
Targeting ErbB3 Receptor in Cancer with Inhibitory Antibodies from Llama

 , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Expression Vectors
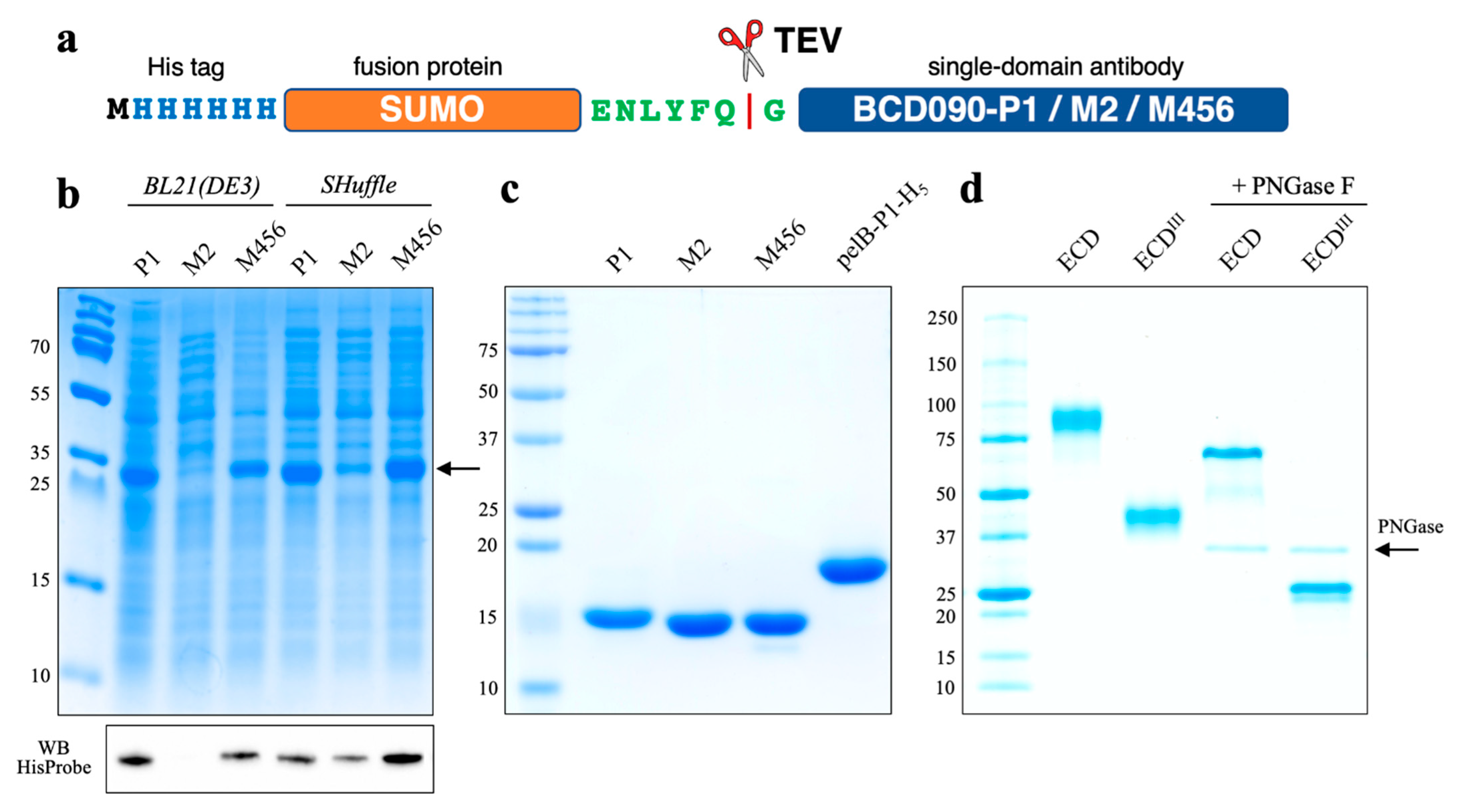
2.3. Cytoplasmic Expression of Single-Domain Antibodies
2.4. Purification and TEV Cleavage of the Single-Domain Antibodies
2.5. Expression and Purification of ErbB3 ECD and ECDIII
2.6. Gel Electrophoresis and Western Blotting
2.7. Size Exclusion Chromatography
2.8. Circular Dichroism Spectroscopy
2.9. Urea Denaturation Curves
2.10. Ellman Assay
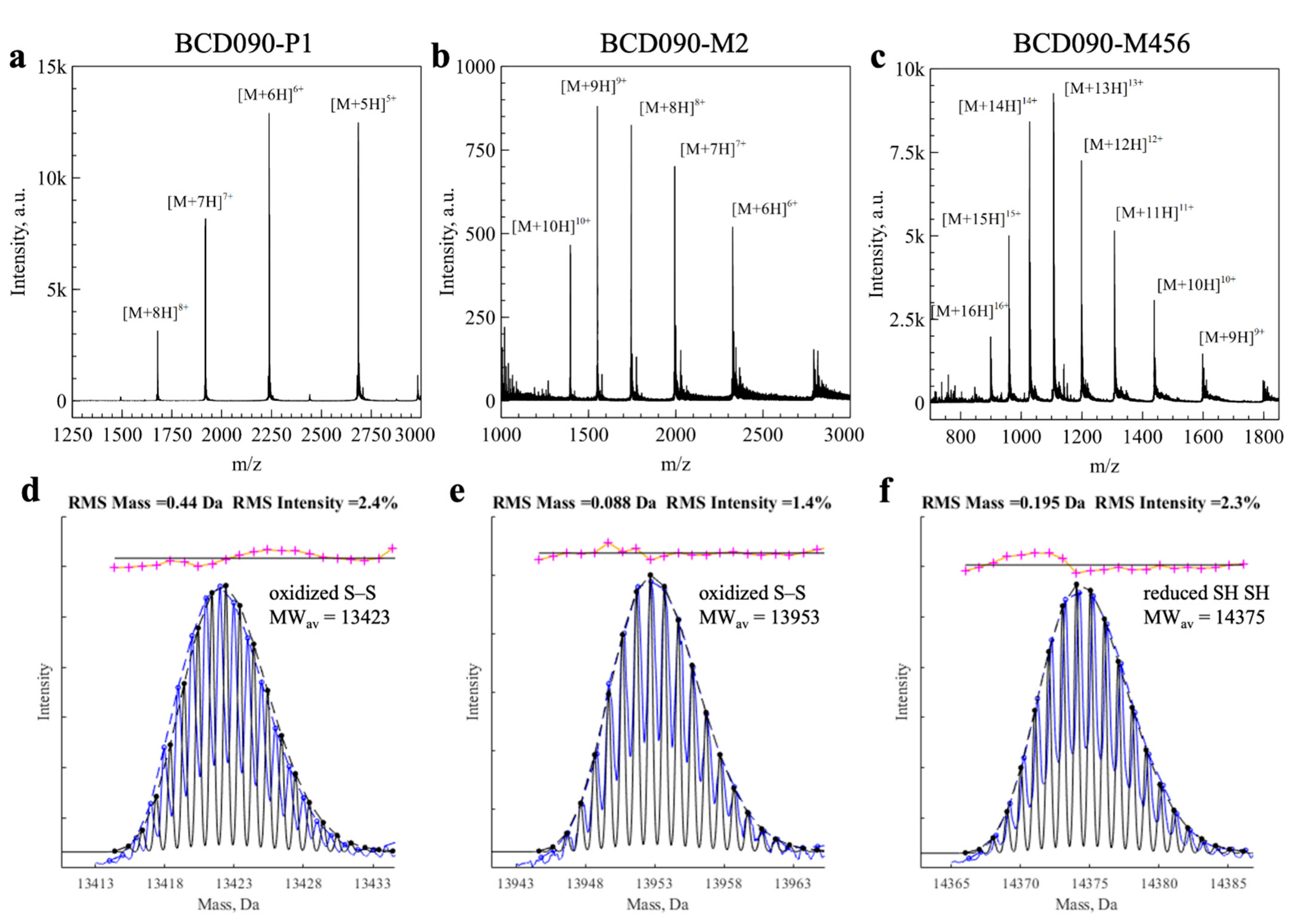
2.11. Mass Spectrometry
2.12. Surface Plasmon Resonance
2.13. Cell Proliferation Assays
2.14. Immunocytochemistry and Confocal Microscopy
3. Results
3.1. Expression and Purification of Single-Domain Antibodies
3.2. Characterization by Analytical Size Exclusion Chromatography
3.3. Secondary Structure Analysis by Circular Dichroism Spectroscopy
3.4. Analysis of Conformational Stability
3.5. Free Thiol and Disulfide Quantitation by Ellman Reagent and NTSB
3.6. Mass Spectrometry Analysis of the Intact Mass and Oxidation State
3.7. Affinity and Kinetics of Binding to ErbB3 ECD and ECDIII
3.8. Epitope Mapping and Competition with Heregulin
3.9. Antiproliferative Action on ErbB3+ Cancer Cells
3.10. Immunofluorescent Staining of ErbB3+ Cancer Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Citri, A.; Yarden, Y. EGF–ERBB signalling: Towards the systems level. Nat. Rev. Mol. Cell Biol. 2006, 7, 505–516. [Google Scholar] [CrossRef]
- Hynes, N.E.; MacDonald, G. ErbB receptors and signaling pathways in cancer. Curr. Opin. Cell Biol. 2009, 21, 177–184. [Google Scholar] [CrossRef]
- Klapper, L.N.; Glathe, S.; Vaisman, N.; Hynes, N.E.; Andrews, G.C.; Sela, M.; Yarden, Y. The ErbB-2/HER2 oncoprotein of human carcinomas may function solely as a shared coreceptor for multiple stroma-derived growth factors. Proc. Natl. Acad. Sci. USA 1999, 96, 4995–5000. [Google Scholar] [CrossRef] [Green Version]
- Guy, P.M.; Platko, J.V.; Cantley, L.; Cerione, R.A.; Carraway, K.L. Insect cell-expressed p180erbB3 possesses an impaired tyrosine kinase activity. Proc. Natl. Acad. Sci. USA 1994, 91, 8132–8136. [Google Scholar] [CrossRef] [Green Version]
