
Stationed or Relocating: The Seesawing EMT/MET Determinants from Embryonic Development to Cancer Metastasis

 ,
,  , ,
, , 
Abstract
:1. Introduction
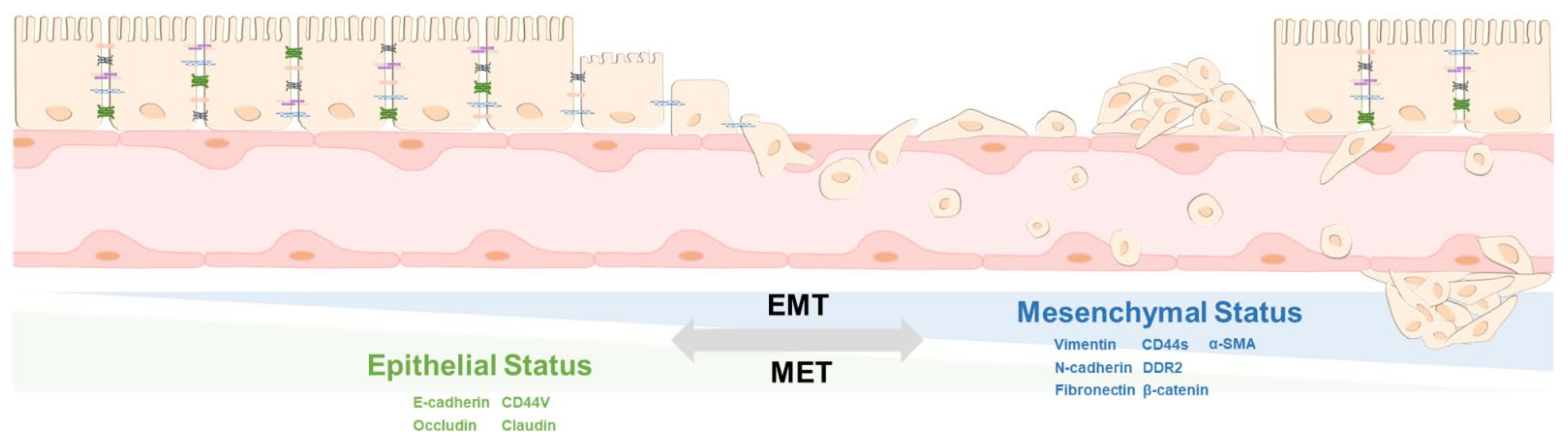
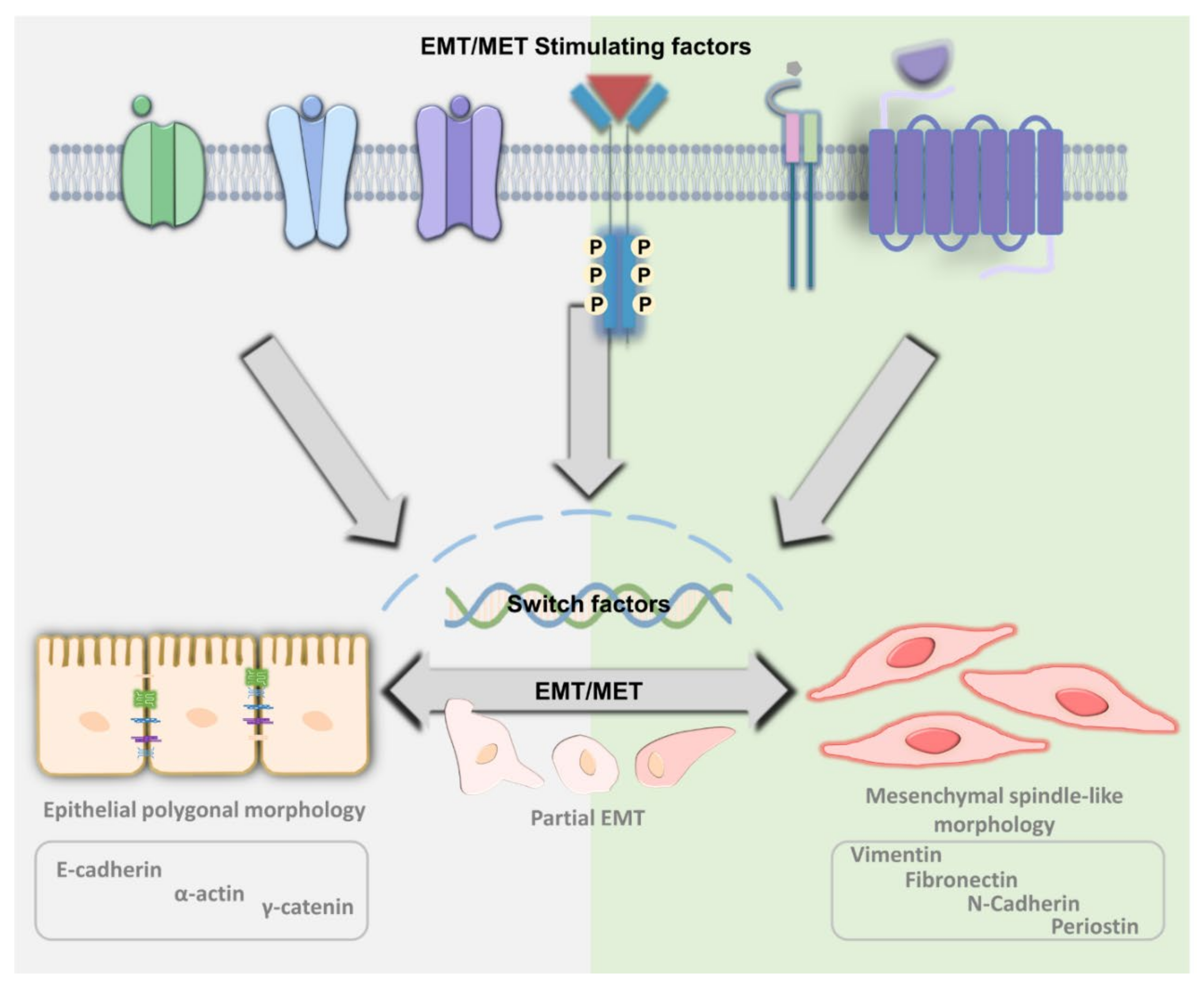
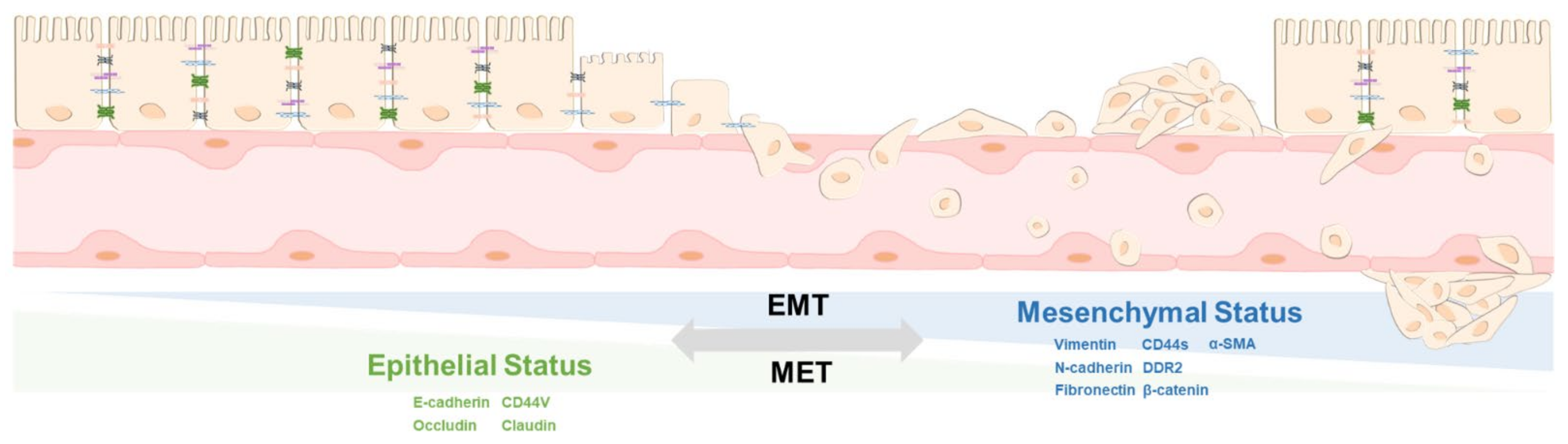
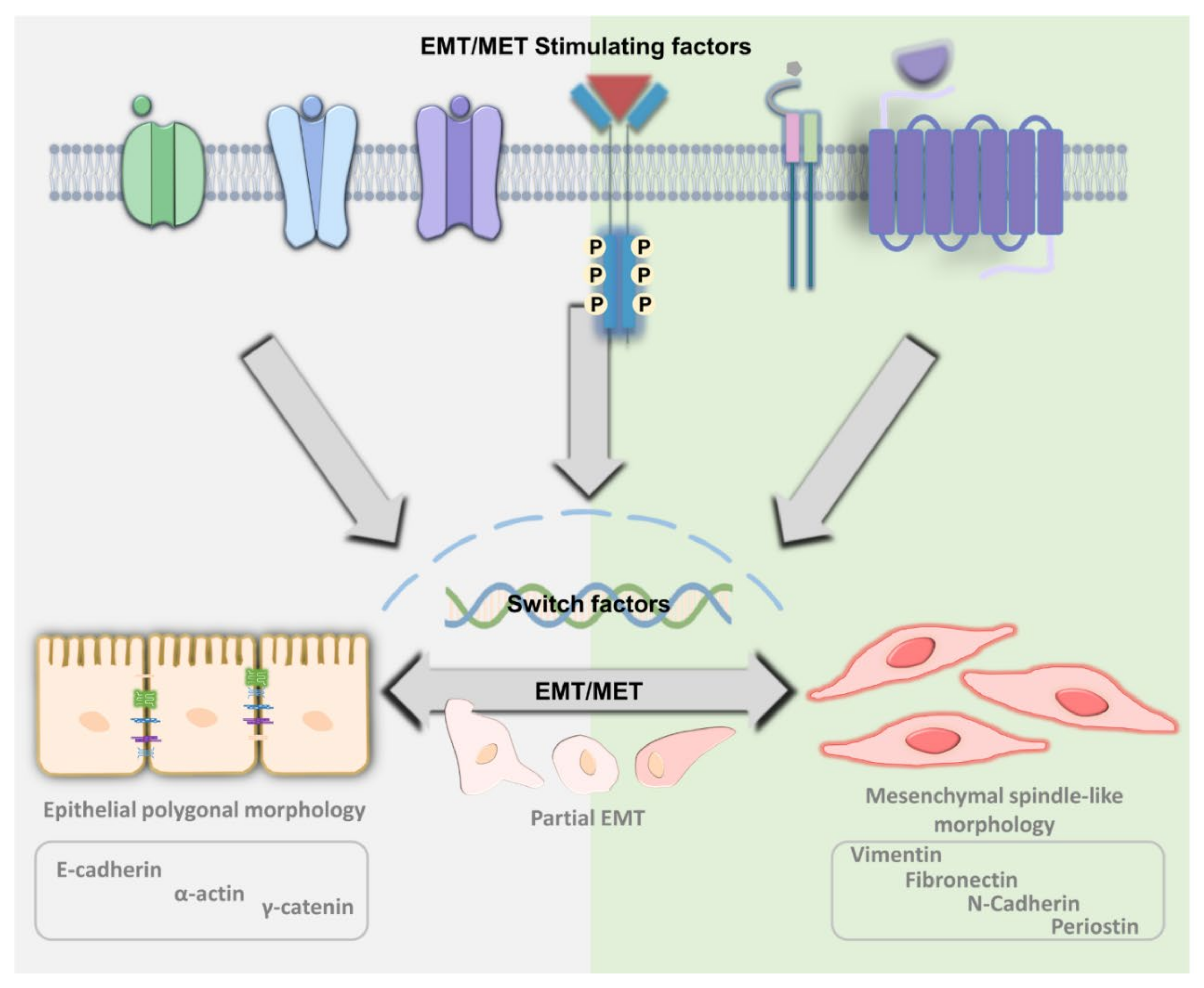
2. The Principle of EMT and MET
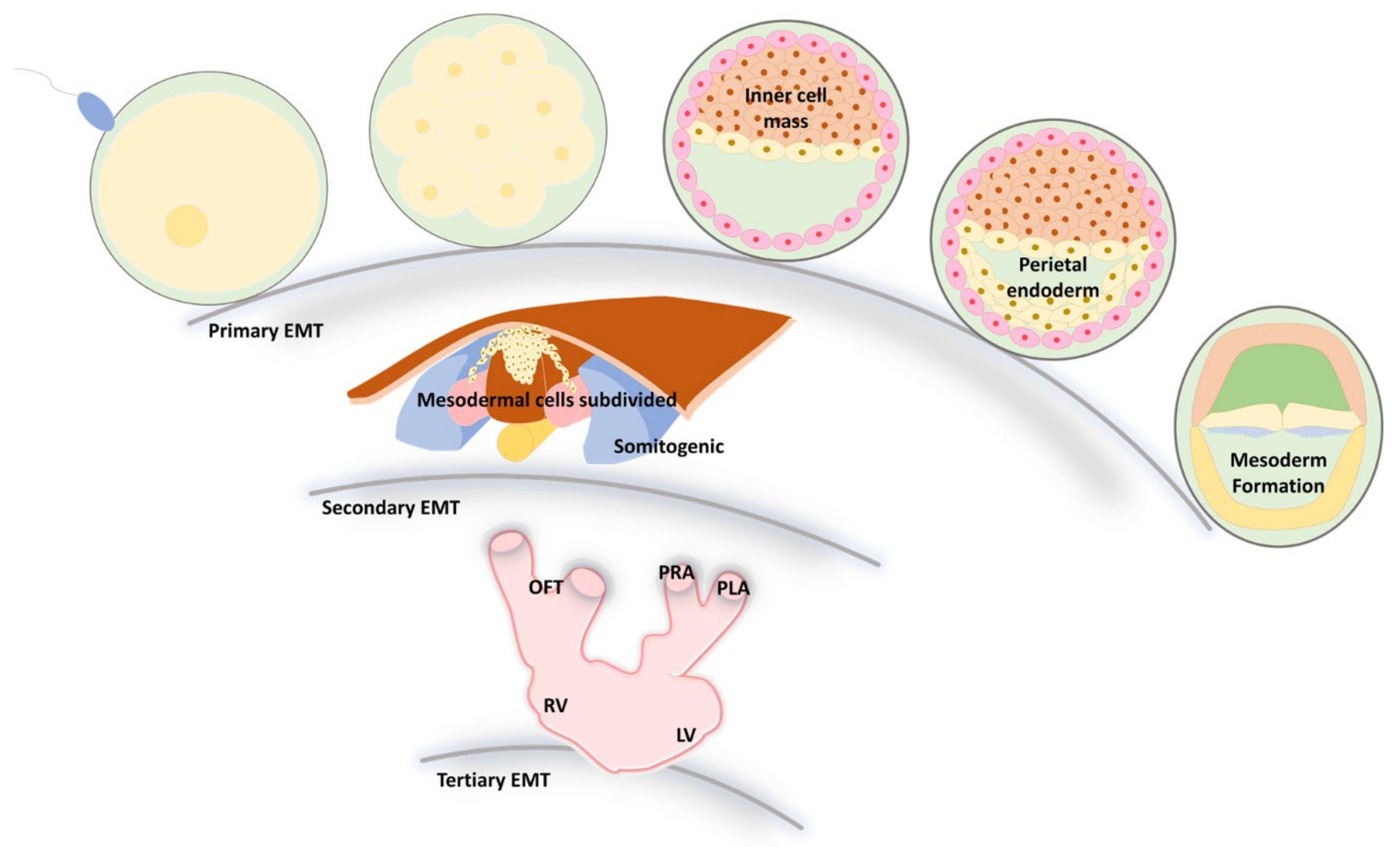
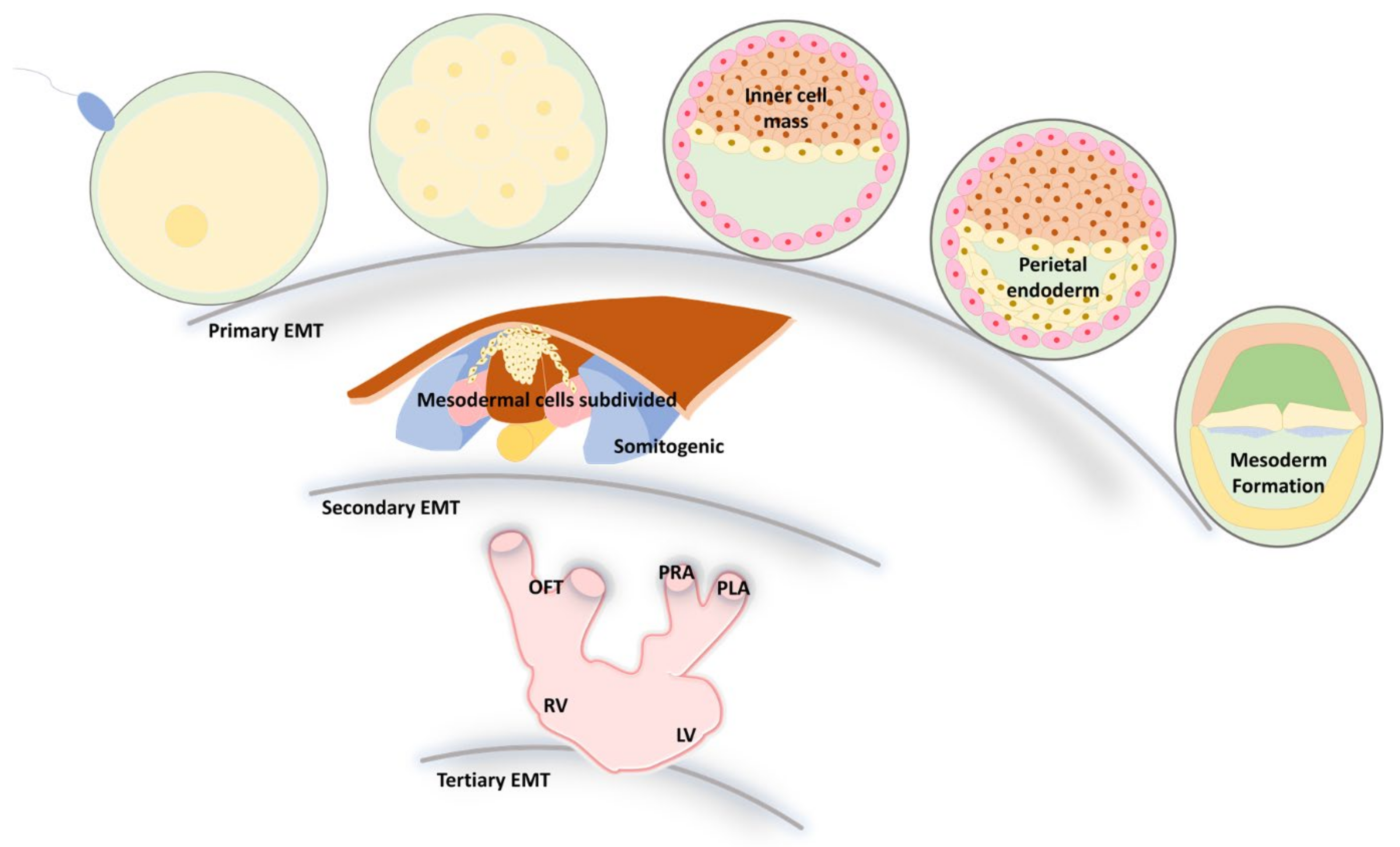
3. EMT and MET during Embryonic Development
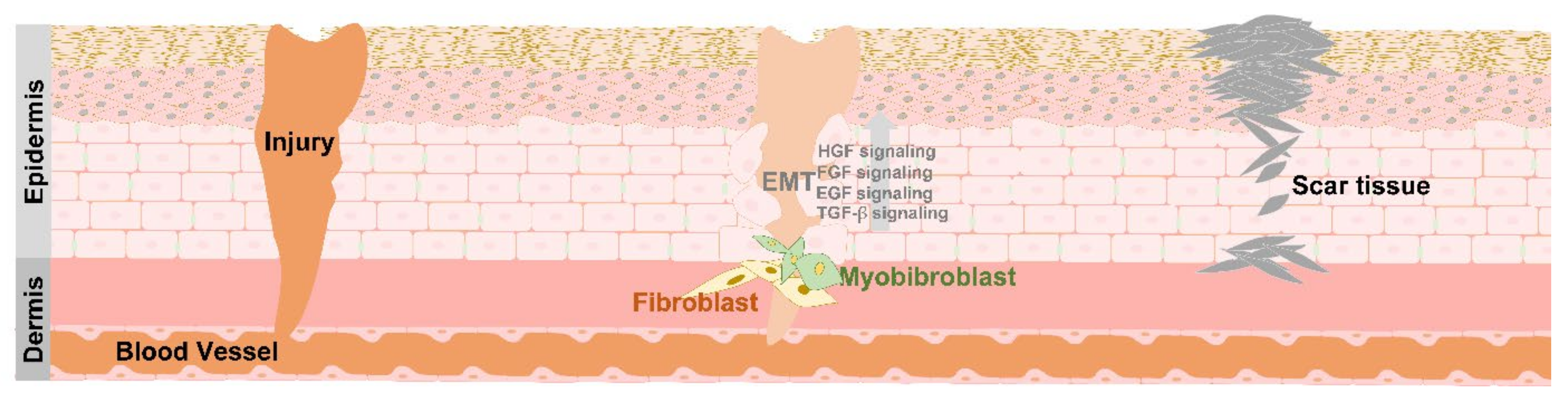
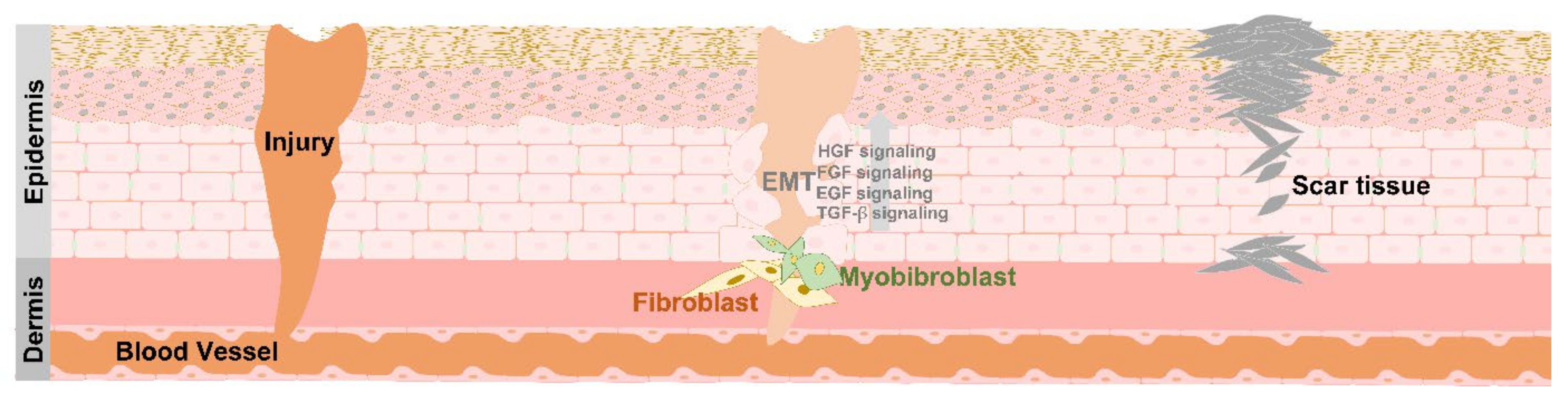
4. EMT in Tissue Repair
5. Differentiation and De-Differentiation between EMT and MET
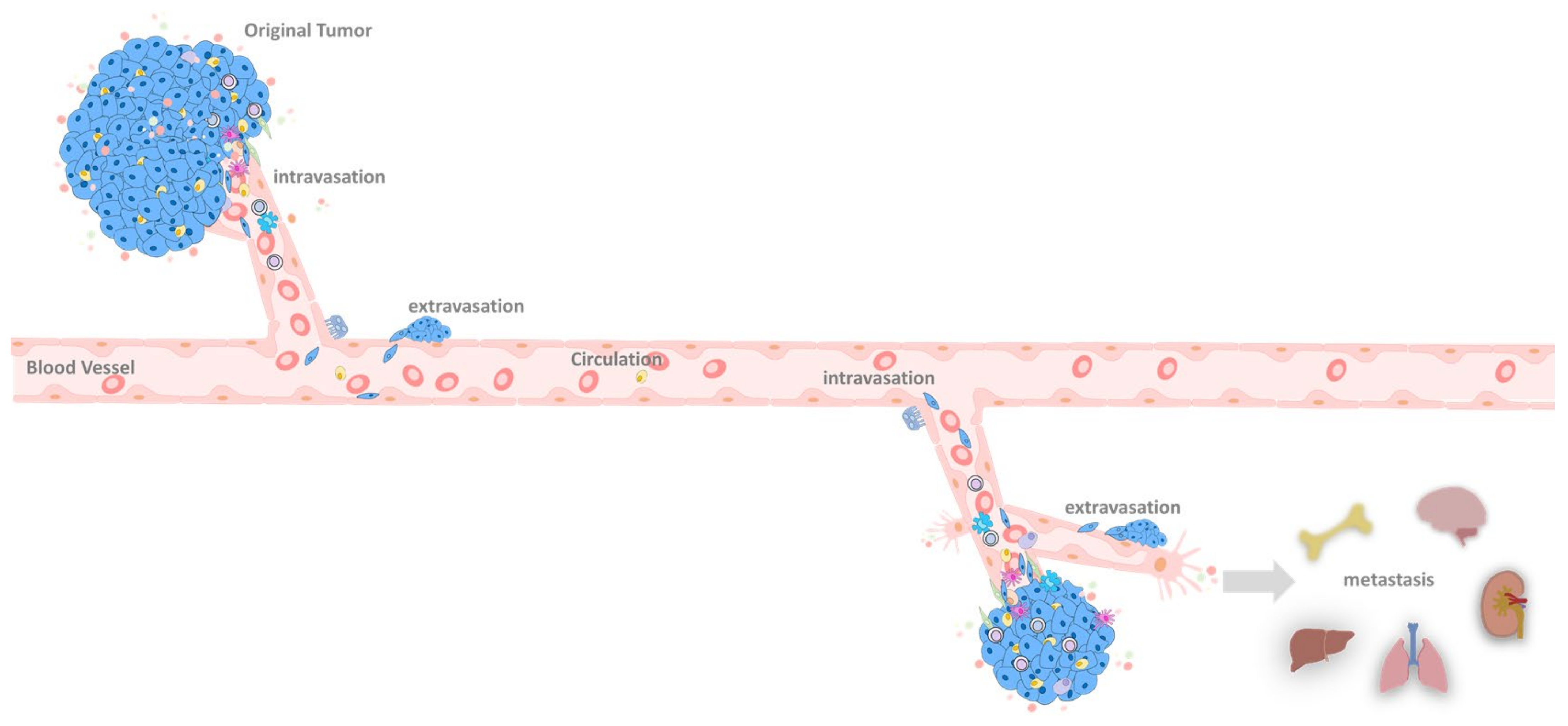
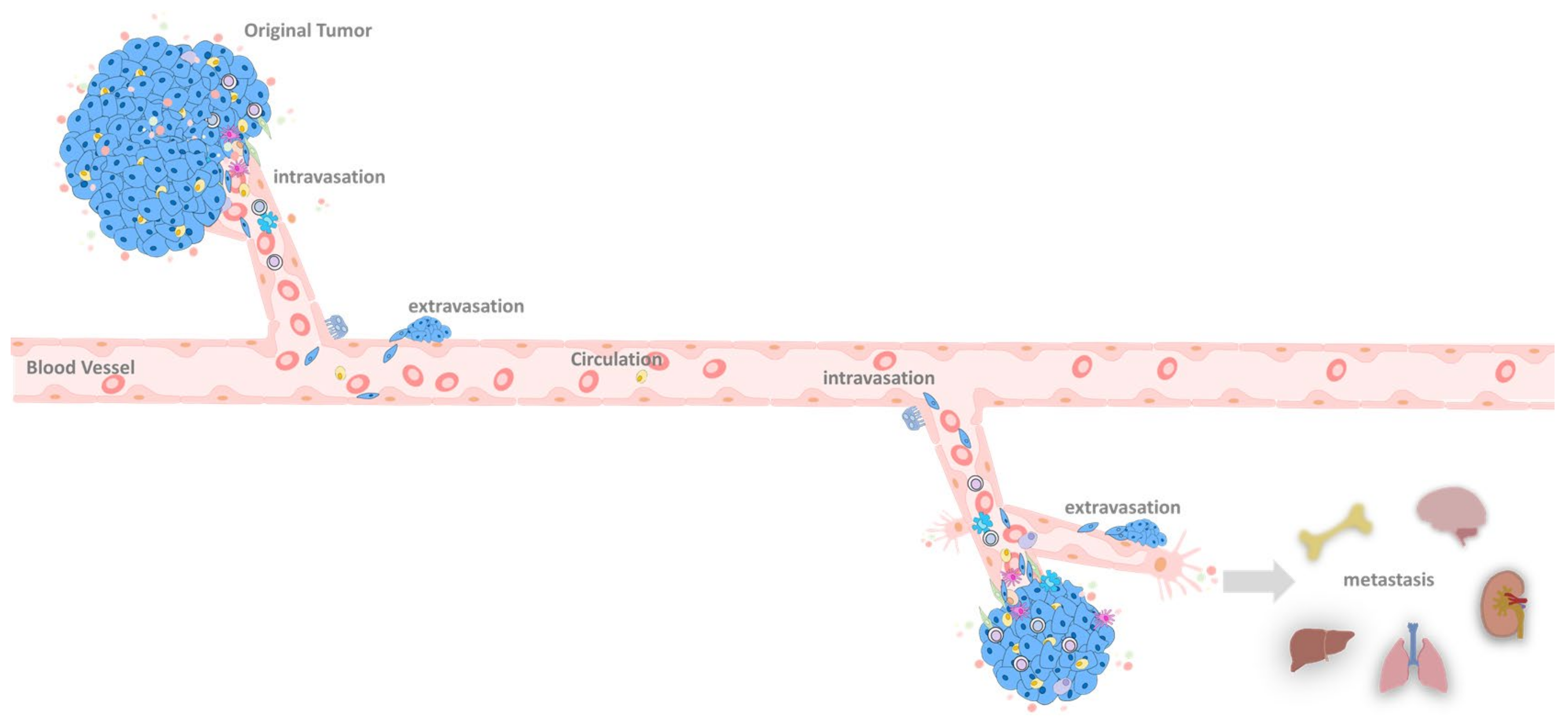
6. EMT at the Primary Tumor Site
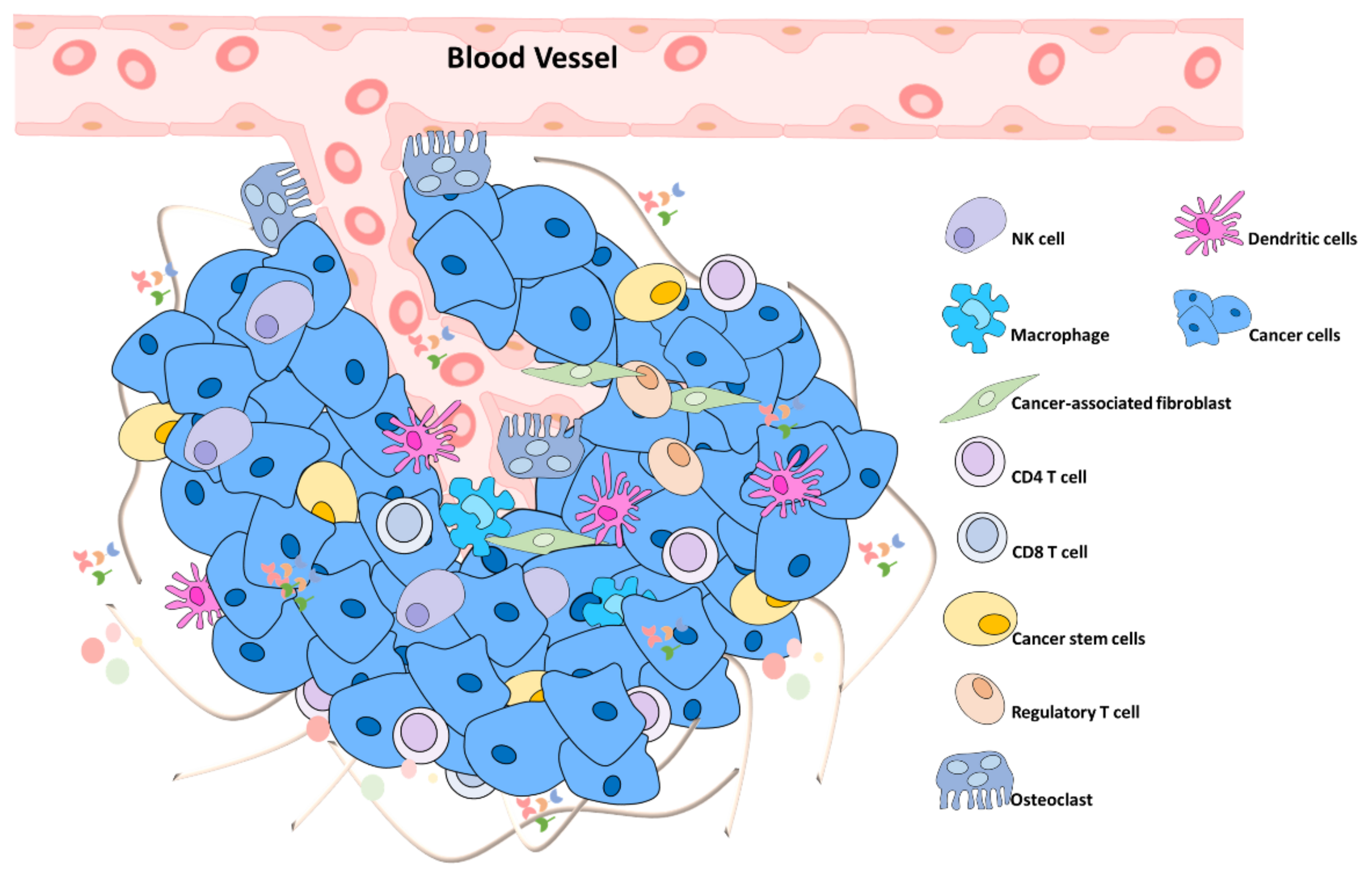
6.1. Primary Tumor
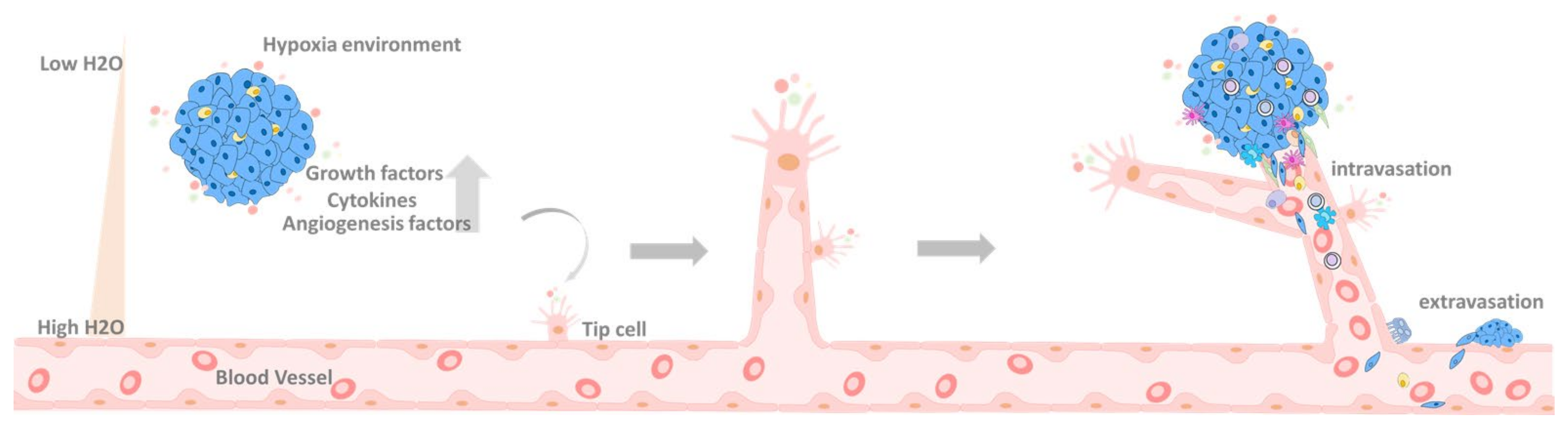
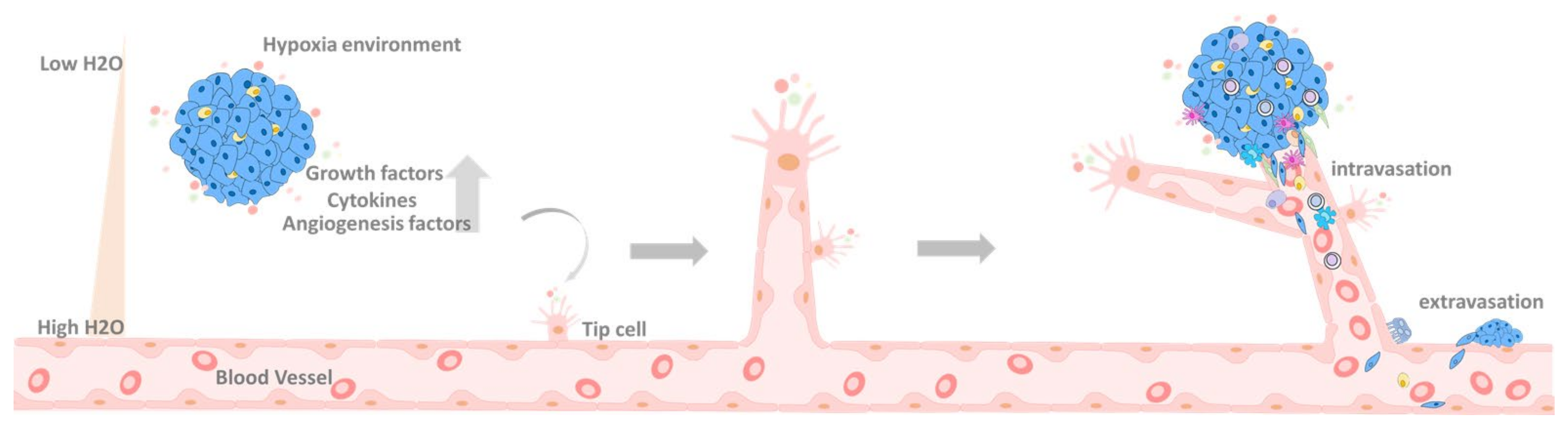
6.2. Primary Tumor Extravasation
7. EMT in the Circulation System
8. EMT in the Metastatic Site
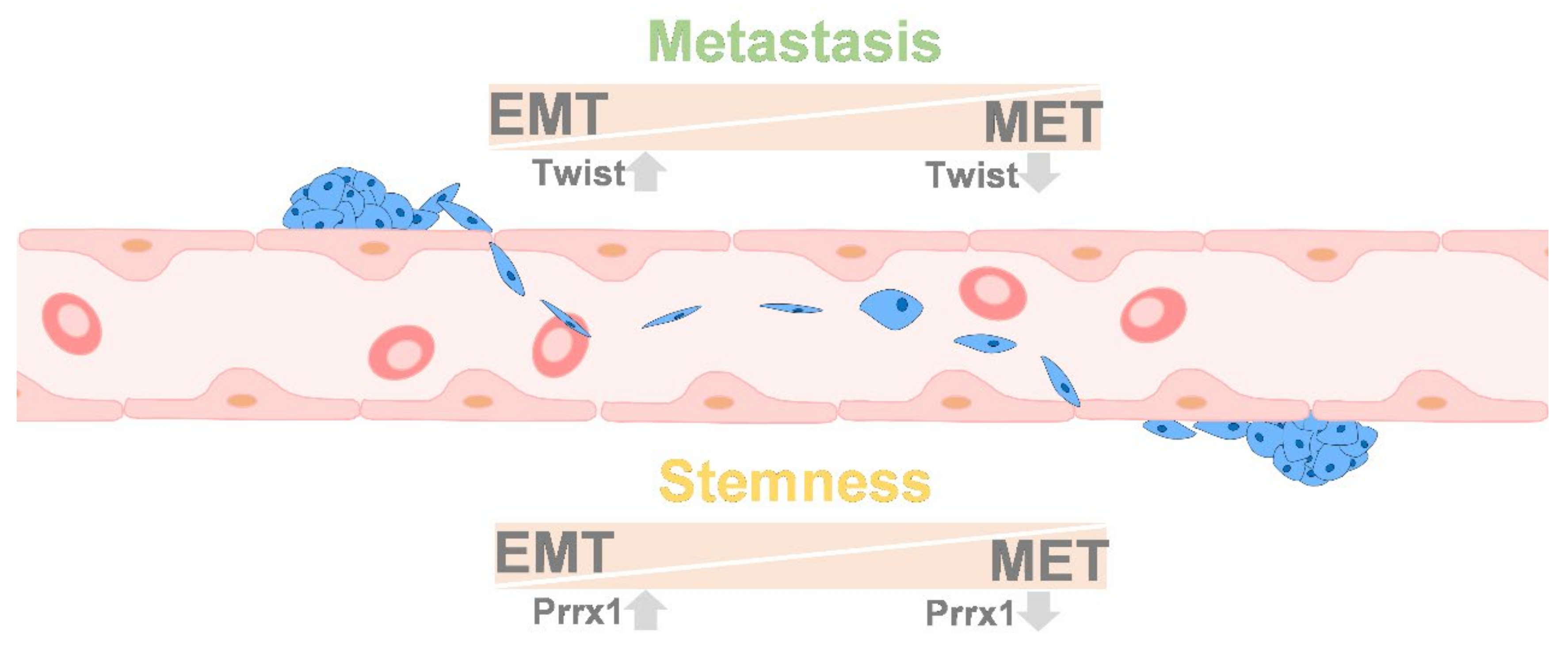
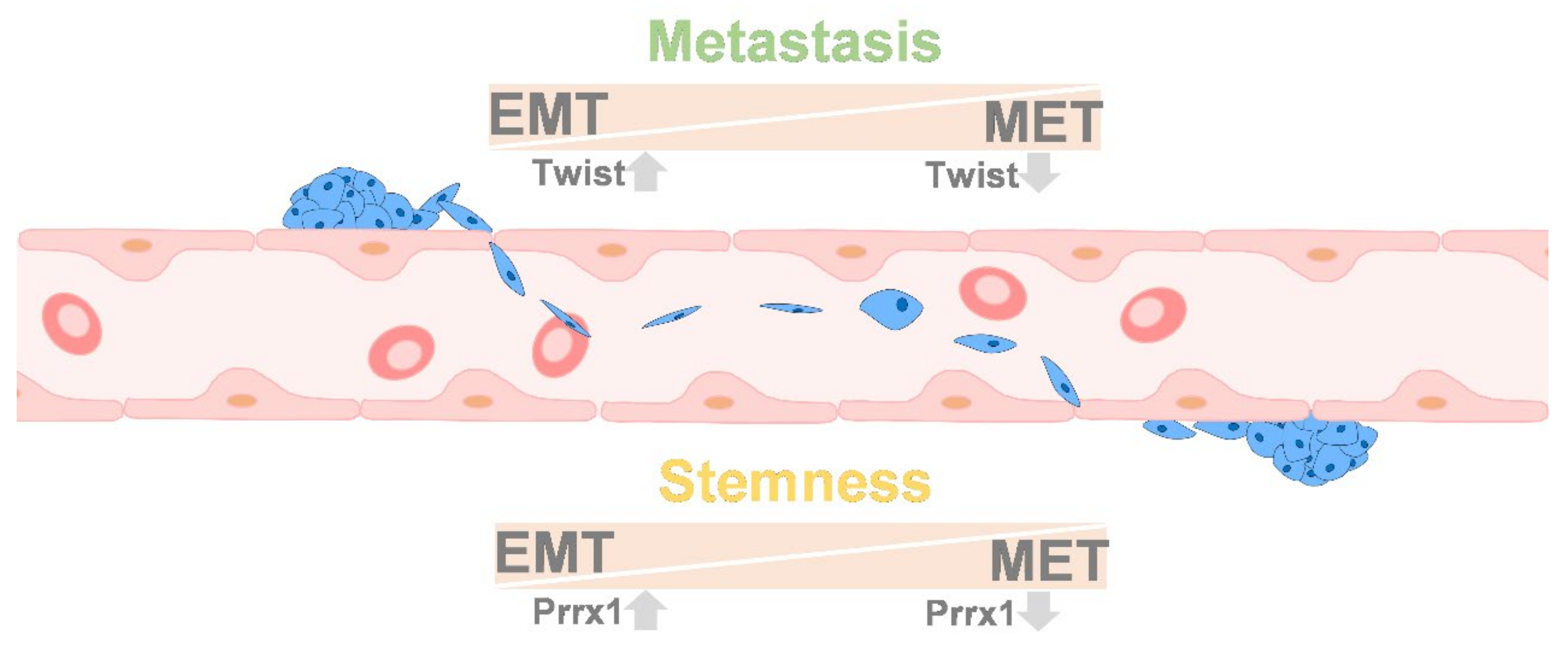
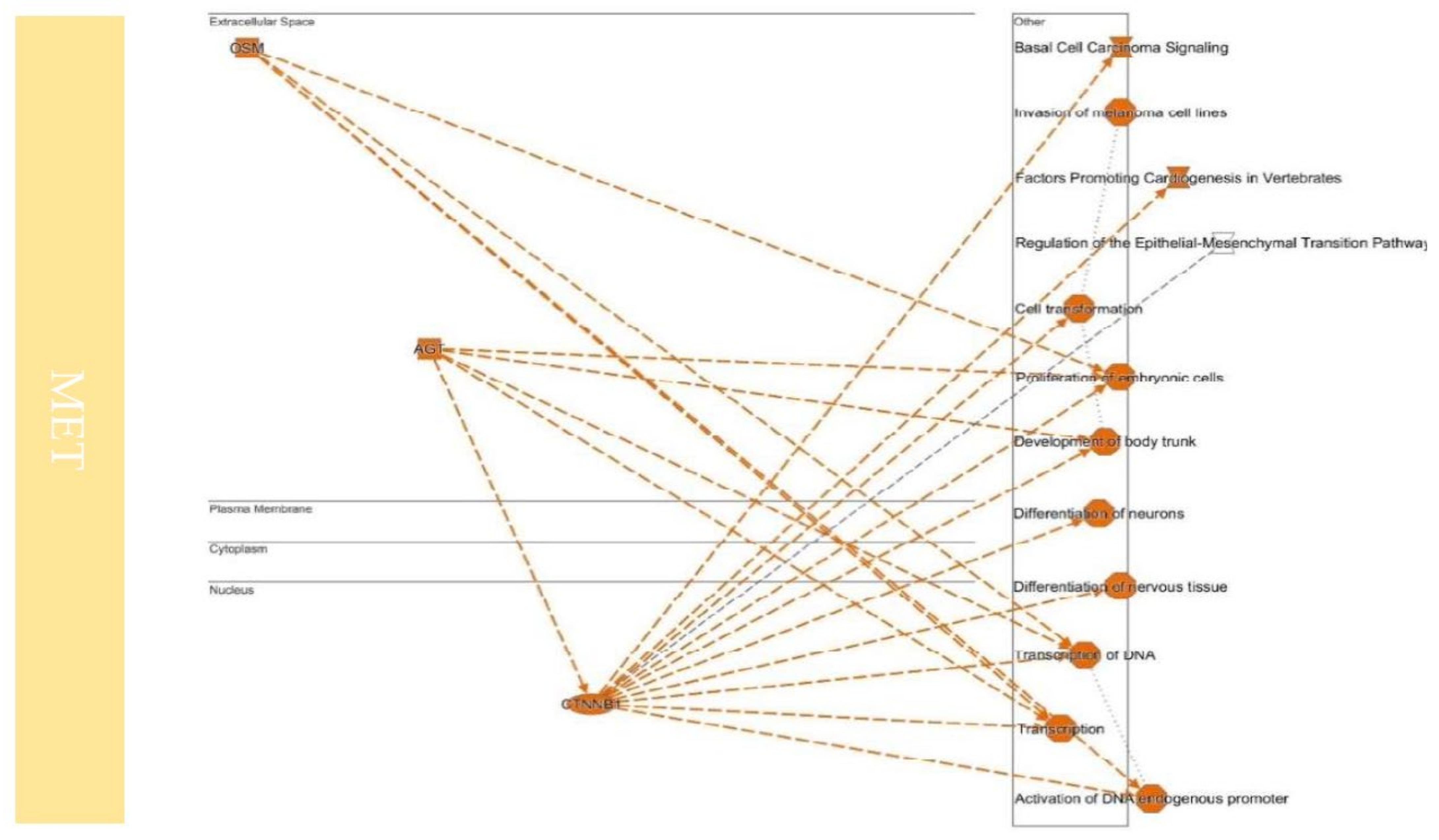
9. Colonization between EMT and MET
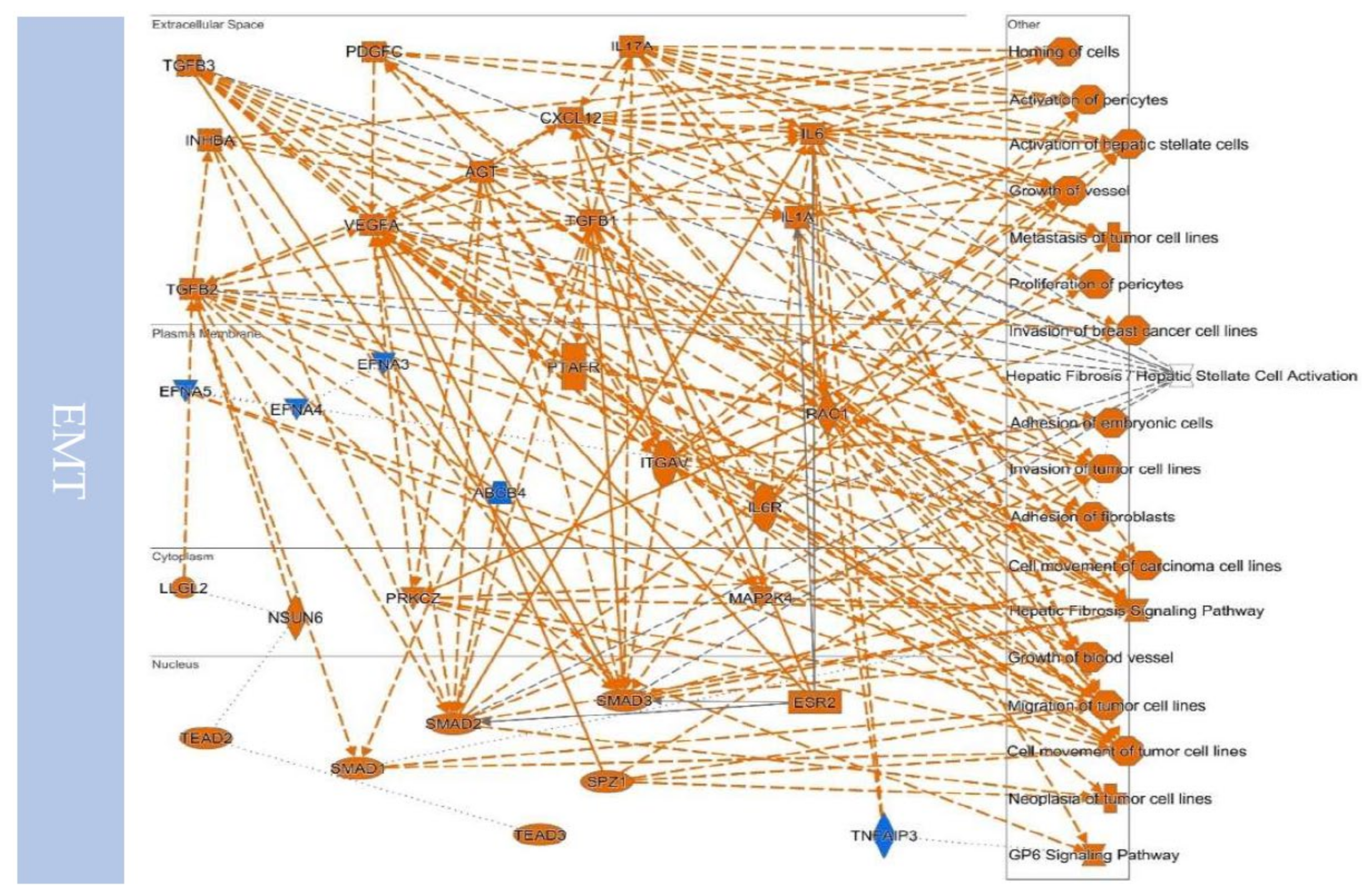
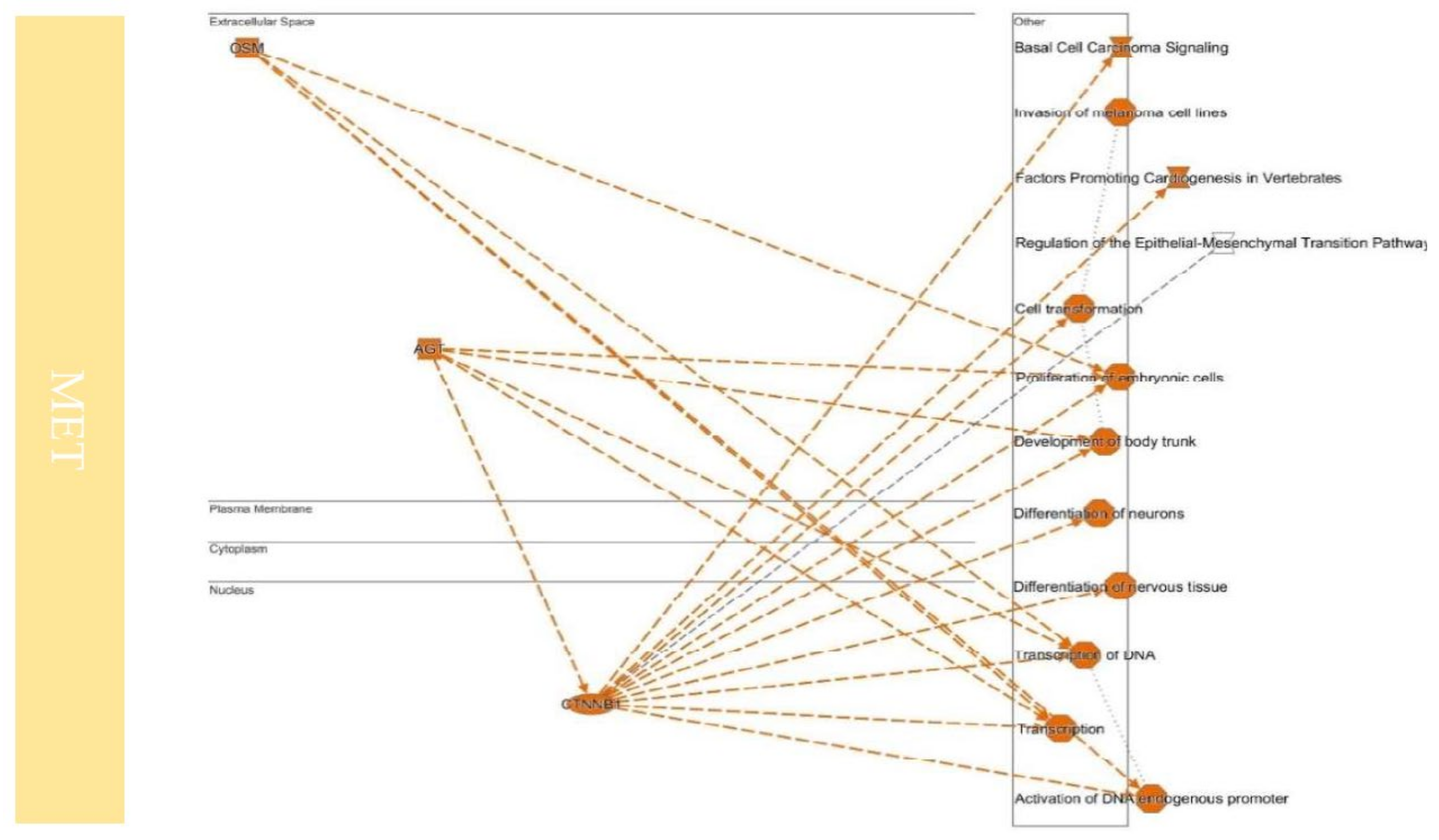
10. Defined EMT Molecules in Cancer
10.1. Cytoskeleton Remodeling
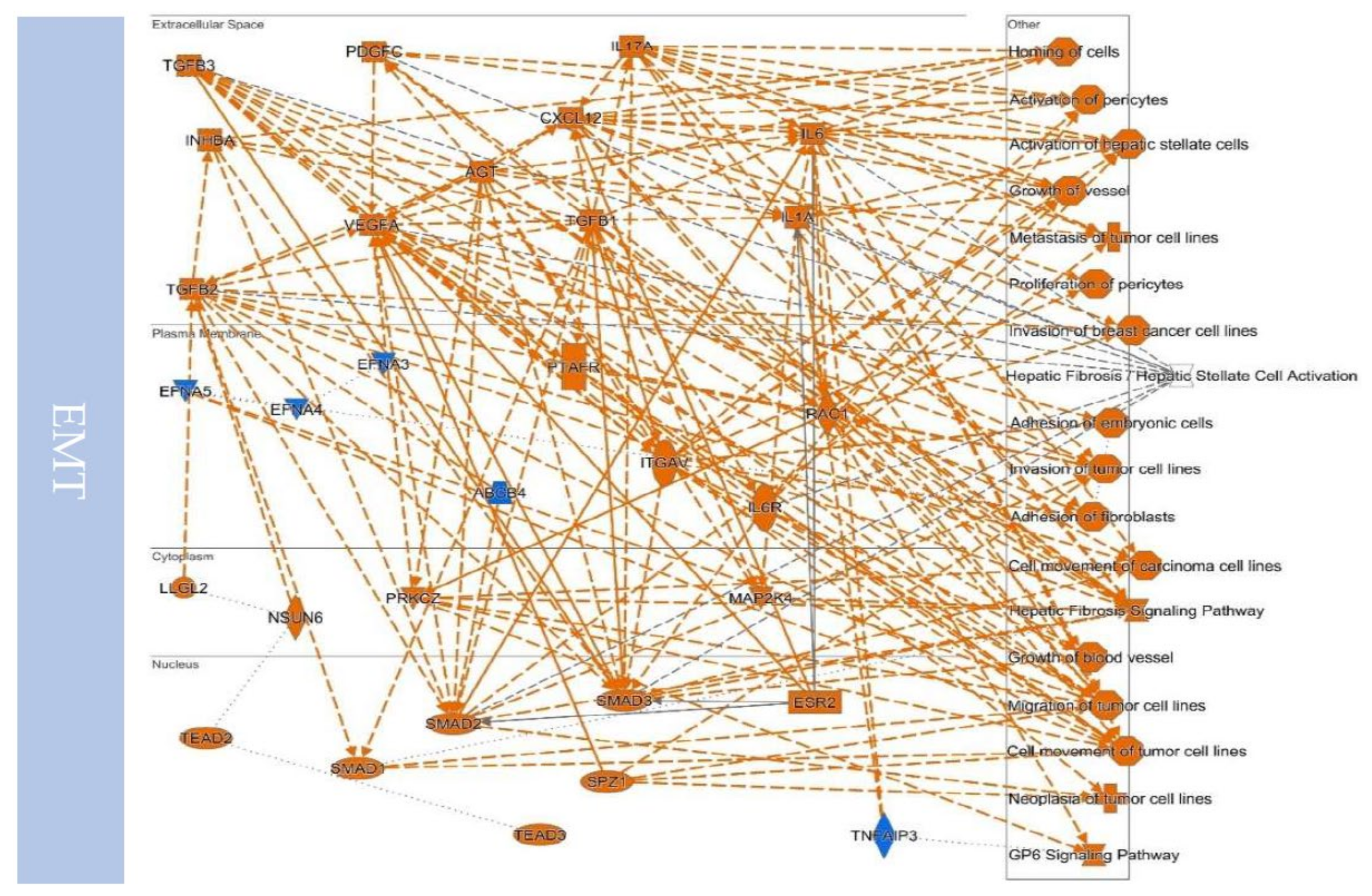
10.2. EMT to Metastasis
10.3. Remodeling the Microenvironment
10.4. Upstream Regulators Contribute to Determining the EMT/MET in Cancers
10.5. Stemness and EMT/MET
11. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Thiery, J.P.; Sleeman, J.P. Complex networks orchestrate epithelial-mesenchymal transitions. Nat. Rev. Mol. Cell. Biol. 2006, 7, 131–142. [Google Scholar] [CrossRef]
- Thomson, J.A.; Itskovitz-Eldor, J.; Shapiro, S.S.; Waknitz, M.A.; Swiergiel, J.J.; Marshall, V.S.; Jones, J.M. Embryonic stem cell lines derived from human blastocysts. Science 1998, 282, 1145–1147. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Hutchins, A.P.; Chen, Y.; Li, S.; Shan, Y.; Liao, B.; Zheng, D.; Shi, X.; Li, Y.; Chan, W.Y.; et al. A sequential EMT-MET mechanism drives the differentiation of human embryonic stem cells towards hepatocytes. Nat. Commun. 2017, 8, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Massri, A.J.; Schiebinger, G.R.; Berrio, A.; Wang, L.; Wray, G.A.; McClay, D.R. Methodologies for Following EMT In Vivo at Single Cell Resolution. Methods Mol. Biol. 2021, 2179, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Osborne, C.C.; Perry, K.J.; Shankland, M.; Henry, J.Q. Ectomesoderm and epithelial-mesenchymal transition-related genes in spiralian development. Dev. Dyn. 2018, 247, 1097–1120. [Google Scholar] [CrossRef] [Green Version]
- Abnave, P.; Aboukhatwa, E.; Kosaka, N.; Thompson, J.; Hill, M.A.; Aboobaker, A.A. Epithelial-mesenchymal transition transcription factors control pluripotent adult stem cell migration in vivo in planarians. Development 2017, 144, 3440–3453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baum, B.; Settleman, J.; Quinlan, M.P. Transitions between epithelial and mesenchymal states in development and disease. Semin. Cell Dev. Biol. 2008, 19, 294–308. [Google Scholar] [CrossRef] [PubMed]
- Magie, C.R.; Daly, M.; Martindale, M.Q. Gastrulation in the cnidarian Nematostella vectensis occurs via invagination not ingression. Dev. Biol. 2007, 305, 483–497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.M.; Dedhar, S.; Kalluri, R.; Thompson, E.W. The epithelial-mesenchymal transition: New insights in signaling, development, and disease. J. Cell Biol. 2006, 172, 973–981. [Google Scholar] [CrossRef]
- Ebnet, K. Organization of multiprotein complexes at cell-cell junctions. Histochem. Cell Biol. 2008, 130, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Lachowicz-Scroggins, M.E.; Gordon, E.D.; Wesolowska-Andersen, A.; Jackson, N.D.; MacLeod, H.J.; Sharp, L.Z.; Sun, M.; Seibold, M.A.; Fahy, J.V. Cadherin-26 (CDH26) regulates airway epithelial cell cytoskeletal structure and polarity. Cell Discov. 2018, 4, 7. [Google Scholar] [CrossRef] [Green Version]
- Wong, M.H.; Hermiston, M.L.; Syder, A.J.; Gordon, J.I. Forced expression of the tumor suppressor adenomatosis polyposis coli protein induces disordered cell migration in the intestinal epithelium. Proc. Natl. Acad. Sci. USA 1996, 93, 9588–9593. [Google Scholar] [CrossRef] [Green Version]
- Fouquet, S.; Lugo-Martinez, V.H.; Faussat, A.M.; Renaud, F.; Cardot, P.; Chambaz, J.; Pincon-Raymond, M.; Thenet, S. Early loss of E-cadherin from cell-cell contacts is involved in the onset of Anoikis in enterocytes. J. Biol. Chem. 2004, 279, 43061–43069. [Google Scholar] [CrossRef] [Green Version]
- Lim, J.; Thiery, J.P. Epithelial-mesenchymal transitions: Insights from development. Development 2012, 139, 3471–3486. [Google Scholar] [CrossRef] [Green Version]
- Rabadan, M.A.; Herrera, A.; Fanlo, L.; Usieto, S.; Carmona-Fontaine, C.; Barriga, E.H.; Mayor, R.; Pons, S.; Marti, E. Delamination of neural crest cells requires transient and reversible Wnt inhibition mediated by Dact1/2. Development 2016, 143, 2194–2205. [Google Scholar] [CrossRef] [Green Version]
- Scully, D.; Keane, E.; Batt, E.; Karunakaran, P.; Higgins, D.F.; Itasaki, N. Hypoxia promotes production of neural crest cells in the embryonic head. Development 2016, 143, 1742–1752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosada, F.M.; Devasthali, V.; Jones, K.A.; Stankunas, K. Wnt/beta-catenin signaling enables developmental transitions during valvulogenesis. Development 2016, 143, 1041–1054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chalamalasetty, R.B.; Garriock, R.J.; Dunty, W.C., Jr.; Kennedy, M.W.; Jailwala, P.; Si, H.; Yamaguchi, T.P. Mesogenin 1 is a master regulator of paraxial presomitic mesoderm differentiation. Development 2014, 141, 4285–4297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pijnenborg, R.; Dixon, G.; Robertson, W.B.; Brosens, I. Trophoblastic invasion of human decidua from 8 to 18 weeks of pregnancy. Placenta 1980, 1, 3–19. [Google Scholar] [CrossRef]
- Camenisch, T.D.; Schroeder, J.A.; Bradley, J.; Klewer, S.E.; McDonald, J.A. Heart-valve mesenchyme formation is dependent on hyaluronan-augmented activation of ErbB2-ErbB3 receptors. Nat. Med. 2002, 8, 850–855. [Google Scholar] [CrossRef]
- Park, I.H.; Zhao, R.; West, J.A.; Yabuuchi, A.; Huo, H.; Ince, T.A.; Lerou, P.H.; Lensch, M.W.; Daley, G.Q. Reprogramming of human somatic cells to pluripotency with defined factors. Nature 2008, 451, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Abad, M.; Mosteiro, L.; Pantoja, C.; Canamero, M.; Rayon, T.; Ors, I.; Grana, O.; Megias, D.; Dominguez, O.; Martinez, D.; et al. Reprogramming in vivo produces teratomas and iPS cells with totipotency features. Nature 2013, 502, 340–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyer, B.; Thiery, J.P. Epithelium-Mesenchyme Interconversion as Example of Epithelial Plasticity. Apmis 1993, 101, 257–268. [Google Scholar] [CrossRef]
- Shen, C.N.; Burke, Z.D.; Tosh, D. Transdifferentiation, metaplasia and tissue regeneration. Organogenesis 2004, 1, 36–44. [Google Scholar] [CrossRef] [Green Version]
- Lin, T.Y.; Gerber, T.; Taniguchi-Sugiura, Y.; Murawala, P.; Hermann, S.; Grosser, L.; Shibata, E.; Treutlein, B.; Tanaka, E.M. Fibroblast dedifferentiation as a determinant of successful regeneration. Dev. Cell 2021, 56, 1541–1551. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, A.C.; Costa, T.F.; Andrade, Z.A.; Medrado, A.R. Wound healing—A literature review. An. Bras. Dermatol. 2016, 91, 614–620. [Google Scholar] [CrossRef] [Green Version]
- Muller, M.; Hermann, P.C.; Liebau, S.; Weidgang, C.; Seufferlein, T.; Kleger, A.; Perkhofer, L. The role of pluripotency factors to drive stemness in gastrointestinal cancer. Stem Cell Res. 2016, 16, 349–357. [Google Scholar] [CrossRef] [Green Version]
- Mittal, V. Epithelial Mesenchymal Transition in Tumor Metastasis. Annu. Rev. Pathol. 2018, 13, 395–412. [Google Scholar] [CrossRef] [PubMed]
- Bakir, B.; Chiarella, A.M.; Pitarresi, J.R.; Rustgi, A.K. EMT, MET, Plasticity, and Tumor Metastasis. Trends Cell Biol. 2020, 30, 764–776. [Google Scholar] [CrossRef] [PubMed]
- Tsai, J.H.; Yang, J. Epithelial-mesenchymal plasticity in carcinoma metastasis. Genes Dev. 2013, 27, 2192–2206. [Google Scholar] [CrossRef] [Green Version]
- Van der Pluijm, G. Epithelial plasticity, cancer stem cells and bone metastasis formation. Bone 2011, 48, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Fares, J.; Fares, M.Y.; Khachfe, H.H.; Salhab, H.A.; Fares, Y. Molecular principles of metastasis: A hallmark of cancer revisited. Signal Transduct. Target. Ther. 2020, 5. [Google Scholar] [CrossRef]
