
First Reported Case of Gabriele-de Vries Syndrome with Spinal Dysraphism
 , , , , ,
, , , , ,
Abstract
1. Introduction
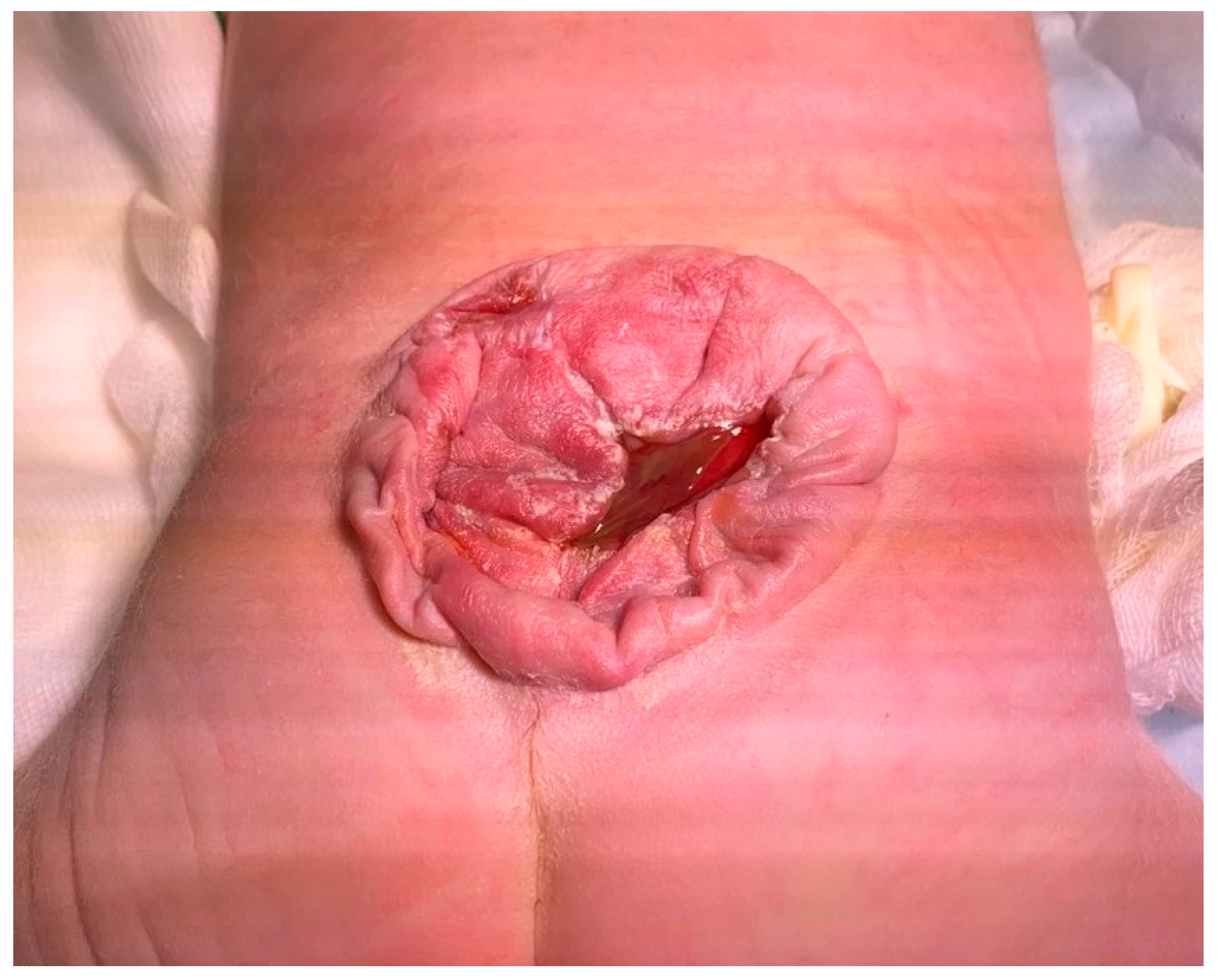
2. Case Report
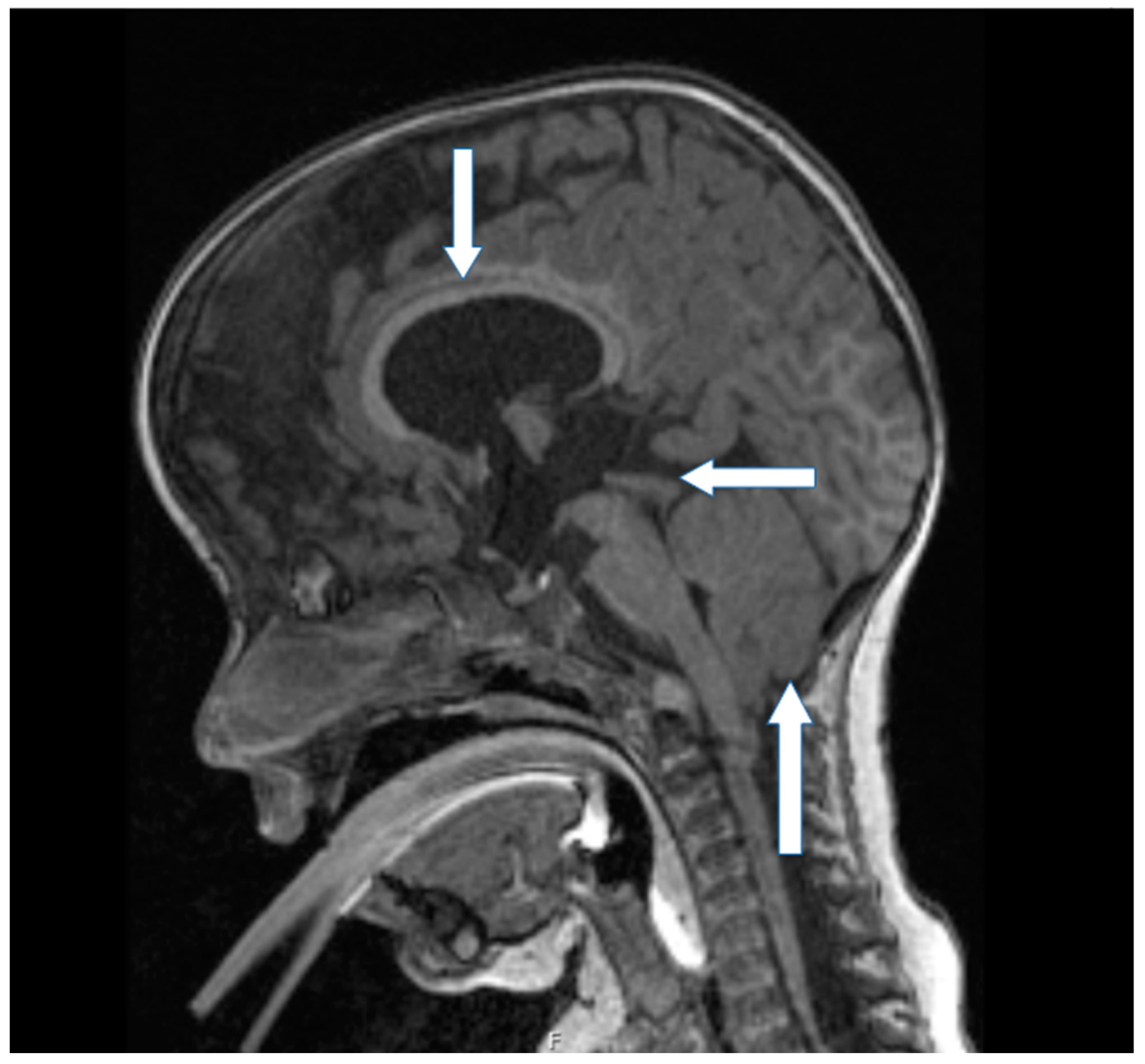
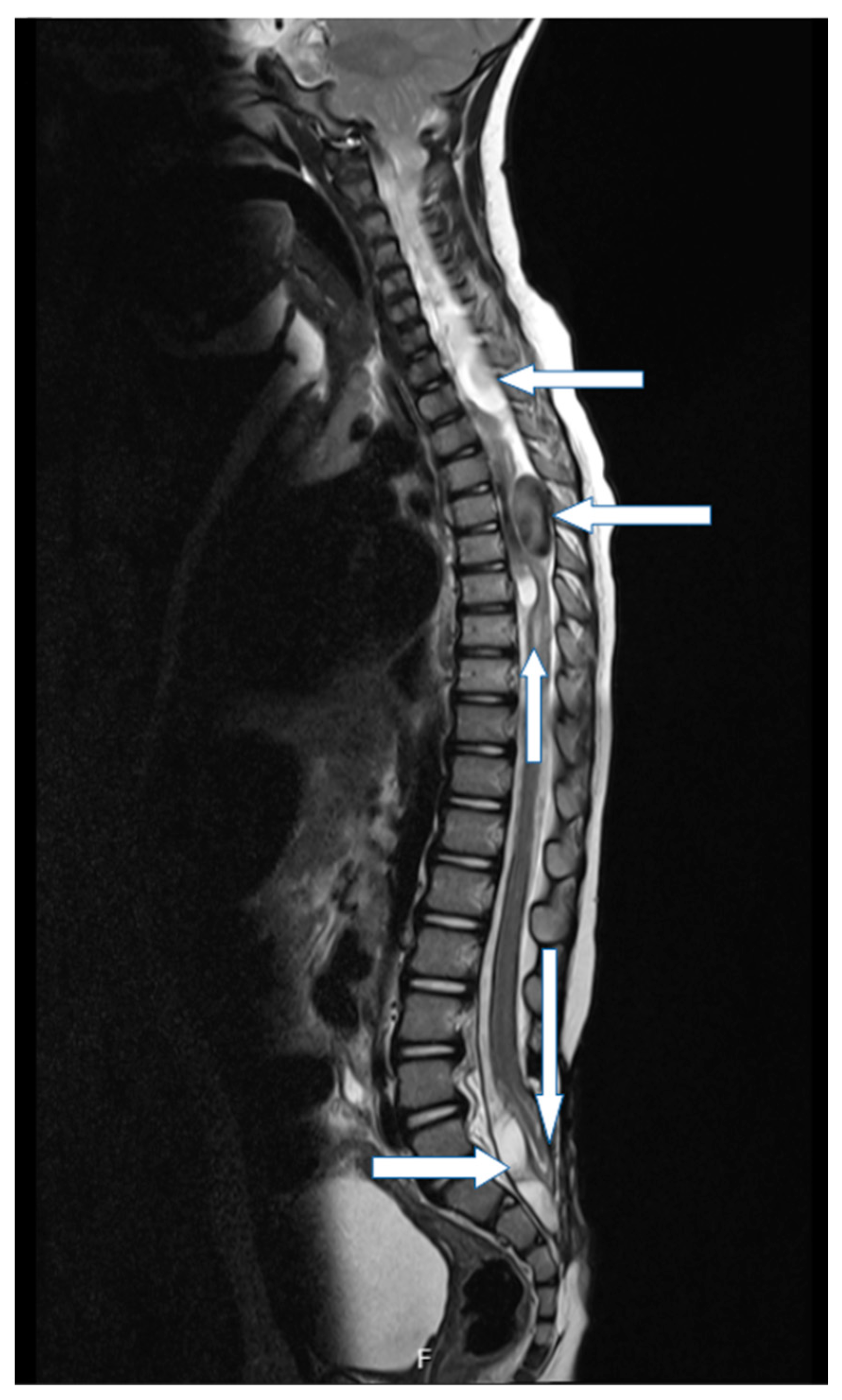
3. Genetic Evaluation and Interpretation of Results
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gabriele, M.; Vulto-van Silfhout, A.T.; Germain, P.-L.; Vitriolo, A.; Kumar, R.; Douglas, E.; Haan, E.; Kosaki, K.; Takenouchi, T.; Rauch, A.; et al. YY1 Haploinsufficiency Causes an Intellectual Disability Syndrome Featuring Transcriptional and Chromatin Dysfunction. Am. J. Hum. Genet. 2017, 100, 907–925. [Google Scholar] [CrossRef] [PubMed]
- Morales-Rosado, J.A.; Kaiwar, C.; Smith, B.E.; Klee, E.W.; Dhamija, R. A Case of YY1-Associated Syndromic Learning Disability or Gabriele-de Vries Syndrome with Myasthenia Gravis. Am. J. Med. Genet. Part A 2018, 176, 2846–2849. [Google Scholar] [CrossRef] [PubMed]
