
Sarcoidosis-like Skin Lesions as the First Manifestation of Ataxia-Telangiectasia
 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
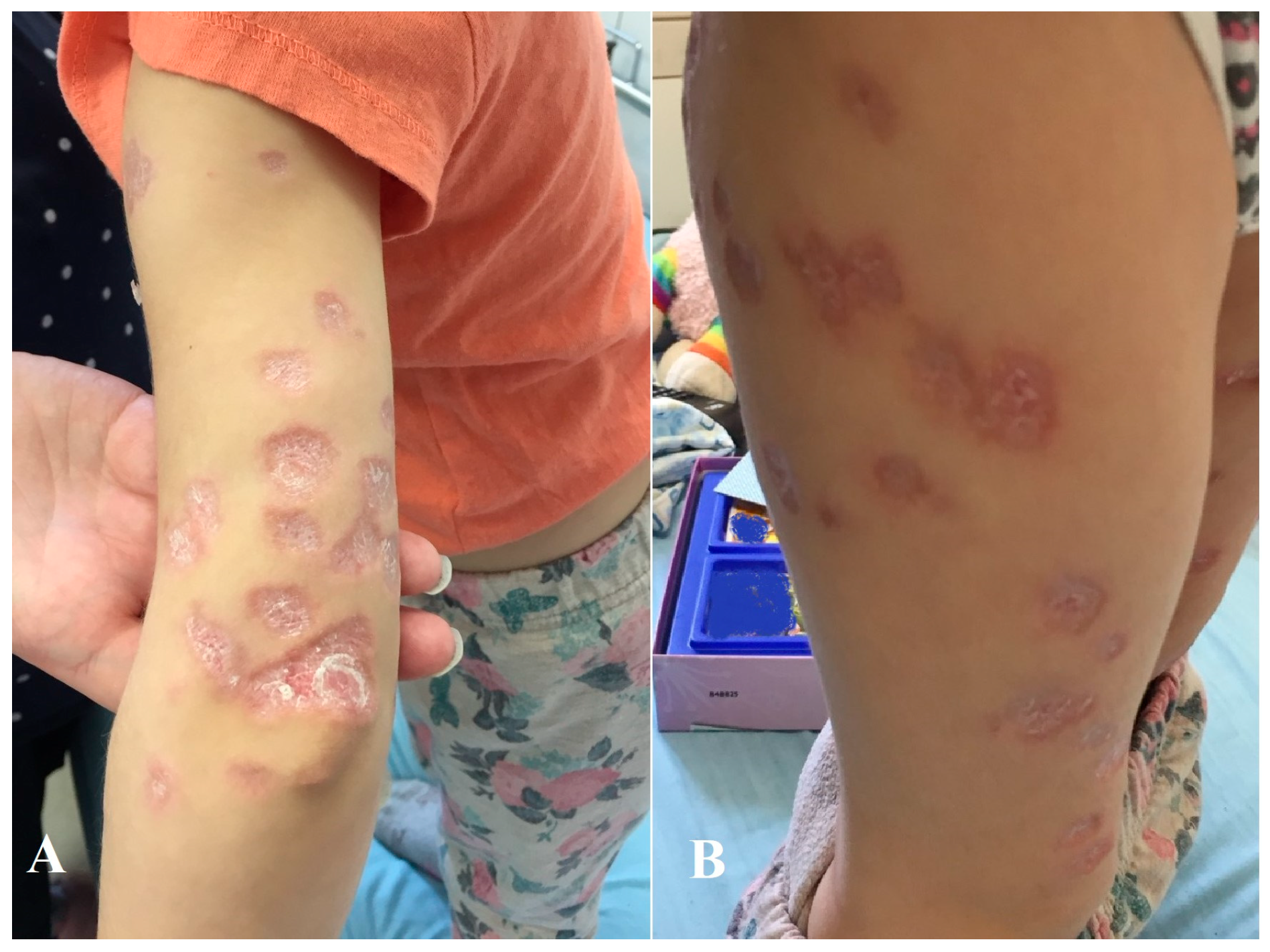
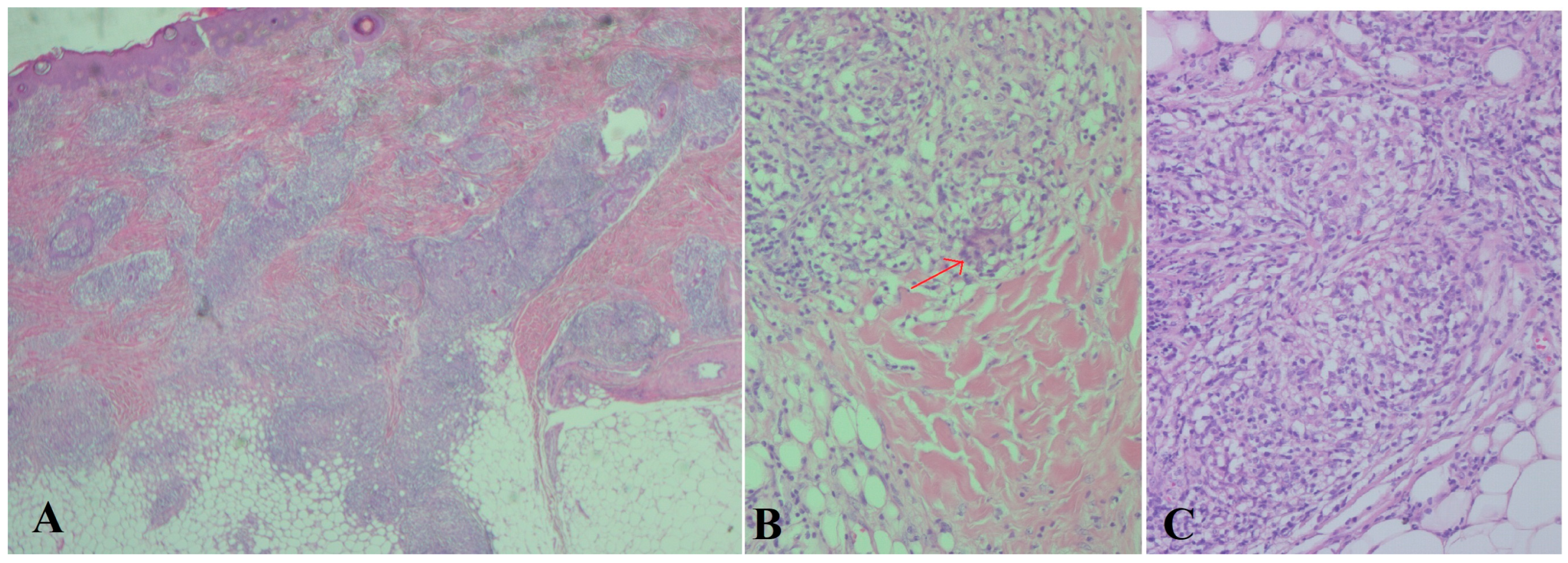
2. Case Description
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ACE | angiotensin-converting enzyme |
| A-T | ataxia-telangiectasia |
| ATM | ataxia-telangiectasia mutated |
| CT | computerized tomography |
| H&E | hematoxylin and eosin |
| Ig | immunoglobulin |
| MRI | magnetic resonance imaging |
| PAS | Periodic acid–Schiff |
References
- Alyasin, S.; Esmaeilzadeh, H.; Ebrahimi, N.; Nabavizadeh, S.H.; Nemati, H. Clinical presentation of ataxia-telangiectasia. Arch. Iran. Med. 2019, 22, 682–686. [Google Scholar] [PubMed]
- Anheim, M.; Tranchant, C.; Koenig, M. The autosomal recessive cerebellar ataxias. N. Engl. J. Med. 2012, 366, 636–646. [Google Scholar] [CrossRef] [PubMed]
