
Immune Thrombocytopenia in Antiphospholipid Syndrome: Is It Primary or Secondary?

 , , , , and
, , , , and 
Abstract
1. Introduction
1.1. Diagnosis of APS
1.2. Diagnosis of Primary ITP
1.3. The Coexistence of APS and ITP
- IgG and IgM antibodies against b2GP1 and IgM antibodies to FVII/VIIa are more common in APS, as well as IgM against phospholipids (aCL, aPC, aPS, aPE);
- IgG antibodies against phospholipids are more common in ITP and generally recognize fewer antigens (<3) than the antibodies detected in APS (>3).
2. APS and Thrombocytopenia
Clinical Significance of APL-Positivity in ITP
- A study by Diz-Kücükkaya et al. found that 45.1% of a cohort of ITP patients who were persistently positive for aPL developed later APS [29];
- A study by Machin et al. showed that the statistically most relevant risk factor for thrombosis in patients with thrombocytopenia is the co-diagnosis of APS, over an average 5-years follow-up [30];
- A study by Hisada et al. found that the combination of aPL and thrombocytopenia doubled the risk of future thrombosis over an average 10-years follow-up [27]
- A study by Funauchi et al. demonstrated that women with ITP and aPL-positivity had increased thrombosis and obstetric complications risks when compared to the aPL-negative group [31];
- An interesting cross-sectional study reported that the platelet counts of patients with a diagnosis of high-risk APS (triple positive: LAC, anti-beta2-GPI, and aCL) decreased earlier before the appearance of a full clinical picture of CAPS [32]. Therefore, the screening for aPL may identify a subgroup of ITP patients at higher risk of thrombosis.
- The already mentioned International Consensus Panel stated that thrombocytopenia occurring in patients with persistent aPL-positivity is associated with increased thrombotic risk and therefore should be considered different from simple ITP [3];
- Data from the Italian Registry of Antiphospholipid Antibodies reported that 40% of the APS patients with moderate thrombocytopenia and 9% of the APS patients with severe thrombocytopenia developed thrombosis [24];
- A review by Frison et al. on the records of 233 outpatients with primary or secondary thrombocytopenia (platelet count < 100 × 109/L) concluded that triple-positive patients had a significantly lower median platelet count compared to other patients with aPL-positivity [33].
3. Clinical Significance of Thrombocytopenia and aPL-Positivity in Patients with Systemic Lupus Erythematosus
3.1. Therapeutic Management of SLE-Associated Thrombocytopenia
3.2. Prevention of aPL-Associated Complications in SLM
4. An Overview of the Treatment of Antiphospholipid Antibodies Syndrome: The Latest Guidelines