- Wallasch, C.; Weiss, F.U.; Niederfellner, G.; Jallal, B.; Issing, W.; Ullrich, A. Heregulin-dependent regulation of HER2/neu oncogenic signaling by heterodimerization with HER3. EMBO J. 1995, 14, 4267–4275. [Google Scholar] [CrossRef]
- Holbro, T.; Beerli, R.R.; Maurer, F.; Koziczak, M.; Barbas, C.F.; Hynes, N.E. The ErbB2/ErbB3 heterodimer functions as an oncogenic unit: ErbB2 requires ErbB3 to drive breast tumor cell proliferation. Proc. Natl. Acad. Sci. USA 2003, 100, 8933–8938. [Google Scholar] [CrossRef] [Green Version]
- Ben-Levy, R.; Paterson, H.; Marshall, C.; Yarden, Y. A single autophosphorylation site confers oncogenicity to the Neu/ErbB-2 receptor and enables coupling to the MAP kinase pathway. EMBO J. 1994, 13, 3302–3311. [Google Scholar] [CrossRef]
- Prigent, S.; Gullick, W. Identification of c-erbB-3 binding sites for phosphatidylinositol 3′-kinase and SHC using an EGF receptor/c-erbB-3 chimera. EMBO J. 1994, 13, 2831–2841. [Google Scholar] [CrossRef] [PubMed]
- Sergina, N.V.; Rausch, M.; Wang, D.; Blair, J.; Hann, B.; Shokat, K.M.; Moasser, M.M. Escape from HER-family tyrosine kinase inhibitor therapy by the kinase-inactive HER3. Nat. Cell Biol. 2007, 445, 437–441. [Google Scholar] [CrossRef] [PubMed]
- Garrett, J.T.; Olivares, M.G.; Rinehart, C.; Granja-Ingram, N.D.; Sanchez, V.; Chakrabarty, A.; Dave, B.; Cook, R.S.; Pao, W.; McKinely, E.; et al. Transcriptional and posttranslational up-regulation of HER3 (ErbB3) compensates for inhibition of the HER2 tyrosine kinase. Proc. Natl. Acad. Sci. USA 2011, 108, 5021–5026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kol, A.; van Scheltinga, A.G.T.; Timmer-Bosscha, H.; Lamberts, L.E.; Bensch, F.; de Vries, E.; Schröder, C.P. HER3, serious partner in crime. Pharmacol. Ther. 2014, 143, 1–11. [Google Scholar] [CrossRef]
- Zhang, N.; Chang, Y.; Rios, A.; An, Z. HER3/ErbB3, an emerging cancer therapeutic target. Acta Biochim. Biophys. Sin. 2015, 48, 39–48. [Google Scholar] [CrossRef] [Green Version]
- Mishra, R.; Patel, H.; Alanazi, S.; Yuan, L.; Garrett, J.T. HER3 signaling and targeted therapy in cancer. Oncol. Rev. 2018, 12, 355. [Google Scholar] [CrossRef] [Green Version]
