Albajara, L.; Gutierrez-Larraya, F. Pulmonary hypertension in bronchopulmonary dysplasia: Clinical findings, cardiovascular anomalies and outcomes. Pediatr. Pulmonol. 2014, 49, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Drossner, D.M.; Kim, D.W.; Maher, K.O.; Mahle, W.T. Pulmonary vein stenosis: Prematurity and associated conditions. Pediatrics 2008, 122, e656–e661. [Google Scholar] [CrossRef] [PubMed]
- Mahgoub, L.; Kaddoura, T.; Kameny, A.R.; Lopez Ortego, P.; Vanderlaan, R.D.; Kakadekar, A.; Dicke, F.; Rebeyka, I.; Calderone, C.A.; Redington, A.; et al. Pulmonary vein stenosis of ex-premature infants with pulmonary hypertension and bronchopulmonary dysplasia, epidemiology, and survival from a multicenter cohort. Pediatr. Pulmonol. 2017, 52, 1063–1070. [Google Scholar] [CrossRef] [PubMed]
- Heching, H.J.; Turner, M.; Farkouh-Karoleski, C.; Krishnan, U. Pulmonary vein stenosis and necrotising enterocolitis: Is there a possible link with necrotising enterocolitis? Arch. Dis. Child. Fetal Neonatal Ed. 2014, 99, F282–F285. [Google Scholar] [CrossRef] [PubMed]
- Acherman, R.J.; Siassi, B.; Pratti-Madrid, G.; Luna, C.; Lewis, A.B.; Ebrahimi, M.; Castillo, W.; Kamat, P.; Ramanathan, R. Systemic to pulmonary collaterals in very low birth weight infants: Color doppler detection of systemic to pulmonary connections during neonatal and early infancy period. Pediatrics 2000, 105, 528–532. [Google Scholar] [CrossRef] [PubMed]
- Abman, S.H.; Hansmann, G.; Archer, S.L.; Ivy, D.D.; Adatia, I.; Chung, W.K.; Hanna, B.D.; Rosenzweig, E.B.; Raj, J.U.; Cornfield, D.; et al. Pediatric Pulmonary Hypertension: Guidelines From the American Heart Association and American Thoracic Society. Circulation 2015, 132, 2037–2099. [Google Scholar] [CrossRef] [PubMed]
- McGoon, M.; Gutterman, D.; Steen, V.; Barst, R.; McCrory, D.C.; Fortin, T.A.; Loyd, J.E.; American College of Chest Physicians. Screening, early detection, and diagnosis of pulmonary arterial hypertension: ACCP evidence-based clinical practice guidelines. Chest 2004, 126, 14S–34S. [Google Scholar] [CrossRef] [PubMed]
- Carlton, E.F.; Sontag, M.K.; Younoszai, A.; DiMaria, M.V.; Miller, J.I.; Poindexter, B.B.; Abman, S.H.; Mourani, P.M. Reliability of Echocardiographic Indicators of Pulmonary Vascular Disease in Preterm Infants at Risk for Bronchopulmonary Dysplasia. J. Pediatr. 2017, 186, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Van de Veerdonk, M.C.; Kind, T.; Marcus, J.T.; Mauritz, G.J.; Heymans, M.W.; Bogaard, H.J.; Boonstra, A.; Marques, K.M.; Westerhof, N.; Vonk-Noordegraaf, A. Progressive right ventricular dysfunction in patients with pulmonary arterial hypertension responding to therapy. J. Am. Coll. Cardiol. 2011, 58, 2511–2519. [Google Scholar] [CrossRef] [PubMed]
- Murata, I.; Kihara, H.; Shinohara, S.; Ito, K. Echocardiographic evaluation of pulmonary arterial hypertension in patients with progressive systemic sclerosis and related syndromes. Jpn. Circ. J. 1992, 56, 983–991. [Google Scholar] [CrossRef] [PubMed]
