
Endothelial Cell Activation by SARS-CoV-2 Spike S1 Protein: A Crosstalk between Endothelium and Innate Immune Cells

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. RT-qPCR Analysis
2.3. Cytokine Analysis
2.4. Western Blot Analysis
2.5. Statistical Analysis
2.6. Materials
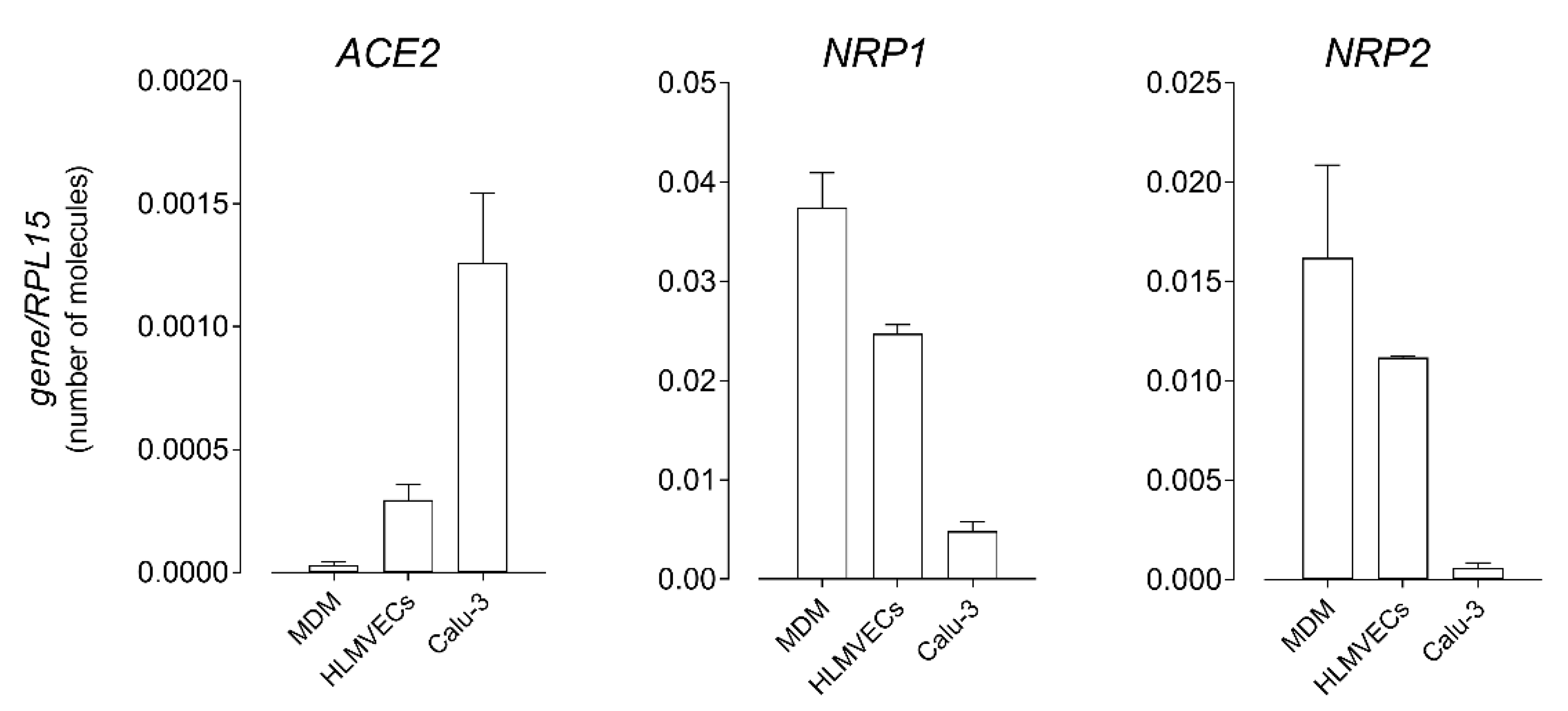
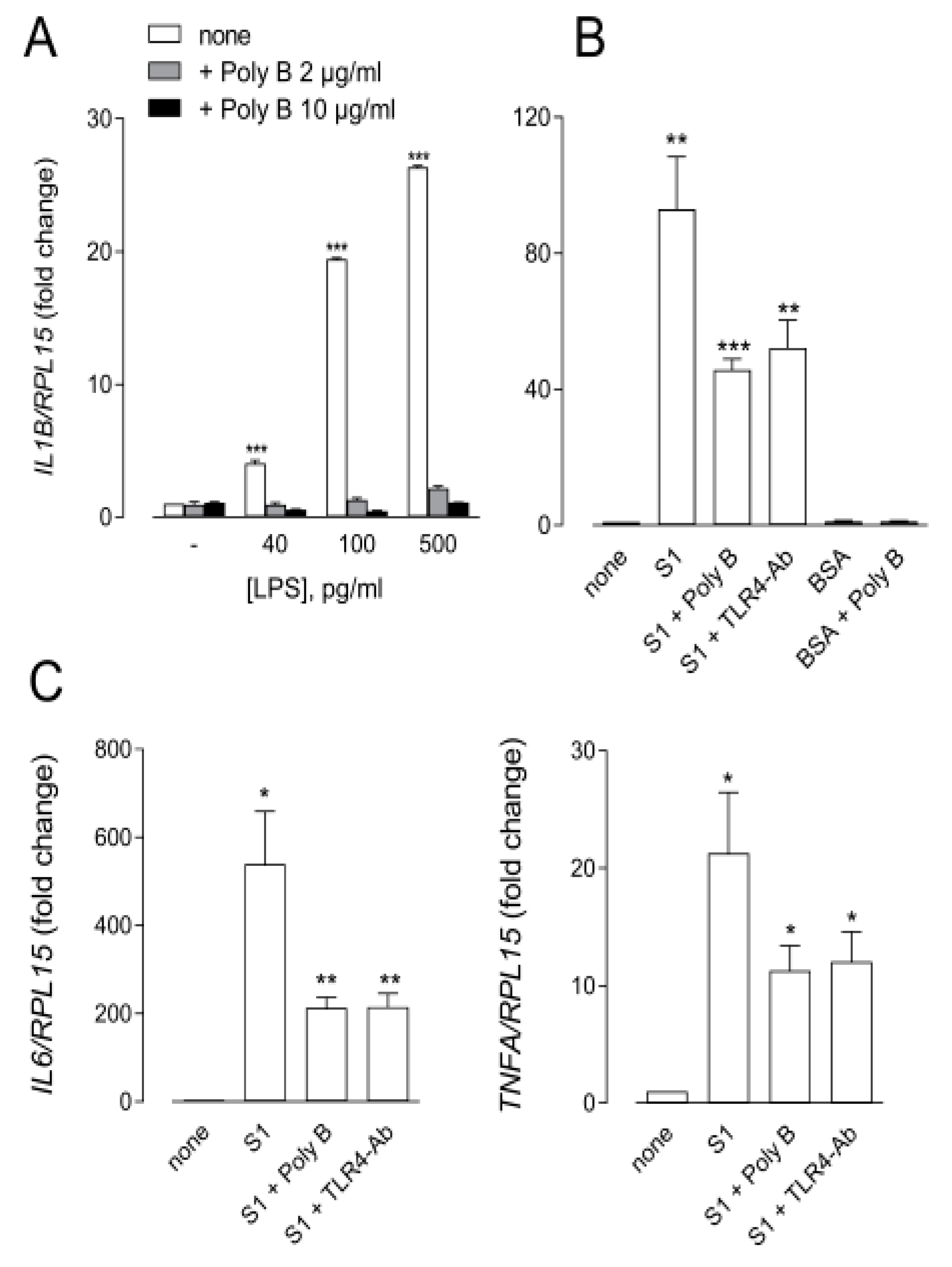
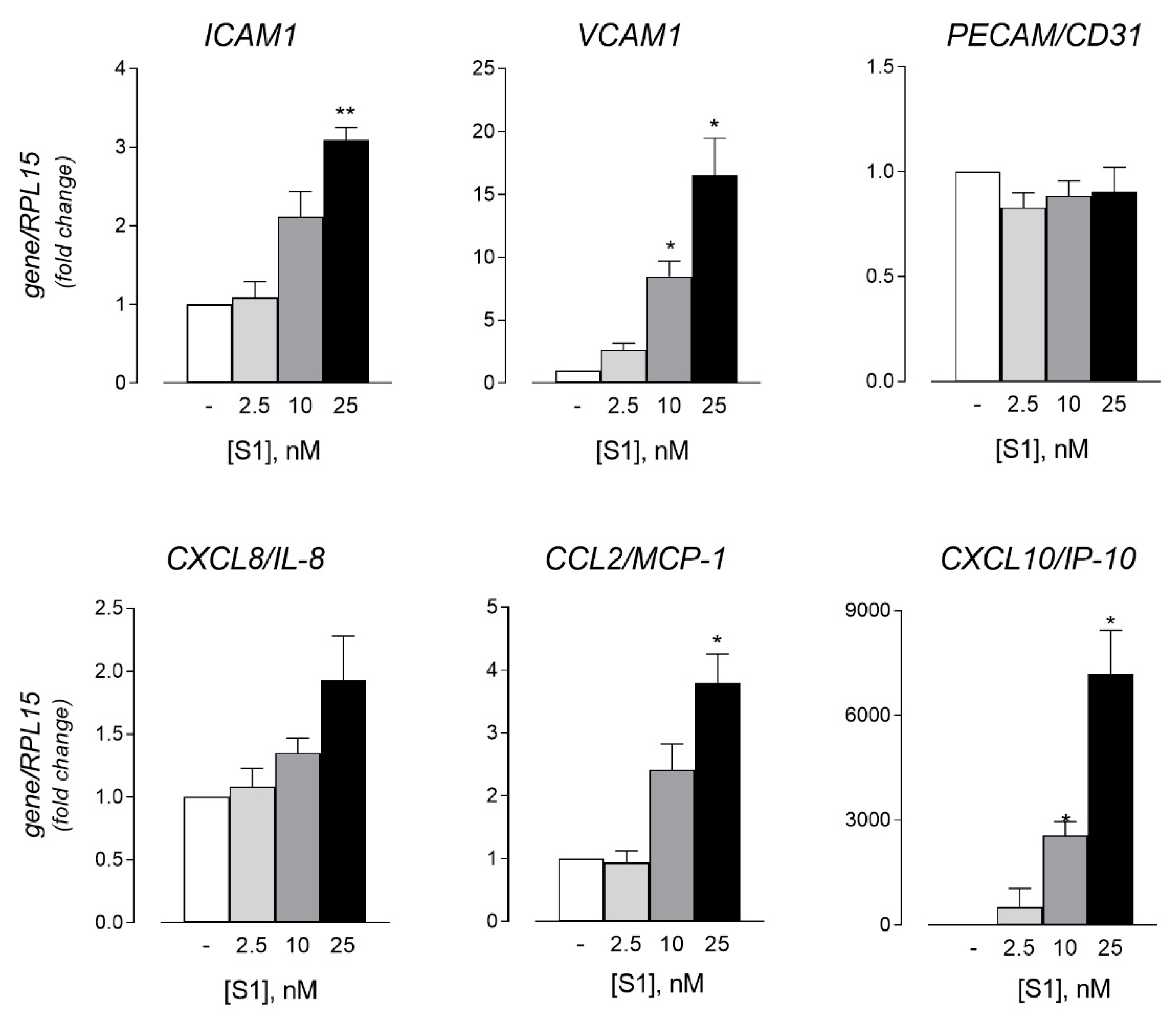
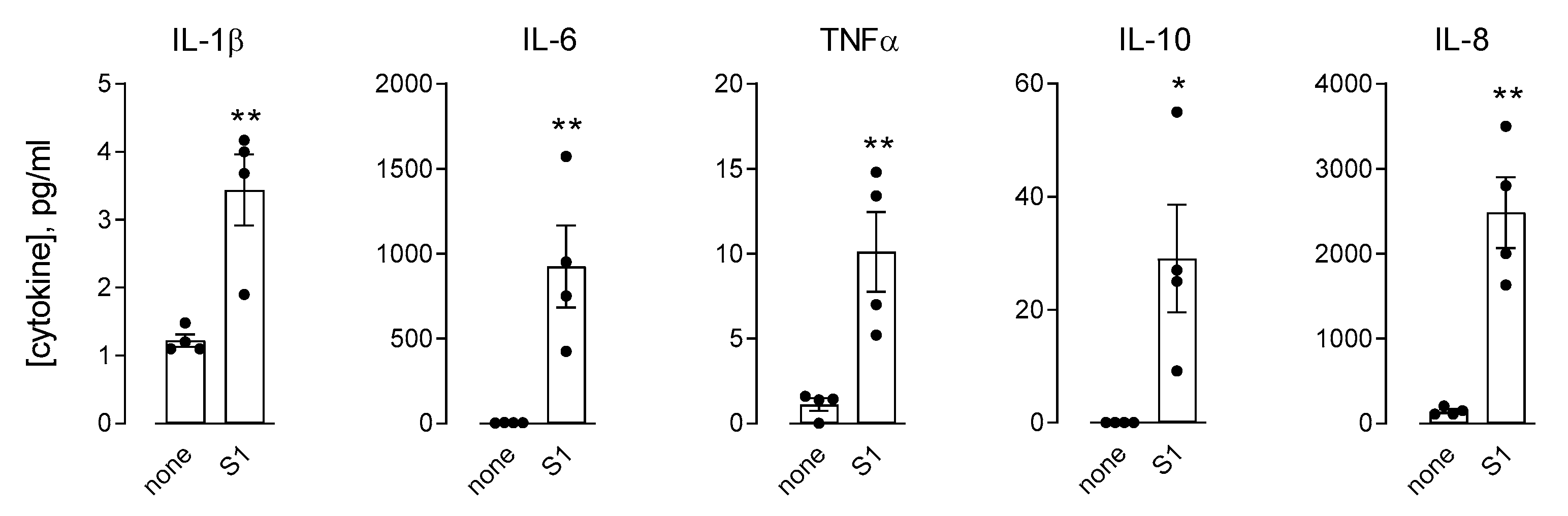
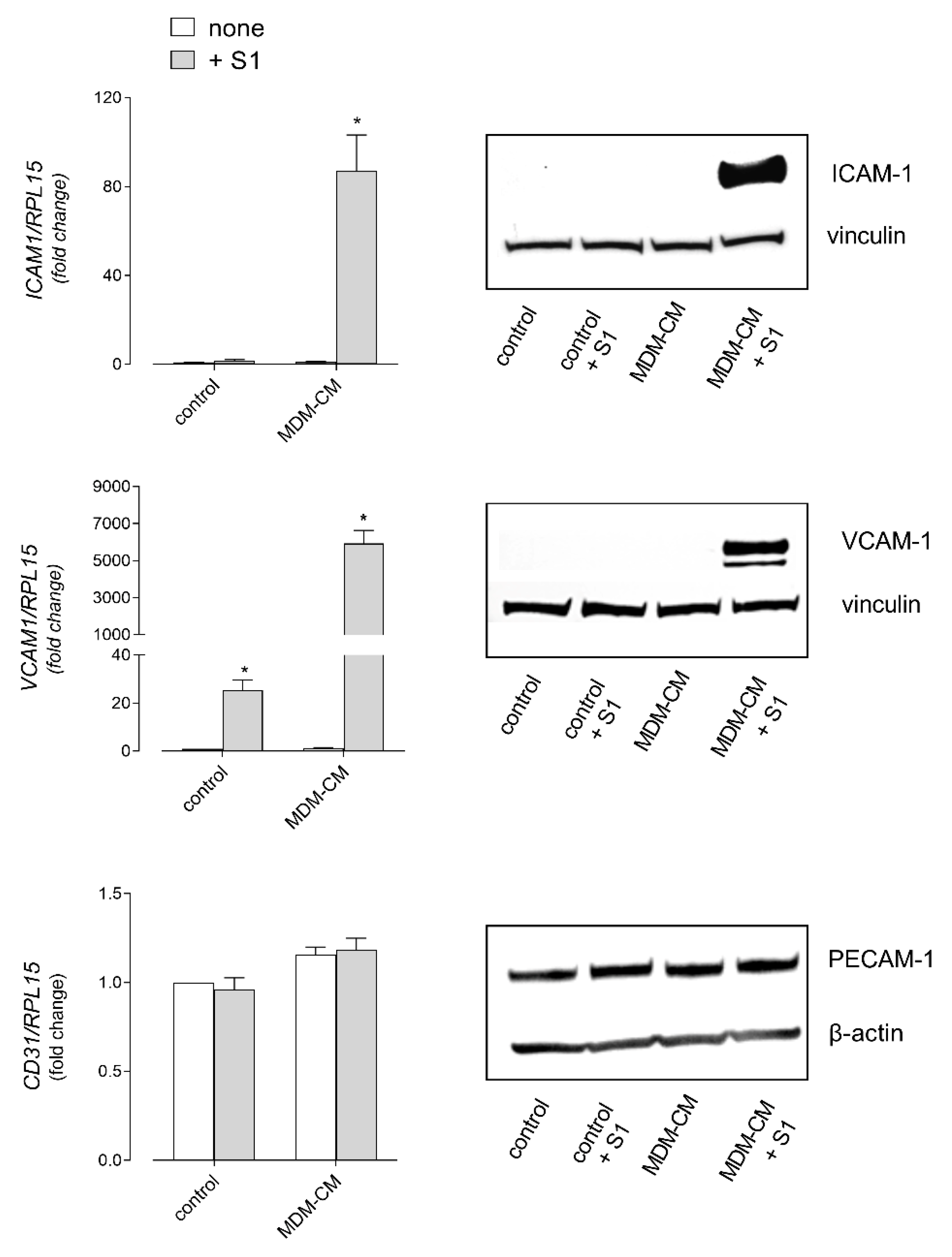
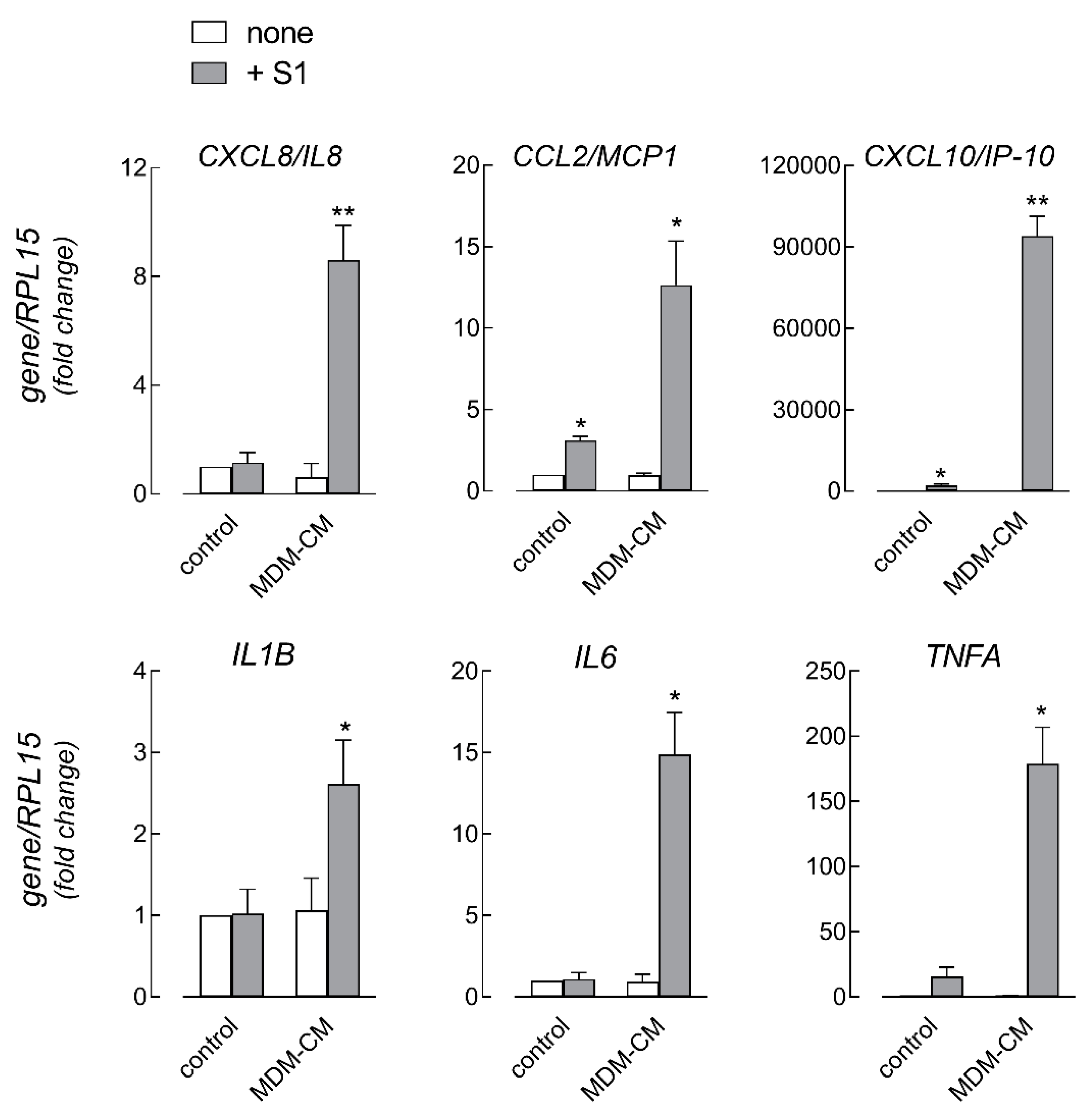
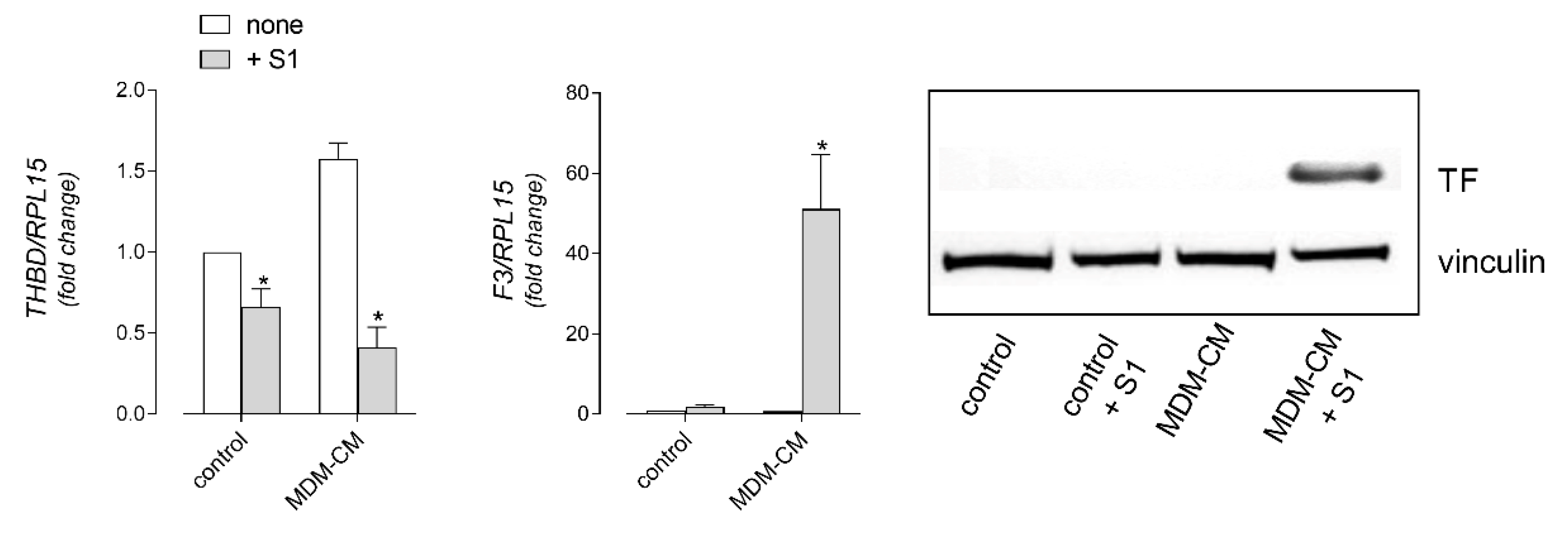
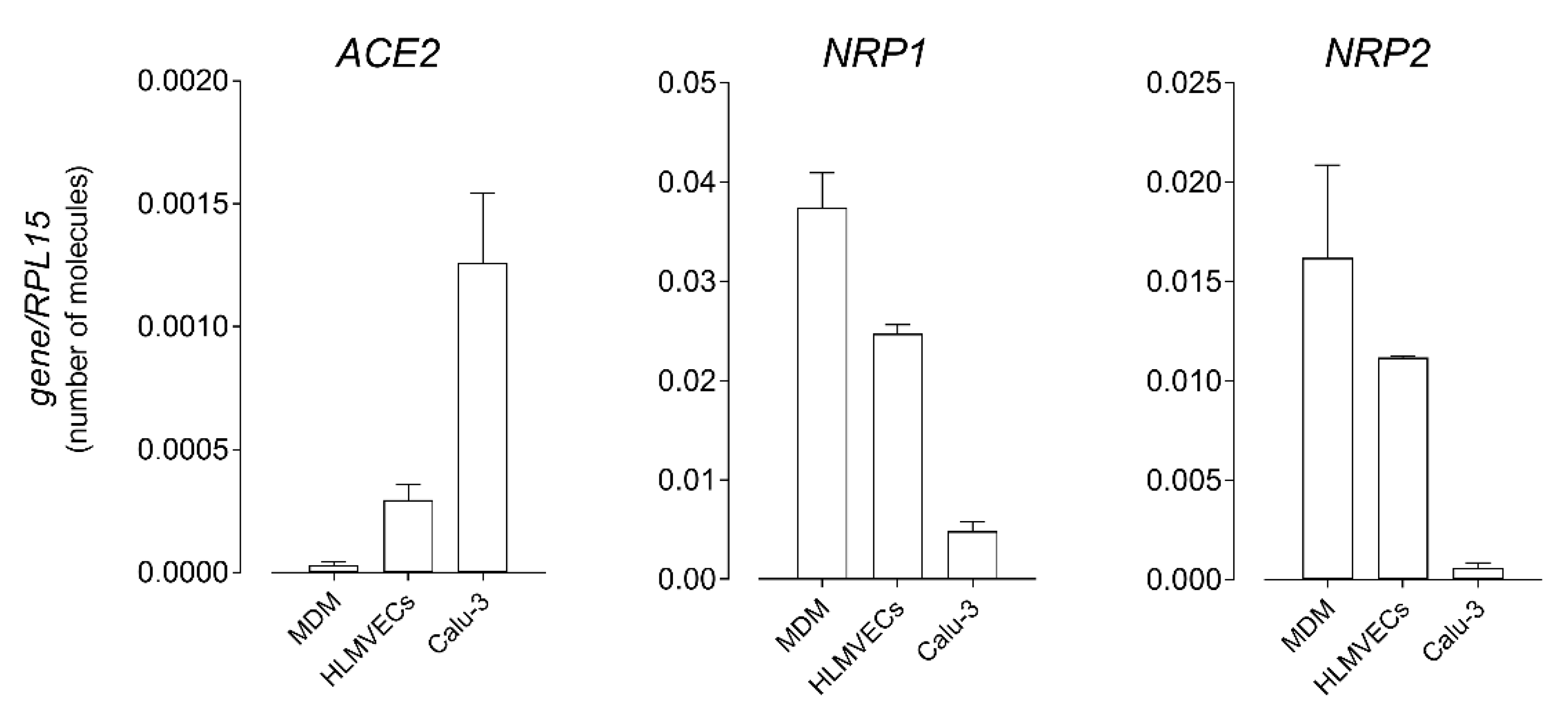
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef] [Green Version]
- Scialo, F.; Daniele, A.; Amato, F.; Pastore, L.; Matera, M.G.; Cazzola, M.; Castaldo, G.; Bianco, A. ACE2: The Major Cell Entry Receptor for SARS-CoV-2. Lung 2020, 198, 867–877. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N. Engl. J. Med. 2020, 383, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Tay, M.Z.; Poh, C.M.; Renia, L.; MacAry, P.A.; Ng, L.F.P. The trinity of COVID-19: Immunity, inflammation and intervention. Nat. Rev. Immunol. 2020, 20, 363–374. [Google Scholar] [CrossRef]
- Wiersinga, W.J.; Rhodes, A.; Cheng, A.C.; Peacock, S.J.; Prescott, H.C. Pathophysiology, Transmission, Diagnosis, and Treatment of Coronavirus Disease 2019 (COVID-19): A Review. JAMA 2020, 324, 782–793. [Google Scholar] [CrossRef]
- Coperchini, F.; Chiovato, L.; Croce, L.; Magri, F.; Rotondi, M. The cytokine storm in COVID-19: An overview of the involvement of the chemokine/chemokine-receptor system. Cytokine Growth Factor Rev. 2020, 53, 25–32. [Google Scholar] [CrossRef]
- Evans, P.C.; Rainger, G.E.; Mason, J.C.; Guzik, T.J.; Osto, E.; Stamataki, Z.; Neil, D.; Hoefer, I.E.; Fragiadaki, M.; Waltenberger, J.; et al. Endothelial dysfunction in COVID-19: A position paper of the ESC Working Group for Atherosclerosis and Vascular Biology, and the ESC Council of Basic Cardiovascular Science. Cardiovasc. Res. 2020, 116, 2177–2184. [Google Scholar] [CrossRef]