- Garner, A.P.; Bialucha, C.U.; Sprague, E.; Garrett, J.T.; Sheng, Q.; Li, S.; Sineshchekova, O.; Saxena, P.; Sutton, C.R.; Chen, D.; et al. An Antibody That Locks HER3 in the Inactive Conformation Inhibits Tumor Growth Driven by HER2 or Neuregulin. Cancer Res. 2013, 73, 6024–6035. [Google Scholar] [CrossRef] [Green Version]
- Mirschberger, C.; Schiller, C.B.; Schräml, M.; Dimoudis, N.; Friess, T.; Gerdes, C.A.; Reiff, U.; Lifke, V.; Hoelzlwimmer, G.; Kolm, I.; et al. RG7116, a Therapeutic Antibody That Binds the Inactive HER3 Receptor and Is Optimized for Immune Effector Activation. Cancer Res. 2013, 73, 5183–5194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Greenlee, E.B.; Amick, J.R.; Ligon, G.F.; Lillquist, J.S.; Natoli, E.J.; Hadari, Y.; Alvarado, D.; Schlessinger, J. Inhibition of ErbB3 by a monoclonal antibody that locks the extracellular domain in an inactive configuration. Proc. Natl. Acad. Sci. USA 2015, 112, 13225–13230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamers-Casterman, C.; Atarhouch, T.; Muyldermans, S.; Robinson, G.; Hammers, C.; Songa, E.B.; Bendahman, N.; Hammers, R. Naturally occurring antibodies devoid of light chains. Nature 1993, 363, 446–448. [Google Scholar] [CrossRef]
- Harmsen, M.M.; De Haard, H.J. Properties, production, and applications of camelid single-domain antibody fragments. Appl. Microbiol. Biotechnol. 2007, 77, 13–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, V.K.; Hamers, R.; Wyns, L.; Muyldermans, S. Camel heavy-chain antibodies: Diverse germline VHH and specific mechanisms enlarge the antigen-binding repertoire. EMBO J. 2000, 19, 921–930. [Google Scholar] [CrossRef]
- Rios, M.D.L.; Criscitiello, M.F.; Smider, V.V. Structural and genetic diversity in antibody repertoires from diverse species. Curr. Opin. Struct. Biol. 2015, 33, 27–41. [Google Scholar] [CrossRef]
- Steeland, S.; Vandenbroucke, R.E.; Libert, C. Nanobodies as therapeutics: Big opportunities for small antibodies. Drug Discov. Today 2016, 21, 1076–1113. [Google Scholar] [CrossRef]
- Beghein, E.; Gettemans, J. Nanobody Technology: A Versatile Toolkit for Microscopic Imaging, Protein–Protein Interaction Analysis, and Protein Function Exploration. Front. Immunol. 2017, 8, 771. [Google Scholar] [CrossRef]