- Borgeson, D.D.; Seward, J.B.; Miller, F.A., Jr.; Oh, J.K.; Tajik, A.J. Frequency of Doppler measurable pulmonary artery pressures. J. Am. Soc. Echocardiogr. 1996, 9, 832–837. [Google Scholar] [CrossRef]
- Mourani, P.M.; Sontag, M.K.; Younoszai, A.; Ivy, D.D.; Abman, S.H. Clinical utility of echocardiography for the diagnosis and management of pulmonary vascular disease in young children with chronic lung disease. Pediatrics 2008, 121, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Arcasoy, S.M.; Christie, J.D.; Ferrari, V.A.; Sutton, M.S.; Zisman, D.A.; Blumenthal, N.P.; Pochettino, A.; Kotloff, R.M. Echocardiographic assessment of pulmonary hypertension in patients with advanced lung disease. Am. J. Respir. Crit. Care Med. 2003, 167, 735–740. [Google Scholar] [CrossRef] [PubMed]
- Latus, H.; Kuehne, T.; Beerbaum, P.; Apitz, C.; Hansmann, G.; Muthurangu, V.; Moledina, S. Cardiac MR and CT imaging in children with suspected or confirmed pulmonary hypertension/pulmonary hypertensive vascular disease. Expert consensus statement on the diagnosis and treatment of paediatric pulmonary hypertension. The European Paediatric Pulmonary Vascular Disease Network, endorsed by ISHLT and DGPK. Heart 2016, 102. [Google Scholar] [CrossRef]
- Van Wolferen, S.A.; Marcus, J.T.; Boonstra, A.; Marques, K.M.; Bronzwaer, J.G.; Spreeuwenberg, M.D.; Postmus, P.E.; Vonk-Noordegraaf, A. Prognostic value of right ventricular mass, volume, and function in idiopathic pulmonary arterial hypertension. Eur. Heart J. 2007, 28, 1250–1257. [Google Scholar] [CrossRef] [PubMed]
- Moledina, S.; Pandya, B.; Bartsota, M.; Mortensen, K.H.; McMillan, M.; Quyam, S.; Taylor, A.M.; Haworth, S.G.; Schulze-Neick, I.; Muthurangu, V. Prognostic significance of cardiac magnetic resonance imaging in children with pulmonary hypertension. Circ. Cardiovasc. Imaging 2013, 6, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Blalock, S.; Chan, F.; Rosenthal, D.; Ogawa, M.; Maxey, D.; Feinstein, J. Magnetic resonance imaging of the right ventricle in pediatric pulmonary arterial hypertension. Pulm. Circ. 2013, 3, 350–355. [Google Scholar] [CrossRef] [PubMed]
- Muthurangu, V.; Atkinson, D.; Sermesant, M.; Miquel, M.E.; Hegde, S.; Johnson, R.; Andriantsimiavona, R.; Taylor, A.M.; Baker, E.; Tulloh, R.; et al. Measurement of total pulmonary arterial compliance using invasive pressure monitoring and MR flow quantification during MR-guided cardiac catheterization. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H1301–H1306. [Google Scholar] [CrossRef] [PubMed]
- Bennett, S.R.; Griffin, S.C. Sevoflurane versus isoflurane in patients undergoing valvular cardiac surgery. J. Cardiothorac. Vasc. Anesth. 2001, 15, 175–178. [Google Scholar] [CrossRef] [PubMed]
- Ortega, R.; Connor, C.; Kim, S.; Djang, R.; Patel, K. Monitoring ventilation with capnography. N. Engl. J. Med. 2012, 367, e27. [Google Scholar] [CrossRef] [PubMed]
- Mehta, R.; Lee, K.J.; Chaturvedi, R.; Benson, L. Complications of pediatric cardiac catheterization: A review in the current era. Catheter. Cardiovasc. Interv. 2008, 72, 278–285. [Google Scholar] [CrossRef] [PubMed]