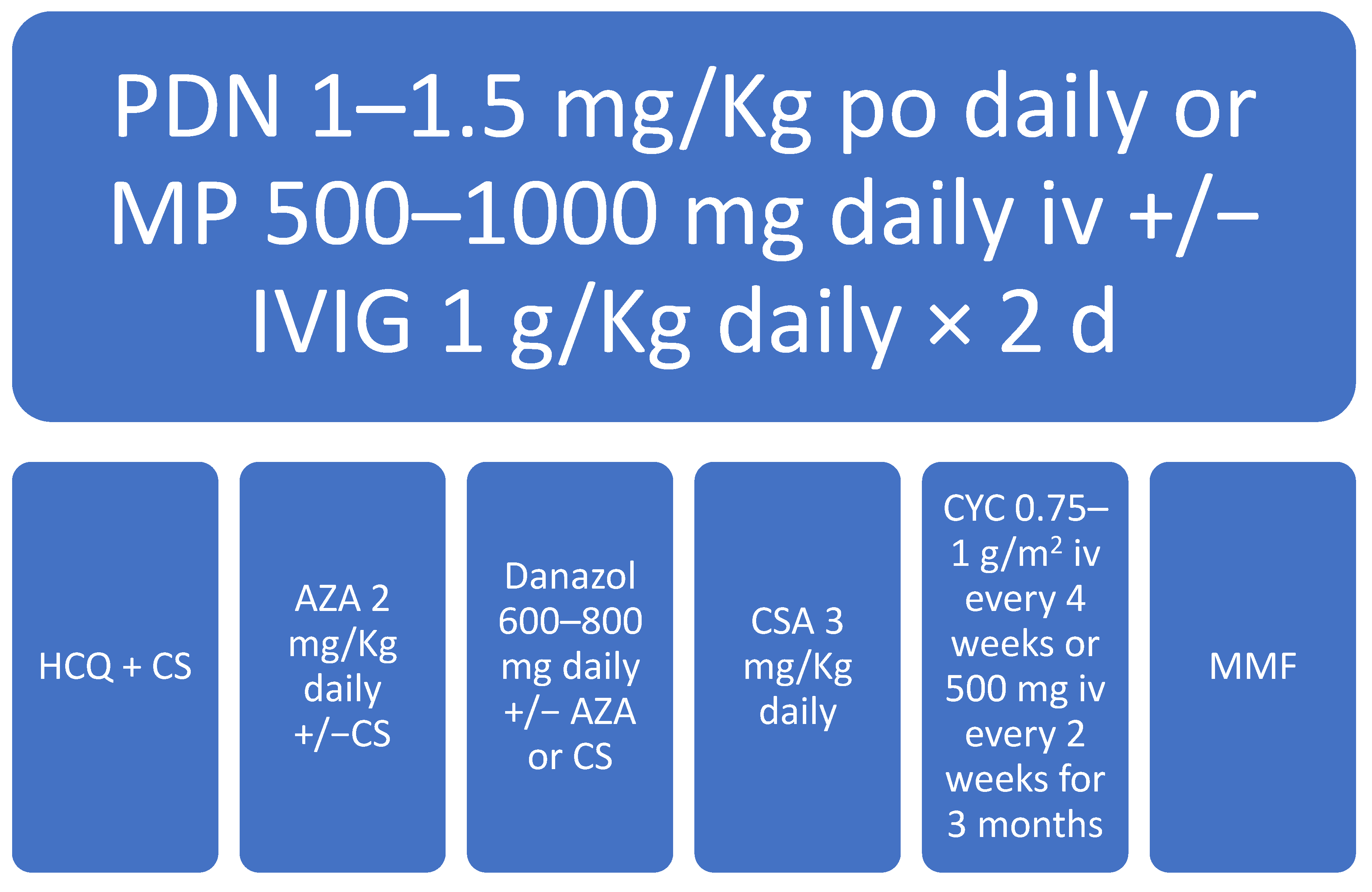
5. Management of aPL-Associated Thrombocytopenia
5.1. Definition of Clinical Response
5.2. The Role of anti-CD20 MoAb in Treating aPL-Associated Thrombocytopenia
5.3. Thrombopoietin Receptor Agonists in the II Line Treatment of Connettive Tissue Disease-Associated Thrombocytopenia
5.4. Hydroxychloroquine as a Possible Second Line Agent in APS-Associated Thrombocytopenia
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Harris, E.N. Syndrome of the Black Swan. Rheumatology 1987, 26, 324–326. [Google Scholar] [CrossRef]
- Wilson, W.A.; Gharavi, A.E.; Koike, T.; Lockshin, M.D.; Branch, D.W.; Piette, J.C.; Brey, R.; Derksen, R.; Harris, E.N.; Hughes, G.R. International consensus statement on preliminary classification criteria for definite antiphospholipid syndrome: Report of an International workshop. Arthritis Rheum. 1999, 42, 1309–1311. [Google Scholar] [CrossRef]
- Miyakis, S.; Lockshin, M.D.; Atsumi, T.; Branch, D.W.; Brey, R.L.; Cervera, R.H.; Derksen, R.H.; De Groot, P.G.; Koike, T.; Meroni, P.L.; et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J. Thromb. Haemost. 2006, 4, 295–306. [Google Scholar] [CrossRef]
- McNeil, H.P.; Simpson, R.J.; Chesterman, C.N.; Krilis, S.A. Antiphospholipid antibodies are directed against a complex antigen that in-cludes lipid binding inhibitor of coagulation: β2 glycoprotein I (apolipoprotein H). Proc. Natl. Acad. Sci. USA 1990, 87, 4120–4124. [Google Scholar] [CrossRef] [PubMed]
- Galli, M.; Barbui, T.; Comfurius, P.; Maassen, C.; de Baets, M.H.; van Breda-Vriesman, P.J.; Barbui, T.; Zwaal, R.F.; Bevers, E.M. Anticardiolipin antibodies (ACA) directed not to cardiolipin but to a plasma protein cofactor. Lancet 1990, 335, 1544–1547. [Google Scholar] [CrossRef]
- Amengual, O.; Atsumi, T.; Koike, T. Antiprothombin antibodies and the diagnosis of antiphospholipid syndrome. Clin. Immunol. 2004, 112, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Permpikul, P.; Rao, L.; Rapaport, S. Functional and binding studies of the roles of prothrombin and beta 2- glycoprotein I in the expression of lupus anticoagulant activity. Blood 1994, 83, 2878–2892. [Google Scholar] [CrossRef]
- Chen, P.P.; Giles, I. Antibodies to Serine Proteases in the Antiphospholipid Syndrome. Curr. Rheumatol. Rep. 2010, 12, 45–52. [Google Scholar] [CrossRef][Green Version]
- Cesarman-Maus, G.; Ríos-Luna, N.P.; Deora, A.B.; Huang, B.; Villa, R.; Cravioto Mdel, C.; Alarcón-Segovia, D.; Sánchez-Guerrero, J.; Hajjar, K.A. Autoantibodies against the fibrinolytic receptor, annexin 2, in antiphospholipid syndrome. Blood 2006, 107, 4375–4382. [Google Scholar] [CrossRef] [PubMed]