- Desmyter, A.; Spinelli, S.; Roussel, A.; Cambillau, C. Camelid nanobodies: Killing two birds with one stone. Curr. Opin. Struct. Biol. 2015, 32, 1–8. [Google Scholar] [CrossRef]
- Moskaleva, O.V.; Ulitin, A.B.; Soloviev, V.V.; Zaripova, D.A.; Vladimirova, A.K.; Nemankin, T.A.; Shmakova, A.P.; Shitikova, V.O.; Artyukhova, M.V.; Diduk, S.V.; et al. Bispecific Anti-HER2/Anti-HER3 Antibodies. Patent RU2653443, 8 May 2018. [Google Scholar]
- Wei, L.; Cai, X.; Qi, Z.; Rong, L.; Cheng, B.; Fan, J. In vivo and in vitro characterization of TEV protease mutants. Protein Expr. Purif. 2012, 83, 157–163. [Google Scholar] [CrossRef]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein identification and analysis tools on the ExPASy server. In The Proteomics Protocols Handbook; Springer: Totowa, NJ, USA, 2005; pp. 571–607. [Google Scholar] [CrossRef]
- Micsonai, A.; Wien, F.; Kernya, L.; Lee, Y.-H.; Goto, Y.; Réfrégiers, M.; Kardos, J. Accurate secondary structure prediction and fold recognition for circular dichroism spectroscopy. Proc. Natl. Acad. Sci. USA 2015, 112, E3095–E3103. [Google Scholar] [CrossRef] [Green Version]
- Micsonai, A.; Wien, F.; Bulyáki, É.; Kun, J.; Moussong, É.; Lee, Y.-H.; Goto, Y.; Réfrégiers, M.; Kardos, J. BeStSel: A web server for accurate protein secondary structure prediction and fold recognition from the circular dichroism spectra. Nucleic Acids Res. 2018, 46, W315–W322. [Google Scholar] [CrossRef]
- Shaw, K.L.; Scholtz, J.M.; Pace, C.N.; Grimsley, R.G. Determining the Conformational Stability of a Protein Using Urea Denaturation Curves. In Advanced Structural Safety Studies; Springer Science and Business Media LLC: Berlin, Germany, 2008; Volume 490, pp. 41–55. [Google Scholar]
- Thannhauser, T.W.; Konishi, Y.; Scheraga, H.A. Sensitive quantitative analysis of disulfide bonds in polypeptides and proteins. Anal. Biochem. 1984, 138, 181–188. [Google Scholar] [CrossRef]
- Rhoads, T.W.; Williams, J.R.; Lopez, N.I.; Morré, J.T.; Bradford, C.S.; Beckman, J.S. Using Theoretical Protein Isotopic Distributions to Parse Small-Mass-Difference Post-Translational Modifications via Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2013, 24, 115–124. [Google Scholar] [CrossRef] [Green Version]