- O'Byrne, M.L.; Glatz, A.C.; Hanna, B.D.; Shinohara, R.T.; Gillespie, M.J.; Dori, Y.; Rome, J.J.; Kawut, S.M. Predictors of Catastrophic Adverse Outcomes in Children With Pulmonary Hypertension Undergoing Cardiac Catheterization: A Multi-Institutional Analysis From the Pediatric Health Information Systems Database. J. Am. Coll. Cardiol. 2015, 66, 1261–1269. [Google Scholar] [CrossRef] [PubMed]
- Ford, S.P.; Leick-Rude, M.K.; Meinert, K.A.; Anderson, B.; Sheehan, M.B.; Haney, B.M.; Leeks, S.R.; Simon, S.D.; Jackson, J.K. Overcoming barriers to oxygen saturation targeting. Pediatrics 2006, 118, S177–S186. [Google Scholar] [CrossRef] [PubMed]
- Hilgendorff, A.; Apitz, C.; Bonnet, D.; Hansmann, G. Pulmonary hypertension associated with acute or chronic lung diseases in the preterm and term neonate and infant. The European Paediatric Pulmonary Vascular Disease Network, endorsed by ISHLT and DGPK. Heart 2016, 102, ii49–ii56. [Google Scholar] [CrossRef] [PubMed]
- Neonatal Inhaled Nitric Oxide Study Group. Inhaled nitric oxide in full-term and nearly full-term infants with hypoxic respiratory failure. N. Engl. J. Med. 1997, 336, 597–604. [Google Scholar]
- Roberts, J.D., Jr.; Fineman, J.R.; Morin, F.C., 3rd; Shaul, P.W.; Rimar, S.; Schreiber, M.D.; Polin, R.A.; Zwass, M.S.; Zayek, M.M.; Gross, I.; et al. Inhaled nitric oxide and persistent pulmonary hypertension of the newborn. The Inhaled Nitric Oxide Study Group. N. Engl. J. Med. 1997, 336, 605–610. [Google Scholar] [CrossRef] [PubMed]
- Balasubramaniam, V.; Maxey, A.M.; Morgan, D.B.; Markham, N.E.; Abman, S.H. Inhaled NO restores lung structure in eNOS-deficient mice recovering from neonatal hypoxia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 291, L119–L127. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, H.H.; Nieves, B.; Chumley, P.; Rivera, A.; Freeman, B.A. Nitric oxide regulation of superoxide-dependent lung injury: Oxidant-protective actions of endogenously produced and exogenously administered nitric oxide. Free Radic. Biol. Med. 1996, 21, 43–52. [Google Scholar] [CrossRef]
- Tang, J.R.; Seedorf, G.; Balasubramaniam, V.; Maxey, A.; Markham, N.; Abman, S.H. Early inhaled nitric oxide treatment decreases apoptosis of endothelial cells in neonatal rat lungs after vascular endothelial growth factor inhibition. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 293, L1271–L1280. [Google Scholar] [CrossRef] [PubMed]
- Field, D.; Elbourne, D.; Truesdale, A.; Grieve, R.; Hardy, P.; Fenton, A.C.; Subhedar, N.; Ahluwalia, J.; Halliday, H.L.; Stocks, J.; et al. Neonatal Ventilation With Inhaled Nitric Oxide Versus Ventilatory Support Without Inhaled Nitric Oxide for Preterm Infants With Severe Respiratory Failure: The INNOVO multicentre randomised controlled trial (ISRCTN 17821339). Pediatrics 2005, 115, 926–936. [Google Scholar] [CrossRef] [PubMed]
- Van Meurs, K.P.; Wright, L.L.; Ehrenkranz, R.A.; Lemons, J.A.; Ball, M.B.; Poole, W.K.; Perritt, R.; Higgins, R.D.; Oh, W.; Hudak, M.L.; et al. Inhaled nitric oxide for premature infants with severe respiratory failure. N. Engl. J. Med. 2005, 353, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Kinsella, J.P.; Cutter, G.R.; Walsh, W.F.; Gerstmann, D.R.; Bose, C.L.; Hart, C.; Sekar, K.C.; Auten, R.L.; Bhutani, V.K.; Gerdes, J.S.; et al. Early inhaled nitric oxide therapy in premature newborns with respiratory failure. N. Engl. J. Med. 2006, 355, 354–364. [Google Scholar] [CrossRef] [PubMed]