- Chiang, S.P.; Cabrera, R.M.; Segall, J.E. Tumor cell intravasation. Am. J. Physiol. Cell Physiol. 2016, 311, C1–C14. [Google Scholar] [CrossRef] [Green Version]
- Quigley, J.P.; Armstrong, P.B. Tumor cell intravasation alu-cidated: The chick embryo opens the window. Cell 1998, 94, 281–284. [Google Scholar] [CrossRef] [Green Version]
- Moens, E.; Veldhoen, M. Epithelial barrier biology: Good fences make good neighbours. Immunology 2012, 135, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Rovensky Yu, A.; Domnina, L.V.; Ivanova, O.; Vasiliev, J.M. Responses of epithelial and fibroblast-like cells to discontinuous configuration of the culture substrate. Membr. Cell Biol. 2001, 14, 617–627. [Google Scholar] [PubMed]
- Brunette, D.M.; Melcher, A.H.; Moe, H.K. Culture and origin of epithelium-like and fibroblast-like cells from porcine periodontal ligament explants and cell suspensions. Arch. Oral. Biol. 1976, 21, 393–400. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [Green Version]
- Gurtner, G.C.; Werner, S.; Barrandon, Y.; Longaker, M.T. Wound repair and regeneration. Nature 2008, 453, 314–321. [Google Scholar] [CrossRef]
- Eming, S.A.; Krieg, T.; Davidson, J.M. Inflammation in wound repair: Molecular and cellular mechanisms. J. Investig. Dermatol. 2007, 127, 514–525. [Google Scholar] [CrossRef] [Green Version]
- Nieto, M.A.; Huang, R.Y.; Jackson, R.A.; Thiery, J.P. Emt: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [Green Version]
- Poncelet, C.; Leblanc, M.; Walker-Combrouze, F.; Soriano, D.; Feldmann, G.; Madelenat, P.; Scoazec, J.Y.; Darai, E. Expression of cadherins and CD44 isoforms in human endometrium and peritoneal endometriosis. Acta Obstet. Gynecol. Scand. 2002, 81, 195–203. [Google Scholar] [CrossRef]
- Zeisberg, M.; Neilson, E.G. Biomarkers for epithelial-mesenchymal transitions. J. Clin. Investig. 2009, 119, 1429–1437. [Google Scholar] [CrossRef] [Green Version]
- Shenoy, S. Cell plasticity in cancer: A complex interplay of genetic, epigenetic mechanisms and tumor micro-environment. Surg. Oncol. 2020, 34, 154–162. [Google Scholar] [CrossRef]
- Illam, S.P.; Narayanankutty, A.; Mathew, S.E.; Valsalakumari, R.; Jacob, R.M.; Raghavamenon, A.C. Epithelial Mesenchymal Transition in Cancer Progression: Prev entive Phytochemicals. Recent Pat. Anticancer Drug Discov. 2017, 12, 234–246. [Google Scholar] [CrossRef] [PubMed]
- Beck, T.N.; Chikwem, A.J.; Solanki, N.R.; Golemis, E.A. Bioinformatic approaches to augment study of epithelial-to-mesenchymal transition in lung cancer. Physiol. Genom. 2014, 46, 699–724. [Google Scholar] [CrossRef] [PubMed]
- Chapman, H.A. Epithelial responses to lung injury: Role of the extracellular matrix. Proc. Am. Thorac. Soc. 2012, 9, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Baronsky, T.; Ruhlandt, D.; Bruckner, B.R.; Schafer, J.; Karedla, N.; Isbaner, S.; Hahnel, D.; Gregor, I.; Enderlein, J.; Janshoff, A.; et al. Cell-Substrate Dynamics of the Epithelial-to-Mesenchymal Transition. Nano Lett. 2017, 17, 3320–3326. [Google Scholar] [CrossRef]
- Park, S.M.; Kim, S.M.; Han, J.H. The role of epithelial-mesenchymal transition in the gastroenterology. Korean J. Gastroenterol. 2010, 56, 69–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Zhang, Y.; Zhu, D. Myostatin promotes the epithelial-to-mesenchymal transition of the dermomyotome during somitogenesis. Dev. Dyn. 2018, 247, 1241–1252. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.J.; Kim, H.H.; Kim, J.G.; Kim, H.J.; Kang, M.; Lee, M.S.; Ryu, J.; Song, H.E.; Nam, S.H.; Lee, D.; et al. TM4SF5 suppression disturbs integrin alpha5-related signalling and muscle development in zebrafish. Biochem. J. 2014, 462, 89–101. [Google Scholar] [CrossRef]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef]
- Peinado, H.; Olmeda, D.; Cano, A. Snail, Zeb and bHLH factors in tumour progression: An alliance against the epithelial phenotype? Nat. Rev. Cancer 2007, 7, 415–428. [Google Scholar] [CrossRef]
- Wu, S.Y.; Ferkowicz, M.; McClay, D.R. Ingression of primary mesenchyme cells of the sea urchin embryo: A precisely timed epithelial mesenchymal transition. Birth Defects Res. Part C Embryo Today Rev. 2007, 81, 241–252. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.Y.; Yang, Y.P.; McClay, D.R. Twist is an essential regulator of the skeletogenic gene regulatory network in the sea urchin embryo. Dev. Biol. 2008, 319, 406–415. [Google Scholar] [CrossRef] [Green Version]
- Oliveri, P.; Tu, Q.; Davidson, E.H. Global regulatory logic for specification of an embryonic cell lineage. Proc. Natl. Acad. Sci. USA 2008, 105, 5955–5962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suyama, K.; Shapiro, I.; Guttman, M.; Hazan, R.B. A signaling pathway leading to metastasis is controlled by N-cadherin and the FGF receptor. Cancer Cell 2002, 2, 301–314. [Google Scholar] [CrossRef] [Green Version]
- Wicki, A.; Lehembre, F.; Wick, N.; Hantusch, B.; Kerjaschki, D.; Christofori, G. Tumor invasion in the absence of epithelial-mesenchymal transition: Podoplanin-mediated remodeling of the actin cytoskeleton. Cancer Cell 2006, 9, 261–272. [Google Scholar] [CrossRef] [PubMed]
- Frieden, L.A.; Townsend, T.A.; Vaught, D.B.; Delaughter, D.M.; Hwang, Y.; Barnett, J.V.; Chen, J. Regulation of heart valve morphogenesis by Eph receptor ligand, ephrin-A1. Dev. Dyn. 2010, 239, 3226–3234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luna-Zurita, L.; Prados, B.; Grego-Bessa, J.; Luxan, G.; del Monte, G.; Benguria, A.; Adams, R.H.; Perez-Pomares, J.M.; de la Pompa, J.L. Integration of a Notch-dependent mesenchymal gene program and Bmp2-driven cell invasiveness regulates murine cardiac valve formation. J. Clin. Investig. 2010, 120, 3493–3507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stankunas, K.; Ma, G.K.; Kuhnert, F.J.; Kuo, C.J.; Chang, C.P. VEGF signaling has distinct spatiotemporal roles during heart valve development. Dev. Biol. 2010, 347, 325–336. [Google Scholar] [CrossRef] [Green Version]
- Manzanares, M.; Locascio, A.; Nieto, M.A. The increasing complexity of the Snail gene superfamily in metazoan evolution. Trends Genet. 2001, 17, 178–181. [Google Scholar] [CrossRef]
- Fischer, A.; Steidl, C.; Wagner, T.U.; Lang, E.; Jakob, P.M.; Friedl, P.; Knobeloch, K.P.; Gessler, M. Combined loss of Hey1 and HeyL causes congenital heart defects because of impaired epithelial to mesenchymal transition. Circ. Res. 2007, 100, 856–863. [Google Scholar] [CrossRef] [Green Version]
- Radisky, D.C.; Kenny, P.A.; Bissell, M.J. Fibrosis and cancer: Do myofibroblasts come also from epithelial cells via EMT? J. Cell Biochem. 2007, 101, 830–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, S.; Dipietro, L.A. Factors affecting wound healing. J. Dent. Res. 2010, 89, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, H.N.; Hardman, M.J. Wound healing: Cellular mechanisms and pathological outcomes. Open Biol. 2020, 10, 200223. [Google Scholar] [CrossRef] [PubMed]
- Imanishi, J.; Kamiyama, K.; Iguchi, I.; Kita, M.; Sotozono, C.; Kinoshita, S. Growth factors: Importance in wound healing and maintenance of transparency of the cornea. Prog. Retin. Eye Res. 2000, 19, 113–129. [Google Scholar] [CrossRef]
- Chen, S.Y.; Xie, C.; Zhu, H.; Shen, Y. Effects of epidermal growth factor on transforming growth factor-beta1-induced epithelial-mesenchymal transition and potential mechanism in human cornea epithelial cells. Int. J. Ophthalmol. 2020, 13, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Qiang, L.; Sample, A.; Liu, H.; Wu, X.; He, Y.Y. Epidermal SIRT1 regulates inflammation, cell migration, and wound healing. Sci. Rep. 2017, 7, 14110. [Google Scholar] [CrossRef] [PubMed]
- Lou, B.S.; Hsieh, J.H.; Chen, C.M.; Hou, C.W.; Wu, H.Y.; Chou, P.Y.; Lai, C.H.; Lee, J.W. Helium/Argon-Generated Cold Atmospheric Plasma Facilitates Cutaneous Wound Healing. Front. Bioeng. Biotechnol. 2020, 8, 683. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Xing, T.; Yang, Z.; Dudek, R.; Lu, Q.; Chen, Y.H. Epithelial Mesenchymal Transition in Embryonic Development, Tissue Repair and Cancer: A Comprehensive Overview. J. Clin. Med. 2017, 7, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plygawko, A.T.; Kan, S.; Campbell, K. Epithelial-mesenchymal plasticity: Emerging parallels between tissue morphogenesis and cancer metastasis. Philos. Trans. R. Soc. B Biol. Sci. 2020, 375, 20200087. [Google Scholar] [CrossRef]
- Wang, H.; Unternaehrer, J.J. Epithelial-mesenchymal Transition and Cancer Stem Cells: At the Crossroads of Differentiation and Dedifferentiation. Dev. Dyn. 2019, 248, 10–20. [Google Scholar] [CrossRef] [Green Version]
- Satoh, A.; Bryant, S.V.; Gardiner, D.M. Regulation of dermal fibroblast dedifferentiation and redifferentiation during wound healing and limb regeneration in the Axolotl. Dev. Growth Differ. 2008, 50, 743–754. [Google Scholar] [CrossRef] [PubMed]