- Rand, J.H.; Wu, X.X.; Quinn, A.S.; Taatjes, D.J. Resistance to annexin A5 anticoagulant activity: A thrombogenic mechanism for the antiphospholipid syndrome. Lupus 2008, 17, 922–930. [Google Scholar] [CrossRef]
- Rodeghiero, F.; Stasi, R.; Gernsheimer, T.; Provan, D.; Arnold, D.M.; Bussel, J.B.; Cines, D.B.; Chong, B.H.; Cooper, N.; Godeau, B.; et al. Standardization of terminology, definitions and outcome criteria in immune thrombocytopenic purpura of adults and children: Report from an international working group. Blood 2009, 113, 2386–2393. [Google Scholar] [CrossRef] [PubMed]
- Bobba, R.S.; Johnson, S.R.; Davis, A.M. A review of the sapporo and revised Sapporo criteria for the classification of antiphos-pholipid syndrome. Where do the revised sapporo criteria add value? J. Rheumatol. 2007, 34, 1522–1527. [Google Scholar] [PubMed]
- Abreu, M.M.; Danowski, A.; Wahl, D.G.; Amigo, M.C.; Tektonidou, M.; Pacheco, M.S.; Fleming, N.; Domingues, V.; Sciascia, S.; Lyra, J.O.; et al. The relevance of “non-criteria” clinical manifestations of antiphospholipid syndrome: 14th International Congress on Antiphospholipid Antibodies Technical Task Force Report on Antiphospholipid Syndrome Clinical Features. Autoimmun. Rev. 2015, 14, 401–414. [Google Scholar] [CrossRef]
- Zuily, S.; Clerc-Urmès, I.; Bauman, C.; Andrade, D.; Sciascia, S.; Pengo, V.; Tektonidou, M.G.; Ugarte, A.; Gerosa, M.; Michael Belmont, H.; et al. Cluster analysis for the identification of clinical phenotypes among antiphospholipid antibody-positive patients from the APS ACTION Registry. Lupus 2020, 29, 1353–1363. [Google Scholar] [CrossRef]
- Espinola, R.G.; Pierangeli, S.S.; Gharavi, A.E.; Harris, E.N. Hydroxychloroquine reverses platelet activation induced by human IgG antiphospholipid antibodies. Thromb. Haemost. 2002, 87, 518–522. [Google Scholar] [CrossRef]
- Harris, E.N.; Gharavi, A.E.; Hegde, U.; Derue, G.; Morgan, S.H.; Englert, H.; Chan, J.K.; Asherson, R.A.; Hughes, G.R. Anticardiolipin antibodies in autoimmune thrombocytopenic purpura. Br. J. Haematol. 1985, 59, 231–234. [Google Scholar] [CrossRef]
- Bidot, C.J.; Jy, W.; Horstman, L.L.; Ahn, E.R.; Jimenez, J.J.; Yaniz, M.; Lander, G.; Ahn, Y.S. Antiphospholipid anti- bodies in immune thrombocytopenic purpura tend to emerge in exacerbation and decline in remission. Br. J. Haematol. 2005, 128, 366–372. [Google Scholar] [CrossRef]
- Stasi, R.; Stipa, E.; Masi, M.; Oliva, F.; Sciarra, A.; Perrotti, A.; Olivieri, M.; Zaccari, G.; Gandolfo, G.M.; Galli, M.; et al. Prevalence and clinical significance of elevated antiphospholipid antibodies in patients with idiopathic thrombocytopenic purpura. Blood 1994, 84, 4203–4208. [Google Scholar] [CrossRef] [PubMed]