- Bernard, I.; Limonta, D.; Mahal, L.K.; Hobman, T.C. Endothelium Infection and Dysregulation by SARS-CoV-2: Evidence and Caveats in COVID-19. Viruses 2020, 13, 29. [Google Scholar] [CrossRef]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef]
- Nicosia, R.F.; Ligresti, G.; Caporarello, N.; Akilesh, S.; Ribatti, D. COVID-19 Vasculopathy: Mounting Evidence for an Indirect Mechanism of Endothelial Injury. Am. J. Pathol. 2021, 191, 1374–1384. [Google Scholar] [CrossRef]
- Liu, L.; Chopra, P.; Li, X.; Bouwman, K.M.; Tompkins, S.M.; Wolfert, M.A.; de Vries, R.P.; Boons, G.J. Heparan Sulfate Proteoglycans as Attachment Factor for SARS-CoV-2. ACS Cent. Sci. 2021, 7, 1009–1018. [Google Scholar] [CrossRef]
- Park, E.J.; Myint, P.K.; Appiah, M.G.; Darkwah, S.; Caidengbate, S.; Ito, A.; Matsuo, E.; Kawamoto, E.; Gaowa, A.; Shimaoka, M. The Spike Glycoprotein of SARS-CoV-2 Binds to beta1 Integrins Expressed on the Surface of Lung Epithelial Cells. Viruses 2021, 13, 645. [Google Scholar] [CrossRef]
- Daly, J.L.; Simonetti, B.; Klein, K.; Chen, K.E.; Williamson, M.K.; Anton-Plagaro, C.; Shoemark, D.K.; Simon-Gracia, L.; Bauer, M.; Hollandi, R.; et al. Neuropilin-1 is a host factor for SARS-CoV-2 infection. Science 2020, 370, 861–865. [Google Scholar] [CrossRef] [PubMed]
- Issitt, T.; Bosseboeuf, E.; De Winter, N.; Dufton, N.; Gestri, G.; Senatore, V.; Chikh, A.; Randi, A.M.; Raimondi, C. Neuropilin-1 Controls Endothelial Homeostasis by Regulating Mitochondrial Function and Iron-Dependent Oxidative Stress. iScience 2019, 11, 205–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Cao, Y.; Mangalam, A.K.; Guo, Y.; LaFrance-Corey, R.G.; Gamez, J.D.; Atanga, P.A.; Clarkson, B.D.; Zhang, Y.; Wang, E.; et al. Neuropilin-1 modulates interferon-gamma-stimulated signaling in brain microvascular endothelial cells. J. Cell Sci. 2016, 129, 3911–3921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantuti-Castelvetri, L.; Ojha, R.; Pedro, L.D.; Djannatian, M.; Franz, J.; Kuivanen, S.; van der Meer, F.; Kallio, K.; Kaya, T.; Anastasina, M.; et al. Neuropilin-1 facilitates SARS-CoV-2 cell entry and infectivity. Science 2020, 370, 856–860. [Google Scholar] [CrossRef]