- Van Caam, A.; Vonk, M.; van den Hoogen, F.; van Lent, P.; van der Kraan, P. Unraveling SSc Pathophysiology; The Myofibroblast. Front. Immunol. 2018, 9, 2452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saadat, S.; Noureddini, M.; Mahjoubin-Tehran, M.; Nazemi, S.; Shojaie, L.; Aschner, M.; Maleki, B.; Abbasi-kolli, M.; Moghadam, H.R.; Alani, B.; et al. Pivotal Role of TGF-beta/Smad Signaling in Cardiac Fibrosis: Non-coding RNAs as Effectual Players. Front. Cardiovasc. Med. 2021, 7. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. Transforming growth factor-beta in tissue fibrosis. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef]
- Anscher, M.S. Targeting the TGF-beta 1 Pathway to Prevent Normal Tissue Injury After Cancer Therapy. Oncologist 2010, 15, 350–359. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.L.; Zhu, R.T.; Sun, Y.L. Epithelial-mesenchymal transition in liver fibrosis. Biomed. Rep. 2016, 4, 269–274. [Google Scholar] [CrossRef] [Green Version]
- Saraswati, S.; Marrow, S.M.W.; Watch, L.A.; Young, P.P. Identification of a pro-angiogenic functional role for FSP1-positive fibroblast subtype in wound healing. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef]
- Bagalad, B.S.; Mohan Kumar, K.P.; Puneeth, H.K. Myofibroblasts: Master of disguise. J. Oral Maxillofac. Pathol. 2017, 21, 462–463. [Google Scholar] [CrossRef]
- Phan, S.H. Biology of fibroblasts and myofibroblasts. Proc. Am. Thorac. Soc. 2008, 5, 334–337. [Google Scholar] [CrossRef] [PubMed]
- Mathison, M.; Singh, V.P.; Sanagasetti, D.; Yang, L.; Pinnamaneni, J.P.; Yang, J.C.; Rosengart, T.K. Cardiac reprogramming factor Gata4 reduces postinfarct cardiac fibrosis through direct repression of the profibrotic mediator snail. J. Thorac. Cardiovasc. Surg. 2017, 154, 1601–1610. [Google Scholar] [CrossRef] [Green Version]
- Song, G.Q.; Pacher, M.; Balakrishnan, A.; Yuan, Q.G.; Tsay, H.C.; Yang, D.K.; Reetz, J.; Brandes, S.; Dai, Z.; Putzer, B.M.; et al. Direct Reprogramming of Hepatic Myofibroblasts into Hepatocytes In Vivo Attenuates Liver Fibrosis. Cell Stem Cell 2016, 18, 797–808. [Google Scholar] [CrossRef] [Green Version]
- Ishimoto, T.; Miyake, K.; Nandi, T.; Yashiro, M.; Onishi, N.; Huang, K.K.; Lin, S.J.; Kalpana, R.; Tay, S.T.; Suzuki, Y.; et al. Activation of Transforming Growth Factor Beta 1 Signaling in Gastric Cancer-associated Fibroblasts Increases Their Motility, via Expression of Rhomboid 5 Homolog 2, and Ability to Induce Invasiveness of Gastric Cancer Cells. Gastroenterology 2017, 153, 191–204. [Google Scholar] [CrossRef]
- Shintani, Y.; Fujiwara, A.; Kimura, T.; Kawamura, T.; Funaki, S.; Minami, M.; Okumura, M. IL-6 Secreted from Cancer-Associated Fibroblasts Mediates Chemoresistance in NSCLC by Increasing Epithelial-Mesenchymal Transition Signaling. J. Thorac. Oncol. 2016, 11, 1482–1492. [Google Scholar] [CrossRef] [Green Version]
- Malfettone, A.; Soukupova, J.; Bertran, E.; Crosas-Molist, E.; Lastra, R.; Fernando, J.; Koudelkova, P.; Rani, B.; Fabra, A.; Serrano, T.; et al. Transforming growth factor-beta-induced plasticity causes a migratory stemness phenotype in hepatocellular carcinoma. Cancer Lett. 2017, 392, 39–50. [Google Scholar] [CrossRef]
- Poiana, C.; Neamtu, M.C.; Avramescu, E.T.; Carsote, M.; Trifanescu, R.; Terzea, D.; Neamtu, O.M.; Miulescu, R.D. The dedifferentiation of neuroendocrine tumor metastases: Myth or reality? Rom. J. Morphol. Embryol 2013, 54, 201–203. [Google Scholar]
- Thewjitcharoen, Y.; Krittiyawong, S.; Butadej, S.; Nakasatien, S.; Polchart, S.; Junyangdikul, P.; Kanchanapituk, A.; Himathongkam, T. De-differentiation of papillary thyroid carcinoma into squamous cell carcinoma in an elderly patient A case report. Medicine 2020, 99. [Google Scholar] [CrossRef] [PubMed]
- Pardee, A.B. Molecules involved in proliferation of normal and cancer cells: Presidential address. Cancer Res. 1987, 47, 1488–1491. [Google Scholar] [PubMed]
- Baghban, R.; Roshangar, L.; Jahanban-Esfahlan, R.; Seidi, K.; Ebrahimi-Kalan, A.; Jaymand, M.; Kolahian, S.; Javaheri, T.; Zare, P. Tumor microenvironment complexity and therapeutic implications at a glance. Cell Commun. Signal. 2020, 18, 59. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro Franco, P.I.; Rodrigues, A.P.; de Menezes, L.B.; Pacheco Miguel, M. Tumor microenvironment components: Allies of cancer progression. Pathol. Res. Pract. 2020, 216, 152729. [Google Scholar] [CrossRef] [PubMed]
- Maishi, N.; Hida, K. Tumor endothelial cells accelerate tumor metastasis. Cancer Sci. 2017, 108, 1921–1926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hida, K.; Maishi, N.; Annan, D.A.; Hida, Y. Contribution of Tumor Endothelial Cells in Cancer Progression. Int. J. Mol. Sci. 2018, 19, 1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohshima, K.; Morii, E. Metabolic Reprogramming of Cancer Cells during Tumor Progression and Metastasis. Metabolites 2021, 11, 28. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Liu, Y.; Xiong, W.; Zhang, L.; Liu, H.; Du, Y.; Li, N. Hypoxia-inducible factor 1alpha-induced epithelial-mesenchymal transition of endometrial epithelial cells may contribute to the development of endometriosis. Hum. Reprod. 2016, 31, 1327–1338. [Google Scholar] [CrossRef] [Green Version]
- Chapman, J.D.; Coia, L.R.; Stobbe, C.C.; Engelhardt, E.L.; Fenning, M.C.; Schneider, R.F. Prediction of tumour hypoxia and radioresistance with nuclear medicine markers. Br. J. Cancer Suppl. 1996, 27, S204–S208. [Google Scholar]
- Revenco, T.; Nicodeme, A.; Pastushenko, I.; Sznurkowska, M.K.; Latil, M.; Sotiropoulou, P.A.; Dubois, C.; Moers, V.; Lemaire, S.; de Maertelaer, V.; et al. Context Dependency of Epithelial-to-Mesenchymal Transition for Metastasis. Cell Rep. 2019, 29, 1458–1468. [Google Scholar] [CrossRef]
- Derksen, P.W.; Liu, X.; Saridin, F.; van der Gulden, H.; Zevenhoven, J.; Evers, B.; van Beijnum, J.R.; Griffioen, A.W.; Vink, J.; Krimpenfort, P.; et al. Somatic inactivation of E-cadherin and p53 in mice leads to metastatic lobular mammary carcinoma through induction of anoikis resistance and angiogenesis. Cancer Cell 2006, 10, 437–449. [Google Scholar] [CrossRef] [Green Version]
- Ruiz, P.; Gunthert, U. The cellular basis of metastasis. World J. Urol. 1996, 14, 141–150. [Google Scholar] [CrossRef]
- Itoh, Y. Membrane-type matrix metalloproteinases: Their functions and regulations. Matrix Biol. 2015, 44-46, 207–223. [Google Scholar] [CrossRef]
- Guo, X.; Rao, J.N.; Liu, L.; Zou, T.; Keledjian, K.M.; Boneva, D.; Marasa, B.S.; Wang, J.Y. Polyamines are necessary for synthesis and stability of occludin protein in intestinal epithelial cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 288, G1159–G1169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Z.; Ghosh, S.; Wang, Z.; Hunter, T. Downregulation of caveolin-1 function by EGF leads to the loss of E-cadherin, increased transcriptional activity of beta-catenin, and enhanced tumor cell invasion. Cancer Cell 2003, 4, 499–515. [Google Scholar] [CrossRef] [Green Version]
- Mierke, C.T. The matrix environmental and cell mechanical properties regulate cell migration and contribute to the invasive phenotype of cancer cells. Rep. Prog. Phys. 2019, 82, 064602. [Google Scholar] [CrossRef]
- Gos, M.; Miloszewska, J.; Przybyszewska, M. Epithelial-mesenchymal transition in cancer progression. Postepy Biochem. 2009, 55, 121–128. [Google Scholar] [PubMed]
- Peng, D.; Fu, L.; Sun, G. Expression analysis of the TGF-beta/SMAD target genes in adenocarcinoma of esophagogastric junction. Open Med. (Wars) 2016, 11, 83–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gambichler, T.; Skrygan, M.; Kaczmarczyk, J.M.; Hyun, J.; Tomi, N.S.; Sommer, A.; Bechara, F.G.; Boms, S.; Brockmeyer, N.H.; Altmeyer, P.; et al. Increased expression of TGF-beta/Smad proteins in basal cell carcinoma. Eur. J. Med. Res. 2007, 12, 509–514. [Google Scholar]
- Stankic, M.; Pavlovic, S.; Chin, Y.; Brogi, E.; Padua, D.; Norton, L.; Massague, J.; Benezra, R. TGF-beta-Id1 signaling opposes Twist1 and promotes metastatic colonization via a mesenchymal-to-epithelial transition. Cell Rep. 2013, 5, 1228–1242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jana, A.; Krett, N.L.; Guzman, G.; Khalid, A.; Ozden, O.; Staudacher, J.J.; Bauer, J.; Baik, S.H.; Carroll, T.; Yazici, C.; et al. NFkB is essential for activin-induced colorectal cancer migration via upregulation of PI3K-MDM2 pathway. Oncotarget 2017, 8, 37377–37393. [Google Scholar] [CrossRef] [Green Version]
- Aiello, N.M.; Maddipati, R.; Norgard, R.J.; Balli, D.; Li, J.; Yuan, S.; Yamazoe, T.; Black, T.; Sahmoud, A.; Furth, E.E.; et al. EMT Subtype Influences Epithelial Plasticity and Mode of Cell Migration. Dev. Cell 2018, 45, 681–695. [Google Scholar] [CrossRef] [Green Version]