- Galli, M.; Daldossi, M.; Barbui, T. Anti-glycoprotein Ib/IX and IIb/IIIa antibodies in patients with antiphospholipid antibodies. Thromb. Haemost. 1994, 71, 571–575. [Google Scholar]
- Campbell, A.L.; Pierangeli, S.S.; Wellhausen, S.; Harris, E.N. Comparison of the effects of anticardiolipin antibodies from patients with the antiphospholipid syndrome and with syphilis on platelet activation and aggregation. Thromb. Haemost. 1995, 73, 529–534. [Google Scholar] [CrossRef] [PubMed]
- Martinuzzo, M.E.; Maclouf, J.; Carreras, L.O.; Lévy-Toledano, S. Antiphospholipid antibodies enhance thrombin-induced platelet activation and thromboxane formation. Thromb. Haemost. 1993, 70, 667–671. [Google Scholar] [CrossRef]
- Macchi, L.; Rispal, P.; Clofent-Sanchez, G.; Pellegrin, J.L.; Nurden, P.; Leng, B.; Nurden, A.T. Anti-platelet antibodies in patients with systemic lupus erythematosus and the primary antiphospholipid antibody syndrome: Their relationship with the observed thrombocytopenia. Br. J. Haematol. 1997, 98, 336–341. [Google Scholar] [CrossRef]
- Panzer, S.; Gschwandtner, M.E.; Hütter, D.; Spitzauer, S.; Pabinger, I. Specificities of platelet autoantibodies in patients with lupus anticoagulants in primary antiphospholipid syndrome. Ann. Hematol. 1997, 74, 239–242. [Google Scholar] [CrossRef]
- Italian Registry of Antiphospholipid Antibodies. Thrombosis and thrombocytopenia in antiphospholipid syndrome (idio-pathic and sec- ondary to SLE): First report from Italian Registry. Haematologica 1993, 78, 313–318. [Google Scholar]
- Cikrikcioglu, M.A.; Hursitoglu, M.; Erkal, H.; Inan, B.; Ozturk, T.; Cakirca, M.; Canpolat, F.; Erkal, S.; Bahtiyar, I.; Cordan, I.; et al. Splenomegaly in Primary Antiphospholipid Syndrome without Accompanying Portal Hypertension or Comorbidity. Pathophysiol. Haemost. Thromb. 2009, 37, 104–109. [Google Scholar] [CrossRef]
- Ng, C.J.; McCrae, K.R.; Ashworth, K.; Sosa, L.J.; Betapudi, V.; Manco-Johnson, M.J.; Liu, A.; Dong, J.-F.; Chung, D.; White Adams, T.C.; et al. Effects of anti-β2GPI antibodies on VWF release from human umbilical vein endothelial cells and ADAMTS13 activity. Res. Pract. Thromb. Haemost. 2018, 2, 380–389. [Google Scholar] [CrossRef]
- Hisada, R.; Kato, M.; Sugawara, E.; Fujieda, Y.; Oku, K.; Bohgaki, T.; Amengual, O.; Yasuda, S.; Atsumi, T. Thrombotic risk stratification by platelet count in patients with antiphospholipid anti-bodies: A longitudinal study. J. Thromb. Haemost. 2017, 15, 1782–1787. [Google Scholar] [CrossRef]