- Mercier, J.C.; Hummler, H.; Durrmeyer, X.; Sanchez-Luna, M.; Carnielli, V.; Field, D.; Greenough, A.; van Overmeire, B.; Jonsson, B.; Hallman, M.; et al. Inhaled nitric oxide for prevention of bronchopulmonary dysplasia in premature babies (EUNO): A randomised controlled trial. Lancet 2010, 376, 346–354. [Google Scholar] [CrossRef]
- Kumar, P.; Committee on Fetus and Newborn. Use of inhaled nitric oxide in preterm infants. Pediatrics 2014, 133, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Soyguder, Z.; Karadag, H.; Nazli, M. Neuronal nitric oxide synthase immunoreactivity in ependymal cells during early postnatal development. J. Chem. Neuroanat. 2004, 27, 3–6. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.T.; Zhang, D.L.; Cao, Y.L.; Zhao, B.L. Developmental expression and activity variation of nitric oxide synthase in the brain of golden hamster. Brain Res. Bull. 2002, 58, 385–389. [Google Scholar] [CrossRef]
- Hintz, S.R.; Van Meurs, K.P.; Perritt, R.; Poole, W.K.; Das, A.; Stevenson, D.K.; Ehrenkranz, R.A.; Lemons, J.A.; Vohr, B.R.; Heyne, R.; et al. Neurodevelopmental outcomes of premature infants with severe respiratory failure enrolled in a randomized controlled trial of inhaled nitric oxide. J. Pediatr. 2007, 151, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Huddy, C.L.; Bennett, C.C.; Hardy, P.; Field, D.; Elbourne, D.; Grieve, R.; Truesdale, A.; Diallo, K.; Group ITC. The INNOVO multicentre randomised controlled trial: Neonatal ventilation with inhaled nitric oxide versus ventilatory support without nitric oxide for severe respiratory failure in preterm infants: Follow up at 4–5 years. Arch. Dis. Child. Fetal Neonatal Ed. 2008, 93, F430–F435. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekar, I.; Eis, A.; Konduri, G.G. Betamethasone attenuates oxidant stress in endothelial cells from fetal lambs with persistent pulmonary hypertension. Pediatr. Res. 2008, 63, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Aikio, O.; Metsola, J.; Vuolteenaho, R.; Perhomaa, M.; Hallman, M. Transient defect in nitric oxide generation after rupture of fetal membranes and responsiveness to inhaled nitric oxide in very preterm infants with hypoxic respiratory failure. J. Pediatr. 2012, 161, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Ball, M.K.; Steinhorn, R.H. Inhaled nitric oxide for preterm infants: A Marksman’s approach. J. Pediatr. 2012, 161, 379–380. [Google Scholar] [CrossRef] [PubMed]
- Afshar, S.; Gibson, L.L.; Yuhanna, I.S.; Sherman, T.S.; Kerecman, J.D.; Grubb, P.H.; Yoder, B.A.; McCurnin, D.C.; Shaul, P.W. Pulmonary NO synthase expression is attenuated in a fetal baboon model of chronic lung disease. Am. J. Physiol. Lung Cell. Mol. Physiol. 2003, 284, L749–L758. [Google Scholar] [CrossRef] [PubMed]