- Mone, P.; Gambardella, J.; Wang, X.; Jankauskas, S.S.; Matarese, A.; Santulli, G. miR-24 Targets the Transmembrane Glycoprotein Neuropilin-1 in Human Brain Microvascular Endothelial Cells. Noncoding RNA 2021, 7, 9. [Google Scholar] [CrossRef]
- Soker, S.; Takashima, S.; Miao, H.Q.; Neufeld, G.; Klagsbrun, M. Neuropilin-1 is expressed by endothelial and tumor cells as an isoform-specific receptor for vascular endothelial growth factor. Cell 1998, 92, 735–745. [Google Scholar] [CrossRef] [Green Version]
- Huertas, A.; Montani, D.; Savale, L.; Pichon, J.; Tu, L.; Parent, F.; Guignabert, C.; Humbert, M. Endothelial cell dysfunction: A major player in SARS-CoV-2 infection (COVID-19)? Eur. Respir. J. 2020, 56, 2001634. [Google Scholar] [CrossRef]
- Rodriguez, C.; Luque, N.; Blanco, I.; Sebastian, L.; Barbera, J.A.; Peinado, V.I.; Tura-Ceide, O. Pulmonary Endothelial Dysfunction and Thrombotic Complications in Patients with COVID-19. Am. J. Respir. Cell Mol. Biol. 2021, 64, 407–415. [Google Scholar] [CrossRef]
- Perico, L.; Benigni, A.; Casiraghi, F.; Ng, L.F.P.; Renia, L.; Remuzzi, G. Immunity, endothelial injury and complement-induced coagulopathy in COVID-19. Nat. Rev. Nephrol. 2021, 17, 46–64. [Google Scholar] [CrossRef]
- Ingoglia, F.; Visigalli, R.; Rotoli, B.M.; Barilli, A.; Riccardi, B.; Puccini, P.; Milioli, M.; Di Lascia, M.; Bernuzzi, G.; Dall’Asta, V. Human macrophage differentiation induces OCTN2-mediated L-carnitine transport through stimulation of mTOR-STAT3 axis. J. Leukoc. Biol. 2017, 101, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Ingoglia, F.; Visigalli, R.; Rotoli, B.M.; Barilli, A.; Riccardi, B.; Puccini, P.; Dall’Asta, V. Functional activity of L-carnitine transporters in human airway epithelial cells. Biochim. Biophys. Acta 2016, 1858, 210–219. [Google Scholar] [CrossRef]