- Abdiche, Y.N.; Malashock, D.S.; Pinkerton, A.; Pons, J. Exploring blocking assays using Octet, ProteOn, and Biacore biosensors. Anal. Biochem. 2009, 386, 172–180. [Google Scholar] [CrossRef]
- Sikora, M.J.; Johnson, M.D.; Lee, A.V.; Oesterreich, S. Endocrine Response Phenotypes Are Altered by Charcoal-Stripped Serum Variability. Endocrinology 2016, 157, 3760–3766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wisdom, G.B. Conjugation of Antibodies to Fluorescein or Rhodamine. In Advanced Structural Safety Studies; Springer Science and Business Media LLC: Berlin, Germany, 2005; Volume 295, pp. 131–134. [Google Scholar]
- Liu, Y.; Huang, H. Expression of single-domain antibody in different systems. Appl. Microbiol. Biotechnol. 2018, 102, 539–551. [Google Scholar] [CrossRef]
- Kipriyanov, S.M.; Moldenhauer, G.; Little, M. High level production of soluble single chain antibodies in small-scale Escherichia coli cultures. J. Immunol. Methods 1997, 200, 69–77. [Google Scholar] [CrossRef]
- Pardon, E.; Laeremans, T.; Triest, S.; Rasmussen, S.; Wohlkonig, A.; Ruf, A.; Muyldermans, S.; Hol, W.G.J.; Kobilka, B.K.; Steyaert, J. A general protocol for the generation of Nanobodies for structural biology. Nat. Protoc. 2014, 9, 674–693. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.K.; Suresh, M.R.; Wuest, F.R. Improved soluble expression of a single-chain antibody fragment in E. coli for targeting CA125 in epithelial ovarian cancer. Protein Expr. Purif. 2014, 102, 27–37. [Google Scholar] [CrossRef]
- Marblestone, J.G.; Edavettal, S.C.; Lim, Y.; Lim, P.; Zuo, X.; Butt, T.R. Comparison of SUMO fusion technology with traditional gene fusion systems: Enhanced expression and solubility with SUMO. Protein Sci. 2006, 15, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Lobstein, J.; Emrich, C.A.; Jeans, C.; Faulkner, M.; Riggs, P.; Berkmen, M. SHuffle, a novel Escherichia coli protein expression strain capable of correctly folding disulfide bonded proteins in its cytoplasm. Microb. Cell Factories 2012, 11, 56. [Google Scholar] [CrossRef] [Green Version]
- Zarschler, K.; Witecy, S.; Kapplusch, F.; Foerster, C.; Stephan, H. High-yield production of functional soluble single-domain antibodies in the cytoplasm of Escherichia coli. Microb. Cell Factories 2013, 12, 97. [Google Scholar] [CrossRef] [Green Version]