- Rothblum-Oviatt, C.; Wright, J.; Lefton-Greif, M.A.; McGrath-Morrow, S.A.; Crawford, T.O.; Lederman, H.M. Ataxia telangiectasia: A review. Orphanet J. Rare Dis. 2016, 11, 159. [Google Scholar] [CrossRef] [PubMed]
- Van Os, N.J.; Jansen, A.F.M.; van Deuren, M.; Haraldsson, A.; van Driel, N.T.M.; Etzioni, A.; van der Flier, M.; Haaxma, C.A.; Morio, T.; Rawat, A.; et al. Ataxia-telangiectasia: Immunodeficiency and survival. Clin. Immunol. 2017, 178, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Takada, S.; Weitering, T.J.; van Os, N.J.; Du, L.; Pico-Knijnenburg, I.; Kuipers, T.B.; Mei, H.; Salzer, E.; Willemsen, M.A.; Weemaes, C.M.; et al. Causative mechanisms and clinical impact of immunoglobulin deficiencies in ataxia telangiectasia. J. Allergy Clin. Immunol. 2024, 153, 1392–1405. [Google Scholar] [CrossRef] [PubMed]
- Riboldi, G.M.; Samanta, D.; Asuncion, R.M.D.; Frucht, S. Ataxia-Telangiectasia; StatPearls Publishing: Treasure Island, FL, USA, 2025. Available online: https://www.ncbi.nlm.nih.gov/books/NBK519542/ (accessed on 9 March 2025).