- Rotoli, B.M.; Barilli, A.; Visigalli, R.; Ferrari, F.; Dall’Asta, V. y+LAT1 and y+LAT2 contribution to arginine uptake in different human cell models: Implications in the pathophysiology of Lysinuric Protein Intolerance. J. Cell Mol. Med. 2020, 24, 921–929. [Google Scholar] [CrossRef] [Green Version]
- Rotoli, B.M.; Barilli, A.; Visigalli, R.; Ingoglia, F.; Milioli, M.; Di Lascia, M.; Riccardi, B.; Puccini, P.; Dall’Asta, V. Downregulation of SLC7A7 Triggers an Inflammatory Phenotype in Human Macrophages and Airway Epithelial Cells. Front. Immunol. 2018, 9, 508. [Google Scholar] [CrossRef] [PubMed]
- Barilli, A.; Visigalli, R.; Ferrari, F.; Borsani, G.; Dall’Asta, V.; Rotoli, B.M. Flagellin From Pseudomonas Aeruginosa Stimulates ATB(0,+) Transporter for Arginine and Neutral Amino Acids in Human Airway Epithelial Cells. Front. Immunol. 2021, 12, 641563. [Google Scholar] [CrossRef]
- Gaiani, F.; Rotoli, B.M.; Ferrari, F.; Barilli, A.; Visigalli, R.; Carra, M.C.; de’Angelis, G.L.; de’Angelis, N.; Dall’Asta, V. Monocytes from infliximab-resistant patients with Crohn’s disease exhibit a disordered cytokine profile. Sci. Rep. 2020, 10, 12238. [Google Scholar] [CrossRef] [PubMed]
- Tseng, C.T.; Tseng, J.; Perrone, L.; Worthy, M.; Popov, V.; Peters, C.J. Apical entry and release of severe acute respiratory syndrome-associated coronavirus in polarized Calu-3 lung epithelial cells. J. Virol. 2005, 79, 9470–9479. [Google Scholar] [CrossRef] [Green Version]
- Petruk, G.; Puthia, M.; Petrlova, J.; Samsudin, F.; Stromdahl, A.C.; Cerps, S.; Uller, L.; Kjellstrom, S.; Bond, P.J.; Schmidtchen, A.A. SARS-CoV-2 spike protein binds to bacterial lipopolysaccharide and boosts proinflammatory activity. J. Mol. Cell Biol. 2021, 12, 916–932. [Google Scholar] [CrossRef]