- Yamaji, H.; Fukuda, H. Long-term cultivation of anchorage-independent animal cells immobilized within reticulated biomass support particles in a circulating bed fermentor. Appl. Microbiol. Biotechnol. 1991, 34, 730–734. [Google Scholar] [CrossRef]
- Paoli, P.; Giannoni, E.; Chiarugi, P. Anoikis molecular pathways and its role in cancer progression. Appl. Microbiol. Biotechnol. 2013, 1833, 3481–3498. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.R.; Lin, H.M.; Biliran, H.; Raz, A. Cell cycle arrest and inhibition of anoikis by galectin-3 in human breast epithelial cells. Cancer Res. 1999, 59, 4148–4154. [Google Scholar]
- Robinson, R.A.; TenEyck, C.J.; Hart, M.N. Establishment and preliminary growth characteristics of a transformed mouse cerebral microvessel endothelial cell line. Lab. Investig. 1986, 54, 579–588. [Google Scholar]
- Pauli, B.U.; Lee, C.L. Organ preference of metastasis. The role of organ-specifically modulated endothelial cells. Lab. Investig. 1988, 58, 379–387. [Google Scholar] [PubMed]
- Evdokimova, V.; Tognon, C.; Ng, T.; Sorensen, P.H. Reduced proliferation and enhanced migration: Two sides of the same coin? Molecular mechanisms of metastatic progression by YB-1. Cell Cycle 2009, 8, 2901–2906. [Google Scholar] [CrossRef] [PubMed]
- Deryugina, E.I.; Kiosses, W.B. Intratumoral Cancer Cell Intravasation Can Occur Independent of Invasion into the Adjacent Stroma. Cell Rep. 2017, 19, 601–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kemp, K.C.; Hows, J.; Donaldson, C. Bone marrow-derived mesenchymal stem cells. Leuk. Lymphoma 2005, 46, 1531–1544. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Li, Q. Cancer stem cells and tumor metastasis (Review). Int. J. Oncol. 2014, 44, 1806–1812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Somarelli, J.A.; Schaeffer, D.; Marengo, M.S.; Bepler, T.; Rouse, D.; Ware, K.E.; Hish, A.J.; Zhao, Y.; Buckley, A.F.; Epstein, J.I.; et al. Distinct routes to metastasis: Plasticity-dependent and plasticity-independent pathways. Oncogene 2016, 35, 4302–4311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearson, G.W. Control of Invasion by Epithelial-to-Mesenchymal Transition Programs during Metastasis. J. Clin. Med. 2019, 8, 646. [Google Scholar] [CrossRef] [Green Version]
- Kalluri, R. EMT: When epithelial cells decide to become mesenchymal-like cells. J. Clin. Investig. 2009, 119, 1417–1419. [Google Scholar] [CrossRef] [Green Version]
- Inan, S.; Hayran, M. Cell Signaling Pathways Related to Epithelial Mesenchymal Transition in Cancer Metastasis. Crit. Rev. Oncog. 2019, 24, 47–54. [Google Scholar] [CrossRef]
- Zhu, Q.Q.; Ma, C.; Wang, Q.; Song, Y.; Lv, T. The role of TWIST1 in epithelial-mesenchymal transition and cancers. Tumor Biol. 2016, 37, 185–197. [Google Scholar] [CrossRef]
- Takano, S.; Reichert, M.; Bakir, B.; Das, K.K.; Nishida, T.; Miyazaki, M.; Heeg, S.; Collins, M.A.; Marchand, B.; Hicks, P.D.; et al. Prrx1 isoform switching regulates pancreatic cancer invasion and metastatic colonization. Genes Dev. 2016, 30, 233–247. [Google Scholar] [CrossRef] [Green Version]
- Brabletz, T. EMT and MET in metastasis: Where are the cancer stem cells? Cancer Cell 2012, 22, 699–701. [Google Scholar] [CrossRef] [Green Version]
- Vasaikar, S.V.; Deshmukh, A.P.; den Hollander, P.; Addanki, S.; Kuburich, N.A.; Kudaravalli, S.; Joseph, R.; Chang, J.T.; Soundararajan, R.; Mani, S.A. EMTome: A resource for pan-cancer analysis of epithelial-mesenchymal transition genes and signatures. Brit. J. Cancer 2021, 124, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Weinberg, R.A. Epithelial-to-mesenchymal transition in cancer: Complexity and opportunities. Front. Med. 2018, 12, 361–373. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Settleman, J. EMT, cancer stem cells and drug resistance: An emerging axis of evil in the war on cancer. Oncogene 2010, 29, 4741–4751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christofori, G.; et al. Guidelines and definitions for research on epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2020, 21, 341–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [Green Version]
- Spiering, D.; Hodgson, L. Dynamics of the Rho-family small GTPases in actin regulation and motility. Cell Adhes. Migr. 2011, 5, 170–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, G.; Feldman, E.L. Signaling mechanisms that regulate actin-based motility processes in the nervous system. J. Neurochem. 2002, 83, 490–503. [Google Scholar] [CrossRef] [Green Version]
- Anton, I.M.; Gomez-Oro, C.; Rivas, S.; Wandosell, F. Crosstalk between WIP and Rho family GTPases. Small GTPases 2020, 11, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Lu, W.; Zou, S.; Wang, H.; Jiang, Y.; Zhang, X.; Li, P.; Songyang, Z.; Wang, L.; Wang, J.; et al. Rho GDP-dissociation inhibitor alpha is a potential prognostic biomarker and controls telomere regulation in colorectal cancer. Cancer Sci. 2017, 108, 1293–1302. [Google Scholar] [CrossRef]
- He, J.; Cai, L.; Chen, Y.; He, Y.; Wang, M.; Tang, J.; Guan, H.; Wang, J.; Peng, X. Antitumor and radiosensitizing effects of SKLB-163, a novel benzothiazole-2-thiol derivative, on nasopharyngeal carcinoma by affecting the RhoGDI/JNK-1 signaling pathway. Radiother. Oncol. 2018, 129, 30–37. [Google Scholar] [CrossRef]
- Yu, Y.; Jin, H.; Xu, J.; Gu, J.; Li, X.; Xie, Q.; Huang, H.; Li, J.; Tian, Z.; Jiang, G.; et al. XIAP overexpression promotes bladder cancer invasion in vitro and lung metastasis in vivo via enhancing nucleolin-mediated Rho-GDIbeta mRNA stability. Int. J. Cancer 2018, 142, 2040–2055. [Google Scholar] [CrossRef] [Green Version]
- Zhu, G.F.; Xu, Y.W.; Li, J.; Niu, H.L.; Ma, W.X.; Xu, J.; Zhou, P.R.; Liu, X.; Ye, D.L.; Liu, X.R.; et al. Mir20a/106a-WTX axis regulates RhoGDIa/CDC42 signaling and colon cancer progression. Nat. Commun. 2019, 10, 112. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.J.; Hwang, Y.S.; Yoon, J.; Lee, M.; Lee, H.G.; Daar, I.O. EphrinB1 promotes cancer cell migration and invasion through the interaction with RhoGDI1. Oncogene 2018, 37, 861–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiorillo, M.; Peiris-Pages, M.; Sanchez-Alvarez, R.; Bartella, L.; Di Donna, L.; Dolce, V.; Sindona, G.; Sotgia, F.; Cappello, A.R.; Lisanti, M.P. Bergamot natural products eradicate cancer stem cells (CSCs) by targeting mevalonate, Rho-GDI-signalling and mitochondrial metabolism. Biochim. Biophys. Acta Bioenerg. 2018, 1859, 984–996. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Ding, G.; Zhou, L.; Shen, T.; Xu, X.; Zhao, T.; Jia, S.; Cao, L. Interferon Gamma Inhibits CXCL8-Induced Proliferation and Migration of Pancreatic Cancer BxPC-3 Cell Line via a RhoGDI2/Rac1/NF-kappaB Signaling Pathway. J. Interf. Cytokine Res. 2018, 38, 413–422. [Google Scholar] [CrossRef] [PubMed]
- Vicente-Manzanares, M.; Choi, C.K.; Horwitz, A.R. Integrins in cell migration—the actin connection (vol 122, pg 199, 2009). J. Cell Sci 2009, 122, 1473. [Google Scholar] [CrossRef] [Green Version]
- Huttenlocher, A.; Horwitz, A.R. Integrins in Cell Migration. Cold Spring Harb. Perspect. Biol. 2011, 3, a005074. [Google Scholar] [CrossRef]
- Blystone, S.D. Integrating an integrin: A direct route to actin. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2004, 1692, 47–54. [Google Scholar] [CrossRef]
- Clark, E.A.; King, W.G.; Brugge, J.S.; Symons, M.; Hynes, R.O. Integrin-mediated signals regulated by members of the Rho family of GTPases. J. Cell Biol. 1998, 142, 573–586. [Google Scholar] [CrossRef] [Green Version]
- Arthur, W.T.; Noren, N.K.; Burridge, K. Regulation of Rho family GTPases by cell-cell and cell-matrix adhesion. Biol. Res. 2002, 35, 239–246. [Google Scholar] [CrossRef]
- Huang, W.; Zhu, J.; Shi, H.; Wu, Q.; Zhang, C. ITGA2 Overexpression Promotes Esophageal Squamous Cell Carcinoma Aggression via FAK/AKT Signaling Pathway. Onco Targets Ther. 2021, 14, 3583–3596. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, Y.; Che, X.; Hou, K.; Wu, J.; Zheng, C.; Cheng, Y.; Liu, Y.; Hu, X.; Zhang, J. Integrin alpha5 promotes migration and invasion through the FAK/STAT3/AKT signaling pathway in icotinib-resistant non-small cell lung cancer cells. Oncol. Lett. 2021, 22, 556. [Google Scholar] [CrossRef]
- Zhong, Y.; Long, T.; Gu, C.S.; Tang, J.Y.; Gao, L.F.; Zhu, J.X.; Hu, Z.Y.; Wang, X.; Ma, Y.D.; Ding, Y.Q.; et al. MYH9-dependent polarization of ATG9B promotes colorectal cancer metastasis by accelerating focal adhesion assembly. Cell Death Differ. 2021. [Google Scholar] [CrossRef]