- Vissers, L.E.; de Ligt, J.; Gilissen, C.; Janssen, I.; Steehouwer, M.; de Vries, P.; van Lier, B.; Arts, P.; Wieskamp, N.; del Rosario, M. A de novo paradigm for mental retardation. Nat. Genet. 2010, 42, 1109–1112. [Google Scholar] [CrossRef] [PubMed]
- ACMG Board of Directors. Direct-to-consumer genetic testing: A revised position statement of the American College of Medical Genetics and Genomics. Genet. Med. 2016, 18, 207–278. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Oza, A.M.; del Castillo, I.; Duzkale, H.; Matsunaga, T.; Pandya, A.; Kang, H.P.; Mar-Heyming, R.; Guha, S.; Moyer, K.; et al. Consensus interpretation of the p.Met34Thr and p.Val37Ile variants in GJB2 by the ClinGen Hearing Loss Expert Panel. Genet. Med. 2019, 21, 2442–2452. [Google Scholar] [CrossRef] [PubMed]
- Minami, S.B.; Mutai, H.; Nakano, A.; Arimoto, Y.; Taiji, H.; Morimoto, N.; Sakata, H.; Adachi, N.; Masuda, S.; Sakamoto, H.; et al. GJB2-associated hearing loss undetected by hearing screening of newborns. Gene 2013, 532, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, S.R.; Piergiorge, R.M.; Rocha, J.; Abdala, B.B.; Gonçalves, A.P.; Pimentel, M.M.G.; Santos-Rebouças, C.B. A de novo YY1 missense variant expanding the Gabriele-de Vries syndrome phenotype and affecting X-chromosome inactivation. Metab. Brain. Dis. 2022, 37, 2431–2440. [Google Scholar] [CrossRef] [PubMed]
- Linglart, A.; Maupetit-Méhouas, S.; Silve, C. GNAS-Related Loss-of-Function Disorders and the Role of Imprinting. Horm. Res. Paediatr. 2013, 79, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Pruitt, K.D.; Brown, G.R.; Hiatt, S.M.; Thibaud-Nissen, F.; Astashyn, A.; Ermolaeva, O.; Farrell, C.M.; Hart, J.; Landrum, M.J.; McGarvey, K.M.; et al. RefSeq: An update on mammalian reference sequences. Nucleic Acids Res. 2014, 42, D756–D763. [Google Scholar] [CrossRef] [PubMed]
- McMullan, P.; Maye, P.; Yang, Q.; Rowe, D.W.; Germain-Lee, E.L. Parental Origin of Gsα Inactivation Differentially Affects Bone Remodeling in a Mouse Model of Albright Hereditary Osteodystrophy. JBMR Plus 2021, 6, e10570. [Google Scholar] [CrossRef] [PubMed]
- Nabais Sá, M.J.; Gabriele, M.; Testa, G.; de Vries, B.B.A. Gabriele-de Vries Syndrome. 2019 May 30. In GeneReviews®; Adam, M.P., Everman, D.B., Mirzaa, G.M., Eds.; University of Washington: Seattle, WA, USA, 2019. Available online: https://www.ncbi.nlm.nih.gov/books/NBK541730/ (accessed on 10 January 2023).