- Tsubery, H.; Ofek, I.; Cohen, S.; Fridkin, M. The functional association of polymyxin B with bacterial lipopolysaccharide is stereospecific: Studies on polymyxin B nonapeptide. Biochemistry 2000, 39, 11837–11844. [Google Scholar] [CrossRef]
- Libby, P.; Luscher, T. COVID-19 is, in the end, an endothelial disease. Eur. Heart J. 2020, 41, 3038–3044. [Google Scholar] [CrossRef] [PubMed]
- Lowenstein, C.J.; Solomon, S.D. Severe COVID-19 Is a Microvascular Disease. Circulation 2020, 142, 1609–1611. [Google Scholar] [CrossRef] [PubMed]
- Jung, F.; Kruger-Genge, A.; Franke, R.P.; Hufert, F.; Kupper, J.H. COVID-19 and the endothelium. Clin. Hemorheol. Microcirc. 2020, 75, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Pons, S.; Fodil, S.; Azoulay, E.; Zafrani, L. The vascular endothelium: The cornerstone of organ dysfunction in severe SARS-CoV-2 infection. Crit. Care 2020, 24, 353. [Google Scholar] [CrossRef] [PubMed]
- Sardu, C.; Gambardella, J.; Morelli, M.B.; Wang, X.; Marfella, R.; Santulli, G. Hypertension, Thrombosis, Kidney Failure, and Diabetes: Is COVID-19 an Endothelial Disease? A Comprehensive Evaluation of Clinical and Basic Evidence. J. Clin. Med. 2020, 9, 1417. [Google Scholar] [CrossRef] [PubMed]
- Amraei, R.; Rahimi, N. COVID-19, Renin-Angiotensin System and Endothelial Dysfunction. Cells 2020, 9, 1652. [Google Scholar] [CrossRef] [PubMed]
- Buzhdygan, T.P.; DeOre, B.J.; Baldwin-Leclair, A.; Bullock, T.A.; McGary, H.M.; Khan, J.A.; Razmpour, R.; Hale, J.F.; Galie, P.A.; Potula, R.; et al. The SARS-CoV-2 spike protein alters barrier function in 2D static and 3D microfluidic in-vitro models of the human blood-brain barrier. Neurobiol. Dis. 2020, 146, 105131. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Zhang, J.; Schiavon, C.R.; He, M.; Chen, L.; Shen, H.; Zhang, Y.; Yin, Q.; Cho, Y.; Andrade, L.; et al. SARS-CoV-2 Spike Protein Impairs Endothelial Function via Downregulation of ACE2. bioRxiv 2020. [Google Scholar] [CrossRef]