- Tetin, S.Y.; Prendergast, F.G.; Venyaminov, S.Y. Accuracy of protein secondary structure determination from circular dichroism spectra based on immunoglobulin examples. Anal. Biochem. 2003, 321, 183–187. [Google Scholar] [CrossRef]
- Eliseev, I.E.; Yudenko, A.N.; Vysochinskaya, V.V.; Svirina, A.A.; Evstratyeva, A.V.; Drozhzhachih, M.S.; Krendeleva, E.A.; Vladi-mirova, A.K.; Nemankin, T.A.; Ekimova, V.M.; et al. Crystal Structures of a Llama VHH Antibody BCD090-M2 Targeting Human ErbB3 Receptor. F1000Res. 2018, 7, 57. [Google Scholar] [CrossRef]
- Eliseev, I.E.; Yudenko, A.N.; Besedina, N.; Ulitin, A.B.; Ekimova, V.M.; Evdokimov, S.R.; Putintceva, J.V.; Yakovlev, P.; Lomovskaya, M.I.; Terterov, I.N.; et al. Thermodynamic Analysis of the Conformational Stability of a Single-Domain Therapeutic Antibody. Tech. Phys. Lett. 2017, 43, 1088–1091. [Google Scholar] [CrossRef]
- Woodward, J.; Tate, J.; Herrmann, P.; Evans, B.R. Comparison of Ellman’s reagent with N-(1-pyrenyl)maleimide for the determination of free sulfhydryl groups in reduced cellobiohydrolase I from Trichoderma reesei. J. Biochem. Biophys. Methods 1993, 26, 121–129. [Google Scholar] [CrossRef]
- Riener, C.K.; Kada, G.; Gruber, H.J. Quick measurement of protein sulfhydryls with Ellman’s reagent and with 4,4′-dithiodipyridine. Anal. Bioanal. Chem. 2002, 373, 266–276. [Google Scholar] [CrossRef] [PubMed]
- Doll, S.; Burlingame, A.L. Mass Spectrometry-Based Detection and Assignment of Protein Posttranslational Modifications. ACS Chem. Biol. 2015, 10, 63–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berthois, Y.; Katzenellenbogen, J.A. Phenol red in tissue culture media is a weak estrogen: Implications concerning the study of estrogen-responsive cells in culture. Proc. Natl. Acad. Sci. USA 1986, 83, 2496–2500. [Google Scholar] [CrossRef] [Green Version]
- Ferraro, D.A.; Gaborit, N.; Maron, R.; Cohen-Dvashi, H.; Porat, Z.; Pareja, F.; Lavi, S.; Lindzen, M.; Ben-Chetrit, N.; Sela, M.; et al. Inhibition of triple-negative breast cancer models by combinations of antibodies to EGFR. Proc. Natl. Acad. Sci. USA 2013, 110, 1815–1820. [Google Scholar] [CrossRef] [Green Version]