- Kao, L.Y.; Chao, W.T. Rab11-mediated focal adhesion turnover in sarcoma cell migration. Chin. J. Physiol. 2021, 64, 43–50. [Google Scholar] [CrossRef]
- Azimian-Zavareh, V.; Dehghani-Ghobadi, Z.; Ebrahimi, M.; Mirzazadeh, K.; Nazarenko, I.; Hossein, G. Wnt5A modulates integrin expression in a receptor-dependent manner in ovarian cancer cells. Sci. Rep. 2021, 11, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Che, P.; Yu, L.; Friedman, G.K.; Wang, M.; Ke, X.; Wang, H.; Zhang, W.; Nabors, B.; Ding, Q.; Han, X. Integrin alphavbeta3 Engagement Regulates Glucose Metabolism and Migration through Focal Adhesion Kinase (FAK) and Protein Arginine Methyltransferase 5 (PRMT5) in Glioblastoma Cells. Cancers 2021, 13, 1111. [Google Scholar] [CrossRef]
- Fu, Y.; Zhang, Y.; Lei, Z.; Liu, T.; Cai, T.; Wang, A.; Du, W.; Zeng, Y.; Zhu, J.; Liu, Z.; et al. Abnormally activated OPN/integrin alphaVbeta3/FAK signalling is responsible for EGFR-TKI resistance in EGFR mutant non-small-cell lung cancer. J. Hematol. Oncol. 2020, 13, 169. [Google Scholar] [CrossRef] [PubMed]
- Afzal, M.S. G proteins: Binary switches in health and disease. Cent. Eur. J. Immunol. 2020, 45, 364–367. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liao, R.; Chen, X.; Ying, X.; Chen, G.; Li, M.; Dong, C. Twist-mediated PAR1 induction is required for breast cancer progression and metastasis by inhibiting Hippo pathway. Cell Death Dis. 2020, 11, 520. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Bai, X.; Jiang, G.; Jin, S.; Wang, Q.; Wang, A.; Peng, R.; Ke, A.; Bai, D. GIT1 overexpression promotes epithelial-mesenchymal transition and predicts poor prognosis in hepatocellular carcinoma. Bioengineered 2021, 12, 30–43. [Google Scholar] [CrossRef]
- Rao, A.; Herr, D.R. G protein-coupled receptor GPR19 regulates E-cadherin expression and invasion of breast cancer cells. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2017, 1864, 1318–1327. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, Q.; Chen, S.; Liu, Y.; Huang, Y.; Chen, P.; Li, X.; Gao, G.; Xu, K.; Fan, S.; et al. APLNR is involved in ATRA-induced growth inhibition of nasopharyngeal carcinoma and may suppress EMT through PI3K-Akt-mTOR signaling. FASEB J. 2019, 33, 11959–11972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arakaki, A.K.S.; Pan, W.A.; Wedegaertner, H.; Roca-Mercado, I.; Chinn, L.; Gujral, T.S.; Trejo, J. alpha-Arrestin ARRDC3 tumor suppressor function is linked to GPCR-induced TAZ activation and breast cancer metastasis. J. Cell Sci. 2021, 134. [Google Scholar] [CrossRef]
- Sommer, A.K.; Falcenberg, M.; Ljepoja, B.; Frohlich, T.; Arnold, G.J.; Wagner, E.; Roidl, A. Downregulation of GRK5 hampers the migration of breast cancer cells. Sci. Rep. 2019, 9, 15548. [Google Scholar] [CrossRef] [Green Version]
- Stein, J.; Stahn, S.; Neudorfl, J.M.; Sperlich, J.; Schmalz, H.G.; Teusch, N. Synthetic Indolactam V Analogues as Inhibitors of PAR2-Induced Calcium Mobilization in Triple-Negative Breast Cancer Cells. ChemMedChem 2018, 13, 147–154. [Google Scholar] [CrossRef]
- Cao, J.; Liu, X.; Yang, Y.; Wei, B.; Li, Q.; Mao, G.; He, Y.; Li, Y.; Zheng, L.; Zhang, Q.; et al. Decylubiquinone suppresses breast cancer growth and metastasis by inhibiting angiogenesis via the ROS/p53/ BAI1 signaling pathway. Angiogenesis 2020, 23, 325–338. [Google Scholar] [CrossRef] [PubMed]
- Paciaroni, N.G.; Norwood, V.M.t.; Ratnayake, R.; Luesch, H.; Huigens, R.W., 3rd. Yohimbine as a Starting Point to Access Diverse Natural Product-Like Agents with Re-programmed Activities against Cancer-Relevant GPCR Targets. Bioorg. Med. Chem. 2020, 28, 115546. [Google Scholar] [CrossRef]
- Wang, X.; Jespers, W.; Bongers, B.J.; Habben Jansen, M.C.C.; Stangenberger, C.M.; Dilweg, M.A.; Gutierrez-de-Teran, H.; AP, I.J.; Heitman, L.H.; van Westen, G.J.P. Characterization of cancer-related somatic mutations in the adenosine A2B receptor. Eur. J. Pharmacol. 2020, 880, 173126. [Google Scholar] [CrossRef]
- Dent, E.W.; Gupton, S.L.; Gertler, F.B. The Growth Cone Cytoskeleton in Axon Outgrowth and Guidance. Cold Spring Harb. Perspect. Biol. 2011, 3, a001800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez, T.M.; Letourneau, P.C. Actin dynamics in growth cone motility and navigation. J. Neurochem. 2014, 129, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Chedotal, A.; Kerjan, G.; Moreau-Fauvarque, C. The brain within the tumor: New roles for axon guidance molecules in cancers. Cell Death Differ. 2005, 12, 1044–1056. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, A.A.; Falcon, P.; Espinoza, N.; Luis, S.R.; Milla, L.A.; Hernandez-SanMiguel, E.; Torres, V.A.; Sanchez-Gomez, P.; Palma, V. The Netrin-4/ Neogenin-1 axis promotes neuroblastoma cell survival and migration. Oncotarget 2017, 8, 9767–9782. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Zeng, Z.; Qiao, L.; Jiang, X.; Ma, J.; Wang, J.; Ye, S.; Ma, Q.; Wei, J.; Wu, M.; et al. Semaphorin 4C Promotes Macrophage Recruitment and Angiogenesis in Breast Cancer. Mol. Cancer Res. 2019, 17, 2015–2028. [Google Scholar] [CrossRef] [Green Version]
- Smeester, B.A.; Slipek, N.J.; Pomeroy, E.J.; Bomberger, H.E.; Shamsan, G.A.; Peterson, J.J.; Crosby, M.R.; Draper, G.M.; Becklin, K.L.; Rahrmann, E.P.; et al. SEMA4C is a novel target to limit osteosarcoma growth, progression, and metastasis. Oncogene 2020, 39, 1049–1062. [Google Scholar] [CrossRef]
- Duquette, P.M.; Lamarche-Vane, N. The calcium-activated protease calpain regulates netrin-1 receptor deleted in colorectal cancer-induced axon outgrowth in cortical neurons. J. Neurochem. 2020, 152, 315–332. [Google Scholar] [CrossRef]
- Abdullah, A.; Herdenberg, C.; Hedman, H. Netrin-1 functions as a suppressor of bone morphogenetic protein (BMP) signaling. Sci. Rep. 2021, 11, 8585. [Google Scholar] [CrossRef]
- Shahi, P.; Wang, C.Y.; Chou, J.; Hagerling, C.; Gonzalez Velozo, H.; Ruderisch, A.; Yu, Y.; Lai, M.D.; Werb, Z. GATA3 targets semaphorin 3B in mammary epithelial cells to suppress breast cancer progression and metastasis. Oncogene 2017, 36, 5567–5575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdullah, A.; Akhand, S.S.; Paez, J.S.P.; Brown, W.; Pan, L.; Libring, S.; Badamy, M.; Dykuizen, E.; Solorio, L.; Andy Tao, W.; et al. Epigenetic targeting of neuropilin-1 prevents bypass signaling in drug-resistant breast cancer. Oncogene 2021, 40, 322–333. [Google Scholar] [CrossRef] [PubMed]
- Shirley, L.A.; McCarty, S.; Yang, M.C.; Saji, M.; Zhang, X.; Phay, J.; Ringel, M.D.; Chen, C.S. Integrin-linked kinase affects signaling pathways and migration in thyroid cancer cells and is a potential therapeutic target. Surgery 2016, 159, 163–170. [Google Scholar] [CrossRef] [Green Version]
- Gagne, D.; Groulx, J.F.; Benoit, Y.D.; Basora, N.; Herring, E.; Vachon, P.H.; Beaulieu, J.F. Integrin-linked kinase regulates migration and proliferation of human intestinal cells under a fibronectin-dependent mechanism. J. Cell Physiol. 2010, 222, 387–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, Y.; Zhong, X.; Flynn, D.C.; Zheng, J.Z.; Qiao, M.; Wu, C.; Dedhar, S.; Shi, X.; Jiang, B.H. ILK mediates actin filament rearrangements and cell migration and invasion through PI3K/Akt/Rac1 signaling. Oncogene 2005, 24, 3154–3165. [Google Scholar] [CrossRef] [Green Version]
- Sternlicht, M.D.; Werb, Z. How matrix metalloproteinases regulate cell behavior. Annu. Rev. Cell Dev. Biol. 2001, 17, 463–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bajbouj, K.; Ramakrishnan, R.K.; Hamid, Q. Role of Matrix Metalloproteinases in Angiogenesis and Its Implications in Asthma. J. Immunol. Res. 2021, 2021. [Google Scholar] [CrossRef] [PubMed]
- Cabral-Pacheco, G.A.; Garza-Veloz, I.; Castruita-De la Rosa, C.; Ramirez-Acuna, J.M.; Perez-Romero, B.A.; Guerrero-Rodriguez, J.F.; Martinez-Avila, N.; Martinez-Fierro, M.L. The Roles of Matrix Metalloproteinases and Their Inhibitors in Human Diseases. Int. J. Mol. Sci. 2020, 21, 9739. [Google Scholar] [CrossRef] [PubMed]
- Nikolov, A.; Popovski, N. Role of Gelatinases MMP-2 and MMP-9 in Healthy and Complicated Pregnancy and Their Future Potential as Preeclampsia Biomarkers. Diagnostics 2021, 11, 480. [Google Scholar] [CrossRef] [PubMed]
- Kessenbrock, K.; Plaks, V.; Werb, Z. Matrix Metalloproteinases: Regulators of the Tumor Microenvironment. Cell 2010, 141, 52–67. [Google Scholar] [CrossRef] [Green Version]
- Mammadova-Bach, E.; Gil-Pulido, J.; Sarukhanyan, E.; Burkard, P.; Shityakov, S.; Schonhart, C.; Stegner, D.; Remer, K.; Nurden, P.; Nurden, A.T.; et al. Platelet glycoprotein VI promotes metastasis through interaction with cancer cell-derived galectin-3. Blood 2020, 135, 1146–1160. [Google Scholar] [CrossRef]
- Schlesinger, M. Role of platelets and platelet receptors in cancer metastasis. J. Hematol. Oncol. 2018, 11, 125. [Google Scholar] [CrossRef] [PubMed]