- Crawford, T.O. Ataxia telangiectasia. Semin. Pediatr. Neurol. 1998, 5, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Sirajwala, A.A.; Khan, S.; Rathod, V.M.; Gevariya, V.C.; Jansari, J.R.; Patel, Y.M.; Patel, Y. Ataxia-telangiectasia: A case report and a brief review. Cureus 2023, 15, e39346. [Google Scholar] [CrossRef] [PubMed]
- Dewang, S.; Awake, P.; Chandravathi, P.L.; Tourani, V. Cutaneous granulomas in a child with Ataxia telangiectasia-A rare association. J. Pak. Assoc. Dermatol. 2020, 30, 511–515. [Google Scholar]
- Chiam, L.Y.T.; Verhagen, M.M.M.; Haraldsson, A.; Wulffraat, N.; Driessen, G.J.; Netea, M.G.; Weemaes, C.; Seyger, M.; van Deuren, M. Cutaneous granulomas in ataxia telangiectasia and other primary immunodeficiencies: Reflection of inappropriate immune regulation. Dermatology 2011, 223, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Naiboğlu, S.; Ulaş, S.; Turan, I.; Çipe, F.E.; Gürbüz, B.Ç.; Aydoğmuş, Ç. A Rare Clinical Finding in Ataxia Telangiectasia: Granulomatous Skin Lesion. Turk. Arch. Pediatr. 2024, 59, 325. [Google Scholar] [CrossRef] [PubMed]
- Hadi Babikir, H.E. Classic ataxia-telangiectasia in a Sudanese boy: Case report and review of the literature. Sudan. J. Paediatr. 2011, 11, 60–63. [Google Scholar] [PubMed]
- Shaikh, A.G.; Marti, S.; Tarnutzer, A.A.; Palla, A.; O Crawford, T.; Straumann, D.; Taylor, A.M.; Zee, D.S. Gaze fixation deficits and their implication in ataxiatelangiectasia. J. Neurol. Neurosurg. Psychiatry 2009, 80, 858–864. [Google Scholar] [CrossRef] [PubMed]
- Nowak-Wegrzyn, A.; O Crawford, T.; A Winkelstein, J.; A Carson, K.; Lederman, H.M. Immunodeficiency and infections in ataxia-telangiectasia. J. Pediatr. 2004, 144, 505–511. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, J.M.; Bush, A. An overview of proactive monitoring and management of respiratory issues in ataxia-telangiectasia in a specialist and shared care pediatric clinic. Front. Pediatr. 2024, 12, 1479620. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, H.; Svensson, E.; Danielsson, A.; Puschmann, A.; Svenningson, P.; Tesi, B.; Paucar, M. The clinical spectrum of ataxia telangiectasia in a cohort in Sweden. Heliyon 2024, 10, e26073. [Google Scholar] [CrossRef] [PubMed]
- De Nardi, L.; Natale, M.F.; Messia, V.; Tomà, P.; De Benedetti, F.; Insalaco, A. A child with polyarthritis and chronic lung disease: A case report of ataxia-telangiectasia. Ital. J. Pediatr. 2023, 49, 111. [Google Scholar] [CrossRef] [PubMed]
- Razzaqi, F.; Albastaki, A.; Sinan, I. Ataxia telangiectasia in a Bahraini child treated with intensive physiotherapy: A case report. Hong Kong Physiother. J. 2025, 45, 1–9. [Google Scholar] [CrossRef]
- Lnu, P.; Sehgal, V.; Kapila, S.; Gulati, N.; Bhalla Sehgal, L. Ataxia Telangiectasia Presenting as Cervical Dystonia. Cureus 2022, 14, e30723. [Google Scholar] [CrossRef] [PubMed]
- Collyer, J.; Rajan, D.S. Ataxia telangiectasia. Semin. Pediatr. Neurol. 2024, 52, 101169. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Milanovic, B.; Vijatov-Djuric, G.; Djuretic, A.; Kesic, J.; Stojanovic, V.; Jaric, M.; Ležakov, O. Sarcoidosis-like Skin Lesions as the First Manifestation of Ataxia-Telangiectasia. Children 2025, 12, 672. https://doi.org/10.3390/children12060672
Milanovic B, Vijatov-Djuric G, Djuretic A, Kesic J, Stojanovic V, Jaric M, Ležakov O. Sarcoidosis-like Skin Lesions as the First Manifestation of Ataxia-Telangiectasia. Children. 2025; 12(6):672. https://doi.org/10.3390/children12060672
Chicago/Turabian StyleMilanovic, Borko, Gordana Vijatov-Djuric, Andrea Djuretic, Jelena Kesic, Vesna Stojanovic, Milica Jaric, and Ognjen Ležakov. 2025. "Sarcoidosis-like Skin Lesions as the First Manifestation of Ataxia-Telangiectasia" Children 12, no. 6: 672. https://doi.org/10.3390/children12060672
APA StyleMilanovic, B., Vijatov-Djuric, G., Djuretic, A., Kesic, J., Stojanovic, V., Jaric, M., & Ležakov, O. (2025). Sarcoidosis-like Skin Lesions as the First Manifestation of Ataxia-Telangiectasia. Children, 12(6), 672. https://doi.org/10.3390/children12060672