- Bland, R.D.; Ling, C.Y.; Albertine, K.H.; Carlton, D.P.; MacRitchie, A.J.; Day, R.W.; Dahl, M.J. Pulmonary vascular dysfunction in preterm lambs with chronic lung disease. Am. J. Physiol. Lung Cell. Mol. Physiol. 2003, 285, L76–L85. [Google Scholar] [CrossRef] [PubMed]
- Farrow, K.N.; Groh, B.S.; Schumacker, P.T.; Lakshminrusimha, S.; Czech, L.; Gugino, S.F.; Russell, J.A.; Steinhorn, R.H. Hyperoxia increases phosphodiesterase 5 expression and activity in ovine fetal pulmonary artery smooth muscle cells. Circ. Res. 2008, 102, 226–233. [Google Scholar] [CrossRef] [PubMed]
- Nyp, M.; Sandritter, T.; Poppinga, N.; Simon, C.; Truog, W.E. Sildenafil citrate, bronchopulmonary dysplasia and disordered pulmonary gas exchange: Any benefits? J. Perinatol. 2012, 32, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Trottier-Boucher, M.N.; Lapointe, A.; Malo, J.; Fournier, A.; Raboisson, M.J.; Martin, B.; Moussa, A. Sildenafil for the Treatment of Pulmonary Arterial Hypertension in Infants with Bronchopulmonary Dysplasia. Pediatr. Cardiol. 2015, 36, 1255–1260. [Google Scholar] [CrossRef] [PubMed]
- Mourani, P.M.; Sontag, M.K.; Ivy, D.D.; Abman, S.H. Effects of long-term sildenafil treatment for pulmonary hypertension in infants with chronic lung disease. J. Pediatr. 2009, 154, 379–384. [Google Scholar] [CrossRef] [PubMed]
- Ladha, F.; Bonnet, S.; Eaton, F.; Hashimoto, K.; Korbutt, G.; Thebaud, B. Sildenafil improves alveolar growth and pulmonary hypertension in hyperoxia-induced lung injury. Am. J. Respir. Crit. Care Med. 2005, 172, 750–756. [Google Scholar] [CrossRef] [PubMed]
- Samiee-Zafarghandy, S.; van den Anker, J.N.; Laughon, M.M.; Clark, R.H.; Smith, P.B.; Hornik, C.P.; Pharmaceuticals for Children Act—Pediatric Trials Network Administrative Core C. Sildenafil and retinopathy of prematurity risk in very low birth weight infants. J. Perinatol. 2016, 36, 137–140. [Google Scholar] [CrossRef] [PubMed]
- Barst, R.J.; Ivy, D.D.; Gaitan, G.; Szatmari, A.; Rudzinski, A.; Garcia, A.E.; Sastry, B.K.; Pulido, T.; Layton, G.R.; Serdarevic-Pehar, M.; et al. A randomized, double-blind, placebo-controlled, dose-ranging study of oral sildenafil citrate in treatment-naive children with pulmonary arterial hypertension. Circulation 2012, 125, 324–334. [Google Scholar] [CrossRef] [PubMed]
- Abman, S.H.; Kinsella, J.P.; Rosenzweig, E.B.; Krishnan, U.; Kulik, T.; Mullen, M.; Wessel, D.L.; Steinhorn, R.; Adatia, I.; Hanna, B.; et al. Implications of the, U.S. Food and Drug Administration warning against the use of sildenafil for the treatment of pediatric pulmonary hypertension. Am. J. Respir. Crit. Care Med. 2013, 187, 572–575. [Google Scholar] [CrossRef] [PubMed]
- Walmrath, D.; Schneider, T.; Pilch, J.; Grimminger, F.; Seeger, W. Aerosolised prostacyclin in adult respiratory distress syndrome. Lancet 1993, 342, 961–962. [Google Scholar] [CrossRef]
- McIntyre, C.M.; Hanna, B.D.; Rintoul, N.; Ramsey, E.Z. Safety of epoprostenol and treprostinil in children less than 12 months of age. Pulm. Circ. 2013, 3, 862–869. [Google Scholar] [CrossRef] [PubMed]