- Au, A.E.; Josefsson, E.C. Regulation of platelet membrane protein shedding in health and disease. Platelets 2017, 28, 342–353. [Google Scholar] [CrossRef]
- Tullemans, B.M.E.; Nagy, M.; Sabrkhany, S.; Griffioen, A.W.; Oude Egbrink, M.G.A.; Aarts, M.; Heemskerk, J.W.M.; Kuijpers, M.J.E. Tyrosine Kinase Inhibitor Pazopanib Inhibits Platelet Procoagulant Activity in Renal Cell Carcinoma Patients. Front. Cardiovasc. Med. 2018, 5, 142. [Google Scholar] [CrossRef] [Green Version]
- Hartzell, C.A.; Jankowska, K.I.; Burkhardt, J.K.; Lewis, R.S. Calcium influx through CRAC channels controls actin organization and dynamics at the immune synapse. Elife 2016, 5. [Google Scholar] [CrossRef]
- Dushek, O.; Mueller, S.; Soubies, S.; Depoil, D.; Caramalho, I.; Coombs, D.; Valitutti, S. Effects of Intracellular Calcium and Actin Cytoskeleton on TCR Mobility Measured by Fluorescence Recovery. PLoS ONE 2008, 3. [Google Scholar] [CrossRef] [Green Version]
- Abedini, A.; Sayed, C.; Carter, L.E.; Boerboom, D.; Vanderhyden, B.C. Non-canonical WNT5a regulates Epithelial-to-Mesenchymal Transition in the mouse ovarian surface epithelium. Sci. Rep. 2020, 10, 1–14. [Google Scholar] [CrossRef]
- Niehrs, C. The complex world of WNT receptor signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 767–779. [Google Scholar] [CrossRef]
- Chen, Y.B.; De Marco, M.A.; Graziani, I.; Gazdar, A.F.; Strack, P.R.; Miele, L.; Bocchetta, M. Oxygen concentration determines the biological effects of NOTCH-1 signaling in adenocarcinoma of the lung. Cancer Res. 2007, 67, 7954–7959. [Google Scholar] [CrossRef] [Green Version]
- Dachs, G.U.; Patterson, A.V.; Firth, J.D.; Ratcliffe, P.J.; Townsend, K.M.S.; Stratford, I.J.; Harris, A.L. Targeting gene expression to hypoxic tumor cells. Nat. Med. 1997, 3, 515–520. [Google Scholar] [CrossRef]
- Muz, B.; de la Puente, P.; Azab, F.; Azab, A.K. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia 2015, 3, 83–92. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.; Zhang, J.; Huo, M.; Gao, J.; Yang, T.; Yin, X.; Wang, P.; Leng, S.; Feng, D.; Chen, Y.; et al. CUL4B Promotes Breast Carcinogenesis by Coordinating with Transcriptional Repressor Complexes in Response to Hypoxia Signaling Pathway. Adv. Sci. (Weinh) 2021, 8, 2001515. [Google Scholar] [CrossRef] [PubMed]
- Tennant, D.A.; Duran, R.V.; Boulahbel, H.; Gottlieb, E. Metabolic transformation in cancer. Carcinogenesis 2009, 30, 1269–1280. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.C.; Chan, Y.C.; Chang, W.M.; Lin, Y.F.; Yang, C.J.; Su, C.Y.; Huang, M.S.; Wu, A.T.H.; Hsiao, M. Feedback regulation of ALDOA activates the HIF-1alpha/MMP9 axis to promote lung cancer progression. Cancer Lett. 2017, 403, 28–36. [Google Scholar] [CrossRef]
- Talreja, J.; Talwar, H.; Bauerfeld, C.; Grossman, L.I.; Zhang, K.; Tranchida, P.; Samavati, L. HIF-1alpha regulates IL-1beta and IL-17 in sarcoidosis. Elife 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Bastid, J.; Dejou, C.; Docquier, A.; Bonnefoy, N. The Emerging Role of the IL-17B/IL-17RB Pathway in Cancer. Front. Immunol. 2020, 11, 718. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.H.; Tsai, L.H.; Huang, C.K.; Hsu, P.H.; Chen, M.Y.; Chen, Y.I.; Hu, C.M.; Shen, C.N.; Lee, C.C.; Chang, M.C.; et al. Characterization of initial key steps of IL-17 receptor B oncogenic signaling for targeted therapy of pancreatic cancer. Sci. Transl. Med. 2021, 13. [Google Scholar] [CrossRef] [PubMed]
- Ho, W.J.; Jaffee, E.M.; Zheng, L. The tumour microenvironment in pancreatic cancer—Clinical challenges and opportunities. Nat. Rev. Clin. Oncol. 2020, 17, 527–540. [Google Scholar] [CrossRef]
- Bussard, K.M.; Mutkus, L.; Stumpf, K.; Gomez-Manzano, C.; Marini, F.C. Tumor-associated stromal cells as key contributors to the tumor microenvironment. Breast Cancer Res. 2016, 18, 84. [Google Scholar] [CrossRef] [Green Version]
- Bremnes, R.M.; Donnem, T.; Al-Saad, S.; Al-Shibli, K.; Andersen, S.; Sirera, R.; Camps, C.; Marinez, I.; Busund, L.T. The role of tumor stroma in cancer progression and prognosis: Emphasis on carcinoma-associated fibroblasts and non-small cell lung cancer. J. Thorac. Oncol. 2011, 6, 209–217. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Yang, Y.; Yang, J.; Zhao, X.; Wei, X. Tumor Microenvironment in Ovarian Cancer: Function and Therapeutic Strategy. Front. Cell Dev. Biol. 2020, 8, 758. [Google Scholar] [CrossRef] [PubMed]
- Fabregat, I.; Caballero-Diaz, D. Transforming Growth Factor-beta-Induced Cell Plasticity in Liver Fibrosis and Hepatocarcinogenesis. Front. Oncol. 2018, 8, 357. [Google Scholar] [CrossRef] [Green Version]
- Hinz, B.; Lagares, D. Evasion of apoptosis by myofibroblasts: A hallmark of fibrotic diseases. Nat. Rev. Rheumatol. 2020, 16, 11–31. [Google Scholar] [CrossRef] [PubMed]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.Y.; Han, C.C.; Wang, S.W.; Fang, P.Q.; Ma, Z.F.; Xu, L.; Yin, R. Cancer-associated fibroblasts: An emerging target of anti-cancer immunotherapy. J. Hematol. Oncol. 2019, 12. [Google Scholar] [CrossRef] [PubMed]
- Apicella, M.; Giannoni, E.; Fiore, S.; Ferrari, K.J.; Fernandez-Perez, D.; Isella, C.; Granchi, C.; Minutolo, F.; Sottile, A.; Comoglio, P.M.; et al. Increased Lactate Secretion by Cancer Cells Sustains Non-cell-autonomous Adaptive Resistance to MET and EGFR Targeted Therapies. Cell Metab. 2018, 28, 848–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosa, M.H.; Michels, B.E.; Menche, C.; Nicolas, A.M.; Darvishi, T.; Greten, F.R.; Farin, H.F. A Wnt-Induced Phenotypic Switch in Cancer-Associated Fibroblasts Inhibits EMT in Colorectal Cancer. Cancer Res. 2020, 80, 5569–5582. [Google Scholar] [CrossRef]
- Lee, C.H. Reversal of Epithelial-Mesenchymal Transition by Natural Anti-Inflammatory and Pro-Resolving Lipids. Cancers 2019, 11, 1841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yadav, P.S.; Feng, S.H.; Cong, Q.; Kim, H.; Liu, Y.C.; Yang, Y.Z. Stat3 loss in mesenchymal progenitors causes Job syndrome-like skeletal defects by reducing Wnt/beta-catenin signaling. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef]
- Lin, W.H.; Chang, Y.W.; Hong, M.X.; Hsu, T.C.; Lee, K.C.; Lin, C.; Lee, J.L. STAT3 phosphorylation at Ser727 and Tyr705 differentially regulates the EMT-MET switch and cancer metastasis. Oncogene 2021, 40, 791–805. [Google Scholar] [CrossRef]
- Aban, C.E.; Lombardi, A.; Neiman, G.; Biani, M.C.; La Greca, A.; Waisman, A.; Moro, L.N.; Sevlever, G.; Miriuka, S.; Luzzani, C. Downregulation of E-cadherin in pluripotent stem cells triggers partial EMT. Sci. Rep. 2021, 11. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, S.; Miyagi, C.; Fukada, T.; Kagara, N.; Che, Y.S.; Hirano, T. Zinc transporter LIVI controls epithelial-mesenchymal transition in zebrafish gastrula organizer. Nature 2004, 429, 298–302. [Google Scholar] [CrossRef]
- Liu, W.H.; Chen, M.T.; Wang, M.L.; Lee, Y.Y.; Chiou, G.Y.; Chien, C.S.; Huang, P.I.; Chen, Y.W.; Huang, M.C.; Chiou, S.H.; et al. Cisplatin-selected resistance is associated with increased motility and stem-like properties via activation of STAT3/Snail axis in atypical teratoid/rhabdoid tumor cells. Oncotarget 2015, 6, 1750–1768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, X.K.; Han, H.Q.; Zhang, H.J.; Xu, M.; Li, L.; Chen, L.; Xiang, T.; Feng, Q.S.; Kang, T.; Qian, C.N.; et al. OVOL2 links stemness and metastasis via fine-tuning epithelial-mesenchymal transition in nasopharyngeal carcinoma. Theranostics 2018, 8, 2202–2216. [Google Scholar] [CrossRef]
- Liu, J.; Wu, Q.; Wang, Y.; Wei, Y.; Wu, H.; Duan, L.; Zhang, Q.; Wu, Y. Ovol2 induces mesenchymal-epithelial transition via targeting ZEB1 in osteosarcoma. Onco Targets Ther. 2018, 11, 2963–2973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murata, M.; Ito, T.; Tanaka, Y.; Yamamura, K.; Furue, K.; Furue, M. OVOL2-Mediated ZEB1 Downregulation May Prevent Promotion of Actinic Keratosis to Cutaneous Squamous Cell Carcinoma. J. Clin. Med. 2020, 9, 618. [Google Scholar] [CrossRef] [Green Version]
- Shen, J.G.; Lv, X.B.; Zhang, L. GRHL2 Acts as an Anti-Oncogene in Bladder Cancer by Regulating ZEB1 in Epithelial-Mesenchymal Transition (EMT) Process (vol 13, pg 2511, 2020). Oncotargets Ther. 2020, 13, 10759–10760. [Google Scholar] [CrossRef]
- Yang, Z.; Wu, D.; Chen, Y.; Min, Z.; Quan, Y. GRHL2 inhibits colorectal cancer progression and metastasis via oppressing epithelial-mesenchymal transition. Cancer Biol. Ther. 2019, 20, 1195–1205. [Google Scholar] [CrossRef]
- Haensel, D.; Sun, P.; MacLean, A.L.; Ma, X.; Zhou, Y.; Stemmler, M.P.; Brabletz, S.; Berx, G.; Plikus, M.V.; Nie, Q.; et al. An Ovol2-Zeb1 transcriptional circuit regulates epithelial directional migration and proliferation. EMBO Rep. 2019, 20. [Google Scholar] [CrossRef] [PubMed]