- George, J.N.; Woolf, S.H.; Raskob, G.E.; Wasser, J.S.; Aledort, L.M.; Ballem, P.J.; Blanchette, V.S.; Bussel, J.B.; Cines, D.B.; Kelton, J.G.; et al. Idiopathic thrombocytopenic purpura: A practice guideline developed by explicit methods for the American Society of Hematology. Blood 1996, 88, 3–40. [Google Scholar] [CrossRef]
- Diz-Küçükkaya, R.; Hacihanefioğlu, A.; Yenerel, M.; Turgut, M.; Keskin, H.; Nalçaci, M.; Inanç, M. Antiphospholipid antibodies and antiphospholipid syndrome in pa-tients presenting with immune thrombocytopenic purpura: A prospective cohort study. Blood 2001, 98, 1760–1764. [Google Scholar] [CrossRef]
- Machin, N.; Ragni, M.V.; Comer, D.M.; Yabes, J.G. Prevalence and correlates of thrombosis in adults with immune thrombocyto-penia: An NIS study. Thromb. Res. 2018, 172, 80–85. [Google Scholar] [CrossRef]
- Funauchi, M.; Hamada, K.; Enomoto, H.; Ikoma, S.; Ohno, M.; Kinoshita, K.; Horiuchi, A. Characteristics of the clinical findings in patients with idiopathic thrombocyto-penic purpura who are positive for antiphospholipid antibodies. Intern. Med. 1997, 36, 882–885. [Google Scholar] [CrossRef] [PubMed]
- Pontara, E.; Banzato, A.; Bison, E.; Cattini, M.G.; Baroni, G.; Denas, G.; Calligaro, A.; Marson, P.; Tison, T.; Ruffatti, A.; et al. Thrombocytopenia in high-risk patients with antiphospholipid syndrome. J. Thromb. Haemost. 2018, 16, 529–532. [Google Scholar] [CrossRef] [PubMed]
- Frison, L.; Lombardi, A.; Caputo, I.; Semenzato, G.; Fabris, F. Vianello FRelevance of antiphospholipid antibody profile in the clinical outcome of ITP: A sin-gle-centre study. Hematology 2019, 24, 134–138. [Google Scholar] [CrossRef]
- Park, J.-M.; Eah, K. Recurrent stroke due to antiphospholipid syndrome remitted by immunotherapy, not by anticoagulation therapy: A case report and literature review. Ann. Indian Acad. Neurol. 2019, 22, 250–252. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Tanaka, Y.; Satoh, T.; Kawai, M.; Hirakata, M.; Kaburaki, J.; Kawakami, Y.; Ikeda, Y.; Kuwana, M. Autoantibody to CD40 ligand in systemic lupus erythematosus: Association with thrombocytopenia but not thromboembolism. Rheumatology 2005, 45, 150–156. [Google Scholar] [CrossRef]
- Galanopoulos, N.; Christoforidou, A.; Bezirgiannidou, Z. Lupus thrombocytopenia: Pathogenesis and therapeutic implications. Mediterr. J. Rheumatol. 2017, 28, 20–26. [Google Scholar] [CrossRef]
- Unlu, O.; Zuily, S.; Erkan, D. The clinical significance of antiphospholipid antibodies in systemic lupus erythematosus. Eur. J. Rheumatol. 2016, 3, 75–84. [Google Scholar] [CrossRef]
- Bernardoff, I.; Picq, A.; Loiseau, P.; Foret, T.; Dufrost, V.; Moulinet, T.; Unlu, O.; Erkan, D.; Wahl, D.; Zuily, S. Antiphospholipid antibodies and the risk of autoimmune hemolytic anemia in patients with systemic lupus erythematosus: A systematic review and meta-analysis. Autoimmun. Rev. 2021, 102913. [Google Scholar] [CrossRef]