- Nascimento Conde, J.; Schutt, W.R.; Gorbunova, E.E.; Mackow, E.R. Recombinant ACE2 Expression Is Required for SARS-CoV-2 To Infect Primary Human Endothelial Cells and Induce Inflammatory and Procoagulative Responses. mBio 2020, 11, 6. [Google Scholar] [CrossRef]
- Raghavan, S.; Kenchappa, D.B.; Leo, M.D. SARS-CoV-2 Spike Protein Induces Degradation of Junctional Proteins That Maintain Endothelial Barrier Integrity. Front. Cardiovasc. Med. 2021, 8, 687783. [Google Scholar] [CrossRef]
- Karwaciak, I.; Salkowska, A.; Karas, K.; Dastych, J.; Ratajewski, M. Nucleocapsid and Spike Proteins of the Coronavirus SARS-CoV-2 Induce IL6 in Monocytes and Macrophages-Potential Implications for Cytokine Storm Syndrome. Vaccines 2021, 9, 54. [Google Scholar] [CrossRef]
- Duarte, C.; Akkaoui, J.; Ho, A.; Garcia, C.; Yamada, C.; Movila, A. Age-dependent effects of the recombinant spike protein/SARS-CoV-2 on the M-CSF- and IL-34-differentiated macrophages in vitro. Biochem. Biophys. Res. Commun. 2021, 546, 97–102. [Google Scholar] [CrossRef]
- Escher, R.; Breakey, N.; Lammle, B. Severe COVID-19 infection associated with endothelial activation. Thromb. Res. 2020, 190, 62. [Google Scholar] [CrossRef]
- Tong, M.; Jiang, Y.; Xia, D.; Xiong, Y.; Zheng, Q.; Chen, F.; Zou, L.; Xiao, W.; Zhu, Y. Elevated Expression of Serum Endothelial Cell Adhesion Molecules in COVID-19 Patients. J. Infect. Dis. 2020, 222, 894–898. [Google Scholar] [CrossRef]
- Chi, Y.; Ge, Y.; Wu, B.; Zhang, W.; Wu, T.; Wen, T.; Liu, J.; Guo, X.; Huang, C.; Jiao, Y.; et al. Serum Cytokine and Chemokine Profile in Relation to the Severity of Coronavirus Disease 2019 in China. J. Infect. Dis. 2020, 222, 746–754. [Google Scholar] [CrossRef] [PubMed]
- Merad, M.; Martin, J.C. Pathological inflammation in patients with COVID-19: A key role for monocytes and macrophages. Nat. Rev. Immunol 2020, 20, 355–362. [Google Scholar] [CrossRef]
- Shirato, K.; Kizaki, T. SARS-CoV-2 spike protein S1 subunit induces pro-inflammatory responses via toll-like receptor 4 signaling in murine and human macrophages. Heliyon 2021, 7, e06187. [Google Scholar] [CrossRef]