- Carpinelli, M.R.; de Vries, M.E.; Auden, A.; Butt, T.; Deng, Z.; Partridge, D.D.; Miles, L.B.; Georgy, S.R.; Haigh, J.J.; Darido, C.; et al. Inactivation of Zeb1 in GRHL2-deficient mouse embryos rescues mid-gestation viability and secondary palate closure. Dis. Model. Mech. 2020, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikolopoulou, E.; Hirst, C.S.; Galea, G.; Venturini, C.; Moulding, D.; Marshall, A.R.; Rolo, A.; De Castro, S.C.P.; Copp, A.J.; Greene, N.D.E. Spinal neural tube closure depends on regulation of surface ectoderm identity and biomechanics by Grhl2. Nat. Commun. 2019, 10, 2487. [Google Scholar] [CrossRef] [Green Version]
- Roca, H.; Hernandez, J.; Weidner, S.; McEachin, R.C.; Fuller, D.; Sud, S.; Schumann, T.; Wilkinson, J.E.; Zaslavsky, A.; Li, H.; et al. Transcription factors OVOL1 and OVOL2 induce the mesenchymal to epithelial transition in human cancer. PLoS ONE 2013, 8, e76773. [Google Scholar] [CrossRef] [PubMed]
- Cieply, B.; Riley, P.t.; Pifer, P.M.; Widmeyer, J.; Addison, J.B.; Ivanov, A.V.; Denvir, J.; Frisch, S.M. Suppression of the epithelial-mesenchymal transition by Grainyhead-like-2. Cancer Res. 2012, 72, 2440–2453. [Google Scholar] [CrossRef] [Green Version]
- Cieply, B.; Farris, J.; Denvir, J.; Ford, H.L.; Frisch, S.M. Epithelial-mesenchymal transition and tumor suppression are controlled by a reciprocal feedback loop between ZEB1 and Grainyhead-like-2. Cancer Res. 2013, 73, 6299–6309. [Google Scholar] [CrossRef] [Green Version]
- Werner, S.; Frey, S.; Riethdorf, S.; Schulze, C.; Alawi, M.; Kling, L.; Vafaizadeh, V.; Sauter, G.; Terracciano, L.; Schumacher, U.; et al. Dual roles of the transcription factor grainyhead-like 2 (GRHL2) in breast cancer. J. Biol. Chem. 2013, 288, 22993–23008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faddaoui, A.; Sheta, R.; Bachvarova, M.; Plante, M.; Gregoire, J.; Renaud, M.C.; Sebastianelli, A.; Gobeil, S.; Morin, C.; Ghani, K.; et al. Suppression of the grainyhead transcription factor 2 gene (GRHL2) inhibits the proliferation, migration, invasion and mediates cell cycle arrest of ovarian cancer cells. Cell Cycle 2017, 16, 693–706. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.H.; Li, Z.; Hu, M.; Yang, Q.J.; Yan, S.; Wu, R.S.; Li, B.A.; Guo, M. Ovol2 gene inhibits the Epithelial-to-Mesenchymal Transition in lung adenocarcinoma by transcriptionally repressing Twist1. Gene 2017, 600, 1–8. [Google Scholar] [CrossRef]
- Tian, X.; Liu, Z.; Niu, B.; Zhang, J.; Tan, T.K.; Lee, S.R.; Zhao, Y.; Harris, D.C.; Zheng, G. E-cadherin/beta-catenin complex and the epithelial barrier. J. Biomed. Biotechnol. 2011, 2011, 567305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Lamouille, S.; Derynck, R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 2009, 19, 156–172. [Google Scholar] [CrossRef] [PubMed]
- Lin, G.; Yu, B.; Liang, Z.; Li, L.; Qu, S.; Chen, K.; Zhou, L.; Lu, Q.; Sun, Y.; Zhu, X. Silencing of c-jun decreases cell migration, invasion, and EMT in radioresistant human nasopharyngeal carcinoma cell line CNE-2R. Onco Targets Ther. 2018, 11, 3805–3815. [Google Scholar] [CrossRef] [Green Version]
- Gluck, C.; Glathar, A.; Tsompana, M.; Nowak, N.; Garrett-Sinha, L.A.; Buck, M.J.; Sinha, S. Molecular dissection of the oncogenic role of ETS1 in the mesenchymal subtypes of head and neck squamous cell carcinoma. PLoS Genet. 2019, 15, e1008250. [Google Scholar] [CrossRef] [Green Version]
- Pires, B.R.B.; Mencalha, A.L.; Ferreira, G.M.; de Souza, W.F.; Morgado-Diaz, J.A.; Maia, A.M.; Correa, S.; Abdelhay, E.S.F.W. NF-kappaB Is Involved in the Regulation of EMT Genes in Breast Cancer Cells. PLoS ONE 2017, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neelakantan, D.; Zhou, H.B.; Oliphant, M.U.J.; Zhang, X.M.; Simon, L.M.; Henke, D.M.; Shaw, C.A.; Wu, M.F.; Hilsenbeck, S.G.; White, L.D.; et al. EMT cells increase breast cancer metastasis via paracrine GLI activation in neighbouring tumour cells (vol 8, 15773, 2017). Nat. Commun. 2018, 9, 4720. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.J.; Sun, M.; Li, S.Q.; Wu, Q.Q.; Ji, L.; Liu, Z.L.; Zhou, G.Z.; Cao, G.; Jin, L.; Xie, H.W.; et al. Upregulation of the long non-coding RNA HOTAIR promotes esophageal squamous cell carcinoma metastasis and poor prognosis. Mol. Carcinog. 2013, 52, 908–915. [Google Scholar] [CrossRef]
- De Angelis, M.L.; Francescangeli, F.; La Torre, F.; Zeuner, A. Stem Cell Plasticity and Dormancy in the Development of Cancer Therapy Resistance. Front. Oncol. 2019, 9. [Google Scholar] [CrossRef] [Green Version]
- Yakisich, J.S.; Azad, N.; Kaushik, V.; Iyer, A.K.V. Cancer Cell Plasticity: Rapid Reversal of Chemosensitivity and Expression of Stemness Markers in Lung and Breast Cancer Tumorspheres. J. Cell Physiol. 2017, 232, 2280–2286. [Google Scholar] [CrossRef]
- Puisieux, A.; Brabletz, T.; Caramel, J. Oncogenic roles of EMT-inducing transcription factors. Nat. Cell Biol. 2014, 16, 488–494. [Google Scholar] [CrossRef]
- Pradella, D.; Naro, C.; Sette, C.; Ghigna, C. EMT and stemness: Flexible processes tuned by alternative splicing in development and cancer progression. Mol. Cancer 2017, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saxena, K.; Jolly, M.K.; Balamurugan, K. Hypoxia, partial EMT and collective migration: Emerging culprits in metastasis. Transl. Oncol. 2020, 13, 100845. [Google Scholar] [CrossRef] [PubMed]
- Papadaki, M.A.; Stoupis, G.; Theodoropoulos, P.A.; Mavroudis, D.; Georgoulias, V.; Agelaki, S. Circulating Tumor Cells with Stemness and Epithelial-to-Mesenchymal Transition Features Are Chemoresistant and Predictive of Poor Outcome in Metastatic Breast Cancer. Mol. Cancer Ther. 2019, 18, 437–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weidenfeld, K.; Barkan, D. EMT and Stemness in Tumor Dormancy and Outgrowth: Are They Intertwined Processes? Front. Oncol. 2018, 8, 381. [Google Scholar] [CrossRef] [PubMed]
- Amini, S.; Fathi, F.; Mobalegi, J.; Sofimajidpour, H.; Ghadimi, T. The expressions of stem cell markers: Oct4, Nanog, Sox2, nucleostemin, Bmi, Zfx, Tcl1, Tbx3, Dppa4, and Esrrb in bladder, colon, and prostate cancer, and certain cancer cell lines. Anat. Cell Biol. 2014, 47, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Ye, S.; Zhang, D.; Cheng, F.; Wilson, D.; Mackay, J.; He, K.; Ban, Q.; Lv, F.; Huang, S.; Liu, D.; et al. Wnt/beta-catenin and LIF-Stat3 signaling pathways converge on Sp5 to promote mouse embryonic stem cell self-renewal. J. Cell Sci. 2016, 129, 269–276. [Google Scholar] [CrossRef] [Green Version]
- Das, B.; Pal, B.; Bhuyan, R.; Li, H.; Sarma, A.; Gayan, S.; Talukdar, J.; Sandhya, S.; Bhuyan, S.; Gogoi, G.; et al. MYC Regulates the HIF2alpha Stemness Pathway via Nanog and Sox2 to Maintain Self-Renewal in Cancer Stem Cells versus Non-Stem Cancer Cells. Cancer Res. 2019, 79, 4015–4025. [Google Scholar] [CrossRef]
- Meskyte, E.M.; Keskas, S.; Ciribilli, Y. MYC as a Multifaceted Regulator of Tumor Microenvironment Leading to Metastasis. Int. J. Mol. Sci. 2020, 21, 7710. [Google Scholar] [CrossRef]
- Krasnapolski, M.A.; Todaro, L.B.; de Kier Joffe, E.B. Is the epithelial-to-mesenchymal transition clinically relevant for the cancer patient? Curr. Pharm. Biotechnol. 2011, 12, 1891–1899. [Google Scholar] [CrossRef]
- Teixeira, A.F.; Ten Dijke, P.; Zhu, H.J. On-Target Anti-TGF-beta Therapies Are Not Succeeding in Clinical Cancer Treatments: What Are Remaining Challenges? Front. Cell Dev. Biol. 2020, 8, 605. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| EMT | MET | ||||
|---|---|---|---|---|---|
| Gene Increased | Transcriptional Factor Increased | Morphology | Gene Increased | Transcriptional Factor Increased | Morphology |
| OPN vimentin matrix metallopro teinase2 | Snail Slug Smuc ZEB1/2 Twist1/2 CDH1 FoxC2 TCF4 XBP1 | Spindle-like | E-cadherin Tight junction proteins Claudin-4 | Ets-1 Pax family members HIF1- α | Epithelium like |
| Canonical Pathways | Related Factors | |
|---|---|---|
| EMT | Hepatic Fibrosis/Hepatic Stellate Cell Activation | ACTA2, CCN2, COL11A1, COL12A1, COL16A1, COL1A1, COL1A2, COL3A1, COL4A1, COL4A2, COL5A1, COL5A2, COL5A3, COL6A2, COL6A3, COL7A1, COL8A2, CXCL8, FAS, FGF2, FN1, IGFBP3, IGFBP4, IL6, LAMA1, MMP1, MMP2, MYL9, PDGFRB, SERPINE1, TGFB1, TIMP1, TNFRSF11B, VCAM1, VEGFA, VEGFC |
| GP6 Signaling Pathway | COL11A1, COL12A1, COL16A1, COL1A1, COL1A2, COL3A1, COL4A1, COL4A2, COL5A1, COL5A2, COL5A3, COL6A2, COL6A3, COL7A1, COL8A2, ITGB3, LAMA1, LAMA2, LAMA3, LAMC1, LAMC2 | |
| Hepatic Fibrosis Signaling Pathway | ACTA2, CCN2, COL1A1, COL1A2, COL3A1, COL5A3, CXCL8, FGF2, FZD8, ITGA2, ITGA5, ITGAV, ITGB1, ITGB3, ITGB5, JUN, LRP1, MMP1, MYL9, MYLK, PDGFRB, RHOB, SERPINE1, SPP1, TGFB1, TGFBR3, TIMP1, TNFRSF11B, VCAM1, VEGFA, VEGFC, WNT5A | |