- Love, P.E. Antiphospholipid Antibodies: Anticardiolipin and the Lupus Anticoagulant in Systemic Lupus Erythematosus (SLE) and in Non-SLE Disorders. Ann. Intern. Med. 1990, 112, 682–698. [Google Scholar] [CrossRef] [PubMed]
- Gadó, K.; Domján, G. Antiphospholipid Syndrome and Thrombocytopenia. Thrombocytopenia 2018, 53. [Google Scholar] [CrossRef]
- Artim-Esen, B.; Diz-Küçükkaya, R.; Inanç, M. The Significance and Management of Thrombocytopenia in Antiphospholipid Syndrome. Curr. Rheumatol. Rep. 2015, 17, 1–10. [Google Scholar] [CrossRef]
- Zuo, Y.; Barbhaiya, M.; Erkan, D. Primary Thrombosis Prophylaxis in Persistently Antiphospholipid Antibody-Positive Individuals: Where Do We Stand in 2018? Curr. Rheumatol. Rep. 2018, 20, 66. [Google Scholar] [CrossRef] [PubMed]
- Erkan, D.; Sciascia, S.; Bertolaccini, M.L.; Cohen, H. Antiphospholipid Syndrome Alliance for Clinical Trials and International Networking (APS ACTION): 10-Year Update. Curr. Rheumatol. Rep. 2021, 23, 1–9. [Google Scholar] [CrossRef]
- Radin, M.; Sciascia, S.; Erkan, D.; Pengo, V.; Tektonidou, M.G.; Ugarte, A.; Meroni, P.; Ji, L.; Belmont, H.M.; Cohen, H.; et al. The adjusted global antiphospholipid syndrome score (aGAPSS) and the risk of recurrent thrombosis: Results from the APS ACTION cohort. Semin. Arthritis Rheum. 2019, 49, 464–468. [Google Scholar] [CrossRef]
- Garcia, D.; Erkan, D. Diagnosis and Management of the Antiphospholipid Syndrome. N. Engl. J. Med. 2018, 378, 2010–2021. [Google Scholar] [CrossRef]
- DOACs not recommended for people with antiphospholipid syndrome. Drug Ther. Bull. 2019, 57, 182. [CrossRef]
- Consolini, R.; Costagliola, G.; Spatafora, D. The Centenary of Immune Thrombocytopenia—Part 2: Revising Diagnostic and Therapeutic Approach. Front. Pediatr. 2017, 5. [Google Scholar] [CrossRef]
- Salama, A. Current treatment options for primary immune thrombocytopenia. Expert Rev. Hematol. 2011, 4, 107–118. [Google Scholar] [CrossRef]
- Neunert, C.; Terrell, D.R.; Arnold, D.M.; Buchanan, G.; Cines, D.B.; Cooper, N.; Cuker, A.; Despotovic, J.M.; George, J.N.; Grace, R.F.; et al. American Society of Hematology 2019 guidelines for immune thrombocytopenia. Blood Adv. 2019, 3, 3829–3866. [Google Scholar] [CrossRef]
- Committee for Medicinal Products for Human Use (CHMP): Guideline on the Clinical Investigation of Human Normal Im-Munoglobulin for Intravenous Administration (IVIg) (EMA/CHMP/BPWP/94033/2007 rev. 2). 2010. Available online: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/10/WC500004766.pdf (accessed on 19 June 2021).