- Panigada, M.; Bottino, N.; Tagliabue, P.; Grasselli, G.; Novembrino, C.; Chantarangkul, V.; Pesenti, A.; Peyvandi, F.; Tripodi, A. Hypercoagulability of COVID-19 patients in intensive care unit: A report of thromboelastography findings and other parameters of hemostasis. J. Thromb. Haemost. 2020, 18, 1738–1742. [Google Scholar] [CrossRef] [PubMed]
- Bautista-Vargas, M.; Bonilla-Abadia, F.; Canas, C.A. Potential role for tissue factor in the pathogenesis of hypercoagulability associated with in COVID-19. J. Thromb. Thrombolysis 2020, 50, 1–5. [Google Scholar] [CrossRef] [PubMed]
- DiNicolantonio, J.J.; McCarty, M. Thrombotic complications of COVID-19 may reflect an upregulation of endothelial tissue factor expression that is contingent on activation of endosomal NADPH oxidase. Open Heart 2020, 7, e001337. [Google Scholar] [CrossRef] [PubMed]
- Hottz, E.D.; Azevedo-Quintanilha, I.G.; Palhinha, L.; Teixeira, L.; Barreto, E.A.; Pao, C.R.R.; Righy, C.; Franco, S.; Souza, T.M.L.; Kurtz, P.; et al. Platelet activation and platelet-monocyte aggregate formation trigger tissue factor expression in patients with severe COVID-19. Blood 2020, 136, 1330–1341. [Google Scholar] [CrossRef] [PubMed]
- Francischetti, I.M.B.; Toomera, K.; Zhangb, Y.; Jayesh Jania, J.; Siddiquic, Z.; Brotmanc, D.J.; Hoopera, J.E.; Kickler, T.S. Upregulation of pulmonary tissue factor, loss of thrombomodulin and immunothrombosis in SARS-CoV-2 infection. E.Clin. Med. 2021, 39, 101069. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene/Protein Name (Gene ID) | Forward Primer | Reverse Primer |
|---|---|---|
| RPL15/RPL15 (6138) | Hs03855120_g1 (TaqMan® Assay, Thermo Fisher Scientific) | |
| ACE2/ACE2 (59272) | ACAGTCCACACTTGCCCAAAT | TGAGAGCACTGAAGACCCATT |
| NRP1/NRP1 (8829) | ACCCAAGTGAAAAATGCGAATG | CCTCCAAATCGAAGTGAGGGTT |
| NRP2/NRP2 (8828) | GCTGGCTATATCACCTCTCCC | TCTCGATTTCAAAGTGAGGGTTG |