| Tumor Microenvironment Pathway | CD44, COL1A1, COL1A2, COL3A1, CXCL12, CXCL8, FAS, FGF2, FN1, IL6, ITGA5, ITGB3, JUN, MMP1, MMP14, MMP2, MMP3, SPP1, TGFB1, TNC, VEGFA, VEGFC | |
| Inhibition of Matrix Metalloproteases | ADAM12, LRP1, MMP1, MMP14, MMP2, MMP3, SDC1, TFPI2, THBS2, TIMP1, TIMP3 | |
| Axonal Guidance Signaling | ADAM12, BDNF, BMP1, CXCL12, FZD8, ITGA2, ITGA5, ITGAV, ITGB1, ITGB3, ITGB5, MMP1, MMP14, MMP2, MMP3, MYL9, PFN2, SLIT2, SLIT3, VEGFA, VEGFC, WIPF1, WNT5A | |
| Leukocyte Extravasation Signaling | ACTA2, CD44, CXCL12, EDIL3, ITGA2, ITGB1, MMP1, MMP14, MMP2, MMP3, THY1, TIMP1, TIMP3, VCAM1, WIPF1 | |
| Regulation of Actin-based Motility by Rho | ACTA2, ITGA2, ITGA5, ITGAV, ITGB1, ITGB3, ITGB5, MYL9, MYLK, PFN2, RHOB, WIPF1 | |
| Regulation Of The Epithelial Mesenchymal Transition By Growth Factors Pathway | CDH2, FGF2, FOXC2, ID2, IL6, JUN, MEST, MMP1, MMP2, PDGFRB, SNAI2, TGFB1, TNFRSF11B, VIM | |
| Signaling by Rho Family GTPases | ACTA2, CDH11, CDH2, CDH6, ITGA2, ITGA5, ITGAV, ITGB1, ITGB3, ITGB5, JUN, MYL9, MYLK, RHOB, VIM, WIPF1 | |
| ILK Signaling | ACTA2, FERMT2, FLNA, FN1, ITGB1, ITGB3, ITGB5, JUN, MYL9, RHOB, SNAI2, VEGFA, VEGFC, VIM | |
| Role of Tissue Factor in Cancer | CCN1, CCN2, CXCL1, CXCL8, ITGAV, ITGB1, ITGB3, MMP1, PLAUR, VEGFA, VEGFC | |
| HIF1α Signaling | FGF2, IL6, JUN, MMP1, MMP14, MMP2, MMP3, SAT1, SERPINE1, TGFB1, VEGFA, VEGFC, VIM | |
| Wnt/β-catenin Signaling | CD44, CDH2, DKK1, FZD8, GJA1, JUN, LRP1, SFRP1, SFRP4, TGFB1, TGFBR3, WNT5A | |
| RhoGDI Signaling | ACTA2, CD44, CDH11, CDH2, CDH6, ITGA2, ITGA5, ITGAV, ITGB1, ITGB3, ITGB5, MYL9, RHOB | |
| Regulation of the Epithelial-Mesenchymal Transition Pathway | CDH2, FGF2, FOXC2, FZD8, ID2, LOX, MMP2, NOTCH2, PDGFRB, SNAI2, TGFB1, WNT5A | |
| Glioma Invasiveness Signaling | CD44, ITGAV, ITGB3, MMP2, PLAUR, RHOB, TIMP1, TIMP3 | |
| Actin Cytoskeleton Signaling | ACTA2, FGF2, FLNA, FN1, ITGA2, ITGA5, ITGAV, ITGB1, ITGB3, ITGB5, MYL9, MYLK, PFN2 | |
| Integrin Signaling | ACTA2, ITGA2, ITGA5, ITGAV, ITGB1, ITGB3, ITGB5, MYL9, MYLK, PFN2, RHOB, WIPF1 | |
| Colorectal Cancer Metastasis Signaling | FZD8, IL6, JUN, LRP1, MMP1, MMP14, MMP2, MMP3, RHOB, TGFB1, VEGFA, VEGFC, WNT5A | |
| IL-17 Signaling | CXCL1, CXCL8, IL15, IL6, JUN, MMP2, MMP3, TGFB1, TNFRSF11B, VEGFA, VEGFC | |
| Bladder Cancer Signaling | CXCL8, FGF2, MMP1, MMP14, MMP2, MMP3, THBS1, VEGFA, VEGFC | |
| MET | Regulation of the Epithelial-Mesenchymal Transition Pathway | FGF10, FGF7, FZD7, HGF, SMO, WNT2B, WNT4, WNT5A, WNT9B |
| Role of NANOG in Mammalian Embryonic Stem Cell Pluripotency | BMP4, FZD7, LIF, SMO, WNT2B, WNT4, WNT5A, WNT9B | |
| Basal Cell Carcinoma Signaling | BMP4, FZD7, SMO, WNT2B, WNT4, WNT5A, WNT9B | |
| Factors Promoting Cardiogenesis in Vertebrates | BMP4, FZD7, SMO, WNT2B, WNT4, WNT5A, WNT9B | |
| Human Embryonic Stem Cell Pluripotency | BMP4, FZD7, SMO, WNT2B, WNT4, WNT5A, WNT9B | |
| Regulation Of The Epithelial Mesenchymal Transition In Development Pathway | FZD7, SMO, WNT2B, WNT4, WNT5A, WNT9B | |
| Colorectal Cancer Metastasis Signaling | FZD7, SMO, STAT1, WNT2B, WNT4, WNT5A, WNT9B | |
| Ovarian Cancer Signaling | FZD7, SMO, WNT2B, WNT4, WNT5A, WNT9B | |
| Glioblastoma Multiforme Signaling | FZD7, SMO, WNT2B, WNT4, WNT5A, WNT9B | |
| WNT/β-catenin Signaling | FZD7, SMO, WNT2B, WNT4, WNT5A, WNT9B | |
| Molecular Mechanisms of Cancer | BMP4, FZD7, SMO, WNT2B, WNT4, WNT5A, WNT9B | |
| Axonal Guidance Signaling | BMP4, FZD7, SMO, WNT2B, WNT4, WNT5A, WNT9B | |
| Mouse Embryonic Stem Cell Pluripotency | BMP4, FZD7, LIF, SMO | |
| Hepatic Fibrosis Signaling Pathway | FZD7, SMO, WNT2B, WNT4, WNT5A, WNT9B | |
| Adipogenesis pathway | BMP4, FZD7, SMO, WNT5A | |
| Tumor Microenvironment Pathway | FGF10, FGF7, HGF, TNC |
| EMT | VDR, CREB3L1, Pou3f1, FEZF1, YBX1, PAX7, BTG2, MEF2D, SOX7, SALL1, SP7, KDM5B, FOSL2, PPP1R13L, TAF4B, MTA2, CITED2, EP300, NKX2-5, MITF, ID3, MEN1, RFX5, ZEB1, MED4, CARM1, ERG, HLTF, TAF6, RCOR1, SMAD2, GSC, FOXQ1, PML, CBFB, BATF2, ZNF580, IRF1, NOSTRIN, RBCK1, MAX, SOX4, HOXA1, GATA3, BRCA2, PAX4, NFIA, TEAD1, HHEX, TSC22D1, HDAC4, BRCA1, ELK1, RUNX3, MECP2, SP100, NFATC4, PDLIM1, ETV3, HIC1, SQSTM1, MTA3, FOXK2, FEV, NFATC1, FOXO4, HES1, ID4, ATF3, TBX5, FOXP3, SPI1, ZNF581, NFIB, RBM14, MDM2, HOXB4, TBXT, LHX4, MAML1, BTG1, NCOR2, CBL, IKZF1, HIF3A, MED7, MIB2, RING1, TFAP4, IRF2BP1, HOXD10, BRMS1, ZFP36L1, FOXM1, ZBTB48, NKX3-1, EZH2, JMY, HTT, HES6, HDAC5, FOXA1, CREB1, HDAC7, PDX1, NEUROG3, FOXA2, SMAD1, ARNT, DDIT3, LCOR, POU2AF1, FOS, PLAG1, MXD4, JUNB, ASCC1, SMARCA4, TSHZ3, FOXL2, ZNF300, BRD7, NFKB2, TEAD3, HTATIP2, MEOX2, STAT5B, ID1, IRF5, ATF2, TFAP2C, SKI, ELF3, KLF17, KLF5, MEF2A, BARX2, APBB1, BORCS8-MEF2B, Gm21596/Hmgb1, ZNF24, ESRRA, IRF3, CEBPG, H2AX, E2F1, TSC22D3, EBF2, SP2, NRIP1, ALX4, ETS2, PMF1/PMF1-BGLAP, DAXX, EHMT2, Ncoa6, ATF1, MTDH, WTIP, ARID5B, TCF12, GLIS2, Yap1, WBP2, MYCN, TBX18, PSMD10, NFIL3, MXD3, TFAP2B, IRF6, CIITA, NUPR1, HIF1A, MED21, LPXN, HOXA4, DTX1, MYBL2, BHLHE40, ID2, TCF4, TRIM28, RB1, MZF1, STAT5A, ZNF350, HOXA9, NFYA, ZNF410, MESP1, ZEB2, CYLD, GRHL2, HEY1, DLX4, XBP1, CEBPA, TFAP2A, PBX1, IRF2, NFKBIB, RUNX2, ERF, ASH1L, BATF3, SALL4, MAFB, NFE2L2, NOTCH4, KLF6, NRF1, HEXIM1, MECOM, E2F2, HDAC3, BHLHE22, TWIST2, ZFP64, IFI16, TFEC, HOXD3, ATF6, PROX1, NFYC, TCF3, SSRP1, NEUROG1, NCOA3, WDR5, BACH1, HOXB5, SOX9, SIM1, TGIF1, SATB2, REL, CEBPD, TEAD2, HOXB8, JUND, ZFP36, PHB2, PURB, SUB1, PRDM5, TCF21, RELB, EGR2, ZNF281, SMARCA5, HNF1B, WWC1, MED24, SRA1, CREBBP, ZNF384, SMAD7, FHL2, TCF7L2, ZBTB16, ATXN1, RFX1, CALR, HEY2, ETV5, HOXB9, NAB2, SIM2, GATA2, NFKBIZ, HAND2, MAF, TAF9, ZMIZ2, ETV4, FOXP1, Cux1, ATN1, TCF20, HOXC5, NFYB, Hmgb1, EBF3, RBM39, NPAS4, CUX1, YAP1, NFKBID, BMI1, RAD21, LDB1, SOX17, FOSB, ELF4, BCL3, ZKSCAN3, CARF, CCND1, SMARCA2, NOTCH2, ZNF613, HOXC8, ZNF740, TOX, SKIL, MKX, KLF12, TGFB1I1, ARRB1, SIN3A, ECSIT, ZBTB7B, FLI1, CRTC1, CEBPZ, ZNF750, SREBF1, NFATC3, EPAS1, HOXC6, ESRRB, PHB, FOXO3, TAF4, POU2F1, ELK3, TBP, RRP1B, SREBF2, STAT6, CDKN2A, UXT, ETV6, EOMES, GATAD2B, DNAJB6, TRPS1, BCL10, HMGA1, HOXB7, ETV1, SUPT16H, BCL11B, NOTCH3, KEAP1, BACH2, MED16, ZFY, FANK1, SNAI2, KLF9, MESP2, E2F3, ZBTB46, TWIST1, SP4, PAX5, POU2F2, ATF4, GPS2, FOXO1, MYBBP1A, FOSL1, RFXANK, PIAS1, MRTFB, SRF, MAZ, PTTG1, PYCARD, GABPA, SIN3B, HMGB2, VAV1, CREB3L4, EVX2, RBL1, SMAD6, MYOCD, VHL, CREM, TFAP2E, HLX, ING2, PRDM1, HDAC6, SF1, RFX2, ZNF224, HMGB1, TRIM24, MEF2C, ARNT2, DEPDC1, KLF3 |
| MET | POU4F2, HOXD11, IKZF2, KMT2D, TBX4, CBX4, HIVEP2, HOXA2, MEOX1, OTX2, SIX5, PAX6, ZC3H8, MEIS2, PAX9, HDAC8, ZMYND8, DLX5, NEUROG2, PAX8, LHX5, GFI1B, MED12, OTX1, FUBP1, PRRX1, DLX6, ZFPM2, HMX2, MEIS1, NSD1, HOXC11, HSF4, IRX1, COMMD3-BMI1, CNOT7, TBX1, EAF1, ZFP90, TFCP2, SHOX2, ASCL1, NKX3-2, ONECUT2, BHLHE41, ARID2, ZBTB32, PTF1A, HES3, PBX3, FOXG1 |
| EMT/MET | CLOCK, ACTN4, IRF4, SMARCD3, PKNOX2, EHF, SS18, KLF11, USF2, HOXA13, FOXC2, SP110, ATOH1, EED, NKX2-3, DACH1, NFIX, SMAD3, SCX, DUX4, FOXE1, ZNF148, SP1, RBPJ, GATA6, HOXC9, PRDM16, FOXC1, SMARCB1, SPZ1, SPDEF, MSX1, NPM1, MYF6, STAT4, HDAC2, MRTFA, FOXF1, ETS1, HNF4A, RELA, TP73, RUNX1, EGR1, HSF1, NFATC2, GFI1, MSX2, USF1, SOX1, KLF4, MYOD1, YY1, SP3, PPARGC1A, WT1, SNAI1, LEF1, PITX2, GLI2, TP53, FOXL1, WWTR1, NFKBIE, SOX3, KLF2, ELF5, HOXA10, LMO2, STAT3, GLI1, GMNN, ASXL1, PAX3, POU5F1, HDAC1, LMX1B, CTNNB1, HOXA5, NFKB1, IRF8, SIRT1, SOX11, SIX1, EBF1, HOXA11, NFAT5, NFKBIA, PCGF2, FOXF2, KLF10, GABPB1, BCL6, SMAD4, UHRF1, GLI3, VAV2, CEBPB, PAX2, NFIC, MYC, LHX2, JUN, KDM3A, Tcf7, NKX2-1, GBX2, NOTCH1, STAT1, ARID1A, SOX2, TP63, GATA4, SMAD5, EAF2, TEAD4, MYB |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, C.-H.; Hsu, T.-I.; Chang, Y.-C.; Chan, M.-H.; Lu, P.-J.; Hsiao, M. Stationed or Relocating: The Seesawing EMT/MET Determinants from Embryonic Development to Cancer Metastasis. Biomedicines 2021, 9, 1265. https://doi.org/10.3390/biomedicines9091265
Li C-H, Hsu T-I, Chang Y-C, Chan M-H, Lu P-J, Hsiao M. Stationed or Relocating: The Seesawing EMT/MET Determinants from Embryonic Development to Cancer Metastasis. Biomedicines. 2021; 9(9):1265. https://doi.org/10.3390/biomedicines9091265
Chicago/Turabian StyleLi, Chien-Hsiu, Tai-I Hsu, Yu-Chan Chang, Ming-Hsien Chan, Pei-Jung Lu, and Michael Hsiao. 2021. "Stationed or Relocating: The Seesawing EMT/MET Determinants from Embryonic Development to Cancer Metastasis" Biomedicines 9, no. 9: 1265. https://doi.org/10.3390/biomedicines9091265
APA StyleLi, C.-H., Hsu, T.-I., Chang, Y.-C., Chan, M.-H., Lu, P.-J., & Hsiao, M. (2021). Stationed or Relocating: The Seesawing EMT/MET Determinants from Embryonic Development to Cancer Metastasis. Biomedicines, 9(9), 1265. https://doi.org/10.3390/biomedicines9091265