- Deshayes, S.; Khellaf, M.; Zarour, A.; Layese, R.; Fain, O.; Terriou, L.; Viallard, J.F.; Cheze, S.; Graveleau, J.; Slama, B.A.; et al. Long-term safety and efficacy of rituximab in 248 adults with immune thrombocytopenia: Results at 5 years from the French prospective registry ITP-ritux. Am. J. Hematol. 2019, 94, 1314–1324. [Google Scholar] [CrossRef]
- Chao, S.-H.; Chang, Y.-L.; Yen, J.-C.; Liao, H.-T.; Wu, T.-H.; Yu, C.-L.; Tsai, C.-Y.; Chou, Y.-C. Efficacy and safety of rituximab in autoimmune and microangiopathic hemolytic anemia: A systematic review and meta-analysis. Exp. Hematol. Oncol. 2020, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Rovin, B.H.; Furie, R.; Latinis, K.; Looney, R.J.; Fervenza, F.C.; Sanchez-Guerrero, J.; Maciuca, R.; Zhang, D.; Garg, J.P.; Brunetta, P.; et al. Efficacy and safety of rituximab in patients with active proliferative lupus nephritis: The lupus nephritis assessment with rituximab study. Arthritis Rheum. 2012, 64, 1215–1226. [Google Scholar] [CrossRef]
- Merrill, J.T.; Neuwelt, C.M.; Wallace, D.J.; Shanahan, J.C.; Latinis, K.M.; Oates, J.C.; Utset, T.O.; Gordon, C.; Isenberg, D.; Hsieh, H.-J.; et al. Efficacy and safety of rituximab in moderately-to-severely active systemic lupus erythematosus: The randomized, double-blind, phase ii/iii systemic lupus erythematosus evaluation of rituximab trial. Arthritis Rheum. 2009, 62, 222–233. [Google Scholar] [CrossRef]
- Terrier, B.; Amoura, Z.; Ravaud, P.; Hachulla, E.; Jouenne, R.; Combe, B.; Bonnet, C.; Cacoub, P.; Cantagrel, A.; de Bandt, M.; et al. Safety and efficacy of rituximab in systemic lupus erythematosus: Results from 136 patients from the French autoimmunity and rituximab registry. Arthritis Rheum. 2010, 62, 2458–2466. [Google Scholar] [CrossRef]
- Erkan, D.; Vega, J.; Ramón, G.; Kozora, E.; Lockshin, M.D. A pilot open-label phase II trial of rituximab for non-criteria manifestations of antiphospholipid syndrome. Arthritis Rheum. 2013, 65, 464–471. [Google Scholar] [CrossRef]
- Wang, J.; Dai, M.; Fu, Q. Eltrombopag for the treatment of refractory thrombocytopenia associated with connective tissue disease. Sci. Rep. 2021, 11, 1–8. [Google Scholar] [CrossRef]
- Martinez, M.J.M.; Gallego, P.; Ramos, M.J.M. Agonista del receptor de trombopoyetina como tratamiento de trombocitopenia asociada a lupus eritematoso sistémico. Reumatol. Clín. 2015, 12, 57. [Google Scholar] [CrossRef]
- Khemichian, S.; Terrault, N.A. Thrombopoietin Receptor Agonists in Patients with Chronic Liver Disease. Semin. Thromb. Hemost. 2020, 46, 682–692. [Google Scholar] [CrossRef]
- Arnal, C.; Piette, J.C.; Léone, J.; Taillan, B.; Hachulla, E.; Roudot-Thoraval, F.; Papo, T.; Schaeffer, A.; Bierling, P.; Godeau, B. Treatment of severe immune thrombocytopenia associated with systemic lupus erythema-tosus: 59 cases. J. Rheumatol. 2002, 29, 75–83. [Google Scholar]
- Khellaf, M.; Chabrol, A.; Mahevas, M.; Roudot-Thoraval, F.; Limal, N.; Languille, L.; Bierling, P.; Michel, M.; Godeau, B. Hydroxychloroquine is a good second-line treatment for adults with immune thrombocytopenia and positive antinuclear antibodies. Am. J. Hematol. 2013, 89, 194–198. [Google Scholar] [CrossRef]