- Khamirani, H.J.; Zoghi, S.; Namdar, Z.M.; Kamal, N.; Dianatpour, M.; Tabei, S.M.B.; Mohammadi, S.; Dehghanian, F.; Farbod, Z.; Dastgheib, S.A. Clinical features of patients with Yin Yang 1 deficiency causing Gabriele-de Vries syndrome: A new case and review of the literature. Ann. Hum. Genet. 2022, 86, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.; Li, Y.; Liu, F.; Huang, Y.; Luo, S.; Zhao, P.; Gu, W.; Lin, J.; Zhou, A.; He, X. A 9-month-old Chinese patient with Gabriele-de Vries syndrome due to novel germline mutation in the YY1 gene. Mol. Genet. Genomic. Med. 2021, 9, e1582. [Google Scholar] [CrossRef] [PubMed]
- Carminho-Rodrigues, M.T.; Steel, D.; Sousa, S.B.; Brandt, G.; Guipponi, M.; Laurent, S.; Fokstuen, S.; Moren, A.; Zacharia, A.; Dirren, E.; et al. Complex movement disorder in a patient with heterozygous YY1 mutation (Gabriele-de Vries syndrome). Am. J. Med. Genet. A 2020, 182, 2129–2132. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Gene | cDNA | Protein | Zygosity | Inheritance | Classification |
|---|---|---|---|---|---|
| GJB2 | c.109G > A | p.Val37lle | Heterozygous | Maternal | Pathogenic |
| YY1 | c.1A > C | p.Met1? | Heterozygous | De novo | Likely pathogenic |
| GNAS | c.1360C > T | p.Gln454* | Heterozygous | Maternal | Likely Pathogenic |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koruga, N.; Pušeljić, S.; Babić, M.; Ćuk, M.; Cvitković Roić, A.; Vrtarić, V.; Soldo Koruga, A.; Rončević, A.; Tomac, V.; Rotim, T.; et al. First Reported Case of Gabriele-de Vries Syndrome with Spinal Dysraphism. Children 2023, 10, 623. https://doi.org/10.3390/children10040623
Koruga N, Pušeljić S, Babić M, Ćuk M, Cvitković Roić A, Vrtarić V, Soldo Koruga A, Rončević A, Tomac V, Rotim T, et al. First Reported Case of Gabriele-de Vries Syndrome with Spinal Dysraphism. Children. 2023; 10(4):623. https://doi.org/10.3390/children10040623
Chicago/Turabian StyleKoruga, Nenad, Silvija Pušeljić, Marko Babić, Mario Ćuk, Andrea Cvitković Roić, Vjenceslav Vrtarić, Anamarija Soldo Koruga, Alen Rončević, Višnja Tomac, Tatjana Rotim, and et al. 2023. "First Reported Case of Gabriele-de Vries Syndrome with Spinal Dysraphism" Children 10, no. 4: 623. https://doi.org/10.3390/children10040623
APA StyleKoruga, N., Pušeljić, S., Babić, M., Ćuk, M., Cvitković Roić, A., Vrtarić, V., Soldo Koruga, A., Rončević, A., Tomac, V., Rotim, T., Turk, T., Kretić, D., Pušeljić, N., Nađ, R., & Serdarušić, I. (2023). First Reported Case of Gabriele-de Vries Syndrome with Spinal Dysraphism. Children, 10(4), 623. https://doi.org/10.3390/children10040623