- Ferdman, D.J.; Rosenzweig, E.B.; Zuckerman, W.A.; Krishnan, U. Subcutaneous treprostinil for pulmonary hypertension in chronic lung disease of infancy. Pediatrics 2014, 134, e274–e278. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, U.; Takatsuki, S.; Ivy, D.D.; Kerstein, J.; Calderbank, M.; Coleman, E.; Rosenzweig, E.B. Effectiveness and safety of inhaled treprostinil for the treatment of pulmonary arterial hypertension in children. Am. J. Cardiol. 2012, 110, 1704–1709. [Google Scholar] [CrossRef] [PubMed]
- Piastra, M.; De Luca, D.; De Carolis, M.P.; Tempera, A.; Stival, E.; Caliandro, F.; Pietrini, D.; Conti, G.; de Rosa, G. Nebulized iloprost and noninvasive respiratory support for impending hypoxaemic respiratory failure in formerly preterm infants: A case series. Pediatr. Pulmonol. 2012, 47, 757–762. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, W.A.; Ismail, M. A randomized, double-blind, placebo-controlled, prospective study of bosentan for the treatment of persistent pulmonary hypertension of the newborn. J. Perinatol. 2012, 32, 608–613. [Google Scholar] [CrossRef] [PubMed]
- Steinhorn, R.H.; Fineman, J.; Kusic-Pajic, A.; Cornelisse, P.; Gehin, M.; Nowbakht, P.; Pierce, C.M.; Beghetti, M.; FUTURE-4 Study Investigators. Bosentan as Adjunctive Therapy for Persistent Pulmonary Hypertension of the Newborn: Results of the Randomized Multicenter Placebo-Controlled Exploratory Trial. J. Pediatr. 2016, 177, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Perez, M.; Lakshminrusimha, S.; Wedgwood, S.; Czech, L.; Gugino, S.F.; Russell, J.A.; Farrow, K.N.; Steinhorn, R.H. Hydrocortisone normalizes oxygenation and cGMP regulation in lambs with persistent pulmonary hypertension of the newborn. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 302, L595–L603. [Google Scholar] [CrossRef] [PubMed]
- Perez, M.; Wedgwood, S.; Lakshminrusimha, S.; Farrow, K.N.; Steinhorn, R.H. Hydrocortisone normalizes phosphodiesterase-5 activity in pulmonary artery smooth muscle cells from lambs with persistent pulmonary hypertension of the newborn. Pulm. Circ. 2014, 4, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Basu, S.; Kumar, A.; Bhatia, B.D.; Satya, K.; Singh, T.B. Role of steroids on the clinical course and outcome of meconium aspiration syndrome—A randomized controlled trial. J. Trop. Pediatr. 2007, 53, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Yeh, T.F.; Srinivasan, G.; Harris, V.; Pildes, R.S. Hydrocortisone therapy in meconium aspiration syndrome: A controlled study. J. Pediatr. 1977, 90, 140–143. [Google Scholar] [CrossRef]
- Kumar, V.H.; Swartz, D.D.; Rashid, N.; Lakshminrusimha, S.; Ma, C.; Ryan, R.M.; Morin, F.C. Prostacyclin and milrinone by aerosolization improve pulmonary hemodynamics in newborn lambs with experimental pulmonary hypertension. J. Appl. Physiol. 2010, 109, 677–684. [Google Scholar] [CrossRef] [PubMed]
- Patel, N. Use of milrinone to treat cardiac dysfunction in infants with pulmonary hypertension secondary to congenital diaphragmatic hernia: A review of six patients. Neonatology 2012, 102, 130–136. [Google Scholar] [CrossRef] [PubMed]