- Tektonidou, M.G.; Andreoli, L.; Limper, M.; Amoura, Z.; Cervera, R.; Costedoat-Chalumeau, N.; Cuadrado, M.J.; Dörner, T.; Ferrer-Oliveras, R.; Hambly, K.; et al. EULAR recommendations for the management of antiphospholipid syndrome in adults. Ann. Rheum. Dis. 2019, 78, 1296–1304. [Google Scholar] [CrossRef] [PubMed]
- Nuri, E.; Taraborelli, M.; Andreoli, L.; Tonello, M.; Gerosa, M.; Calligaro, A.; Argolini, L.M.; Kumar, R.; Pengo, V.; Meroni, P.L.; et al. Long-term use of hydroxychloroquine reduces antiphospholipid antibodies levels in patients with primary antiphospholipid syndrome. Immunol. Res. 2016, 65, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Miranda, S.; Billoir, P.; Damian, L.; Thiebaut, P.A.; Schapman, D.; Le Besnerais, M.; Jouen, F.; Galas, L.; Levesque, H.; Le Cam-Duchez, V.; et al. Hydroxychloroquine reverses the prothrombotic state in a mouse model of antiphospholipid syndrome: Role of reduced inflammation and endothelial dysfunction. PLoS ONE 2019, 14, e0212614. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Clinical and Laboratory Criteria | |
|---|---|
| Vascular thrombosis |
|
| Pregnancy morbidity |
|
| Laboratory criteria |
|
| Pathogenesis Hypothesis | Pathway |
|---|---|
| Secondary Immune Thrombocytopenia |
|
| Decreased platelet production |
|
| Increased platelet pooling |
|
| Increased platelet consumption |
|
| Patient Group | Clinical History | I Line Therapy |
|---|---|---|
| non-triple-positive aPL carrier | No thrombotic events | No prophylaxis required |
| triple-positive aPL carrier | No thrombotic events | Primary prophylaxis with LDA may be considered |
| thrombotic APS | VTE | Secondary prophylaxis with LT-VKA (target INR 2.5, range 2–3) |
| thrombotic APS | Arterial thrombosis | Secondary prophylaxis with LT-VKA (target INR 3.5, range 3–4) or LDA |
| obstetric APS | VTE/arterial thrombosis | LDA + LMWH |
| CAPS | Without secondary CTD * | Anticoagulation + glucocorticoids + HD-IVIG + PEX |
| CAPS | Secondary CTD | Anticoagulation + glucocorticoids + HD-IVIG + PEX + cyclophosphamide |
| Thrombotic APS/CAPS | Refractory disease; Microangiopathic hemolytic anemia | Anticoagulation + glucocorticoids + HD-IVIG + PEX + rituximab |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tomasello, R.; Giordano, G.; Romano, F.; Vaccarino, F.; Siragusa, S.; Lucchesi, A.; Napolitano, M. Immune Thrombocytopenia in Antiphospholipid Syndrome: Is It Primary or Secondary? Biomedicines 2021, 9, 1170. https://doi.org/10.3390/biomedicines9091170
Tomasello R, Giordano G, Romano F, Vaccarino F, Siragusa S, Lucchesi A, Napolitano M. Immune Thrombocytopenia in Antiphospholipid Syndrome: Is It Primary or Secondary? Biomedicines. 2021; 9(9):1170. https://doi.org/10.3390/biomedicines9091170
Chicago/Turabian StyleTomasello, Riccardo, Giulio Giordano, Francesco Romano, Federica Vaccarino, Sergio Siragusa, Alessandro Lucchesi, and Mariasanta Napolitano. 2021. "Immune Thrombocytopenia in Antiphospholipid Syndrome: Is It Primary or Secondary?" Biomedicines 9, no. 9: 1170. https://doi.org/10.3390/biomedicines9091170
APA StyleTomasello, R., Giordano, G., Romano, F., Vaccarino, F., Siragusa, S., Lucchesi, A., & Napolitano, M. (2021). Immune Thrombocytopenia in Antiphospholipid Syndrome: Is It Primary or Secondary? Biomedicines, 9(9), 1170. https://doi.org/10.3390/biomedicines9091170