| IL1B/IL-1β (3553) | Hs99999029_m1 (TaqMan® Assay, Thermo Fisher Scientific) | |
| IL6/IL-6 [27] (3569) | AACCTGAACCTTCCAAAGATGG | TCTGGCTTGTTCCTCACTACT |
| CXCL8/IL-8 [27] (3576) | ACTGAGAGTGATTGAGAGTGGAC | AACCCTCTGCACCCAGTTTTC |
| CXCL10/IP-10 (3627) | GTGGCATTCAAGGAGTACCTC | TGATGGCCTTCGATTCTGGATT |
| CCL2/MCP1 [28] (6347) | CAGCCAGATGCAATCAATGCC | TGGAATCCTGAACCCACTTCT |
| TNFA/TNFα [28] (7124) | ATGAGCACTGAAAGCATGATCC | GAGGGCTGATTAGAGAGAGGTC |
| ICAM1/ICAM-1 (3383) | TGAACCCCACAGTCACCTATG | CTCGTCCTCTGCGGTCAC |
| VCAM1/VCAM-1 (7412) | GGGAAGATGGTCGTGATCCTT | TCTGGGGTGGTCTCGATTTTA |
| PECAM1/CD31 (5175) | GCTGACCCTTCTGCTCTGTT | ATCTGGTGCTGAGGCTTGAC |
| THBD/THBD (7056) | ACCTTCCTCAATGCCAGTCA | AAGGAAATGACATCGGCAGC |
| F3/TF (2152) | GGCGCTTCAGGCACTACAA | TTGATTGACGGGTTTGGGTTC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rotoli, B.M.; Barilli, A.; Visigalli, R.; Ferrari, F.; Dall’Asta, V. Endothelial Cell Activation by SARS-CoV-2 Spike S1 Protein: A Crosstalk between Endothelium and Innate Immune Cells. Biomedicines 2021, 9, 1220. https://doi.org/10.3390/biomedicines9091220
Rotoli BM, Barilli A, Visigalli R, Ferrari F, Dall’Asta V. Endothelial Cell Activation by SARS-CoV-2 Spike S1 Protein: A Crosstalk between Endothelium and Innate Immune Cells. Biomedicines. 2021; 9(9):1220. https://doi.org/10.3390/biomedicines9091220
Chicago/Turabian StyleRotoli, Bianca Maria, Amelia Barilli, Rossana Visigalli, Francesca Ferrari, and Valeria Dall’Asta. 2021. "Endothelial Cell Activation by SARS-CoV-2 Spike S1 Protein: A Crosstalk between Endothelium and Innate Immune Cells" Biomedicines 9, no. 9: 1220. https://doi.org/10.3390/biomedicines9091220
APA StyleRotoli, B. M., Barilli, A., Visigalli, R., Ferrari, F., & Dall’Asta, V. (2021). Endothelial Cell Activation by SARS-CoV-2 Spike S1 Protein: A Crosstalk between Endothelium and Innate Immune Cells. Biomedicines, 9(9), 1220. https://doi.org/10.3390/biomedicines9091220