- James, A.T.; Bee, C.; Corcoran, J.D.; McNamara, P.J.; Franklin, O.; El-Khuffash, A.F. Treatment of premature infants with pulmonary hypertension and right ventricular dysfunction with milrinone: A case series. J. Perinatol. 2015, 35, 268–273. [Google Scholar] [CrossRef] [PubMed]
- McNamara, P.J.; Shivananda, S.P.; Sahni, M.; Freeman, D.; Taddio, A. Pharmacology of milrinone in neonates with persistent pulmonary hypertension of the newborn and suboptimal response to inhaled nitric oxide. Pediatr. Crit. Care Med. 2013, 14, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Gurakan, B.; Kayiran, P.; Ozturk, N.; Kayiran, S.M.; Dindar, A. Therapeutic combination of sildenafil and iloprost in a preterm neonate with pulmonary hypertension. Pediatr. Pulmonol. 2011, 46, 617–620. [Google Scholar] [CrossRef] [PubMed]
- Landry, J.S.; Tremblay, G.M.; Li, P.Z.; Wong, C.; Benedetti, A.; Taivassalo, T. Lung Function and Bronchial Hyperresponsiveness in Adults Born Prematurely: A Cohort Study. Ann. Am. Thorac. Soc. 2016, 13, 17–24. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Dosage/Route of Administration | Common Adverse Effects |
|---|---|---|
| Nitric Oxide | Inhaled 5–20 ppm (OI > 20); Wean iNO—FiO2 < 60%; PaO2 > 60 mmHg; Keep SpO2 ≥ 91; Infants on chronic iNO therapy—wean last 5 ppm gradually to ↓ rebound PH | Monitor methemoglobin during use |
| PDE5 Inhibitor—Sildenafil | Oral—0.5 mg/kg q8–6 ↑ to 2 mg/kg q8–6 over 2 weeks IV (continuous infusion): 0.4 mg/kg over 3 h (LD); Infusion—0.07 mg/kg/h | Systemic hypotension; watch for worsening oxygenation due to vasodilation of unventilated areas of the lung; flushing, diarrhea, nasal congestion, priapism |
| Prostanoids *—Epoprostenol | IV/continuous Aerosolization—2 ng/kg/min ↑ to 20–50 ng/kg/min | Systemic hypotension, nausea, vomiting, flushing, diarrhea, thrombocytopenia, bloodstream infection |
| Treprostinil | Subcutaneous—1.5 ng/kg/min ↑ to 20–40 ng/kg/min; Inhaled—3–9 breaths (6 µg/breath) q6 | Infusion site pain, site infection, flushing, diarrhea, nausea, jaw pain, bloodstream infection |
| Iloprost | Inhalation: 1–2.5 µg/kg q2–4 h | Cough, syncope, hypotension, flushing, headache, trismus |
| PDE3 Inhibitor—Milrinone | IV—50 µg/kg (LD) over 1–2 h; Infusion—20–75 µg/kg/min | Hypotension, tachycardia, arrhythmias, thrombocytopenia, low potassium, bronchospasm |
| Endothelial Receptor Antagonist—Bosentan | Oral: 1 mg/kg q12 | Hypotension, flushing, hepatotoxicity, anemia, thrombocytopenia, teratogenesis |
© 2017 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumar, V.H.S. Diagnostic Approach to Pulmonary Hypertension in Premature Neonates. Children 2017, 4, 75. https://doi.org/10.3390/children4090075
Kumar VHS. Diagnostic Approach to Pulmonary Hypertension in Premature Neonates. Children. 2017; 4(9):75. https://doi.org/10.3390/children4090075
Chicago/Turabian StyleKumar, Vasantha H.S. 2017. "Diagnostic Approach to Pulmonary Hypertension in Premature Neonates" Children 4, no. 9: 75. https://doi.org/10.3390/children4090075
APA StyleKumar, V. H. S. (2017). Diagnostic Approach to Pulmonary Hypertension in Premature Neonates. Children, 4(9), 75. https://doi.org/10.3390/children4090075