- Ben-Kasus, T.; Schechter, B.; Lavi, S.; Yarden, Y.; Sela, M. Persistent elimination of ErbB-2/HER2-overexpressing tumors using combinations of monoclonal antibodies: Relevance of receptor endocytosis. Proc. Natl. Acad. Sci. USA 2009, 106, 3294–3299. [Google Scholar] [CrossRef] [Green Version]
- Gaborit, N.; Abdul-Hai, A.; Mancini, M.; Lindzen, M.; Lavi, S.; Leitner, O.; Mounier, L.; Chentouf, M.; Dunoyer, S.; Ghosh, M.; et al. Examination of HER3 targeting in cancer using monoclonal antibodies. Proc. Natl. Acad. Sci. USA 2015, 112, 839–844. [Google Scholar] [CrossRef] [Green Version]
- Göstring, L.; Malm, M.; Höidén-Guthenberg, I.; Frejd, F.Y.; Ståhl, S.; Lofblom, J.; Gedda, L. Cellular Effects of HER3-Specific Affibody Molecules. PLoS ONE 2012, 7, e40023. [Google Scholar] [CrossRef] [Green Version]
- Kronqvist, N.; Malm, M.; Göstring, L.; Gunneriusson, E.; Nilsson, M.; Guthenberg, I.H.; Gedda, L.; Frejd, F.Y.; Ståhl, S.; Löfblom, J. Combining phage and staphylococcal surface display for generation of ErbB3-specific Affibody molecules. Protein Eng. Des. Sel. 2011, 24, 385–396. [Google Scholar] [CrossRef] [Green Version]
- Capone, E.; Giansanti, F.; Ponziani, S.; Lamolinara, A.; Iezzi, M.; Cimini, A.; Angelucci, F.; La Sorda, R.; De Laurenzi, V.; Natali, P.G.; et al. EV20-Sap, a novel anti-HER-3 antibody-drug conjugate, displays promising antitumor activity in melanoma. Oncotarget 2017, 8, 95412–95424. [Google Scholar] [CrossRef] [Green Version]
- Van Hoecke, L.; Roose, K. How mRNA therapeutics are entering the monoclonal antibody field. J. Transl. Med. 2019, 17, 54. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | ka 10−5, M−1s−1 | kd ⋅103, s−1 | KD, nM from Kinetics | KD, nM from Equilibrium |
|---|---|---|---|---|
| BCD090-P1 | 19 ± 4 | 3.0 ± 1.0 | 1.6 ± 0.2 | 1.61 ± 0.06 |
| BCD090-M2 | 2.2 ± 0.4 | 3.6 ± 0.1 | 16.5 ± 2.0 | 15 ± 1 |
| BCD090-M456 | 0.0072 ± 0.0002 | 2.2 ± 0.2 | 3100 ± 200 | 8600 ± 800 |
| pelB-BCD090-P1-His5 | 8.7 ± 2.0 | 4.4 ± 0.2 | 5.1 ± 0.9 | 6.5 ± 0.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eliseev, I.E.; Ukrainskaya, V.M.; Yudenko, A.N.; Mikushina, A.D.; Shmakov, S.V.; Afremova, A.I.; Ekimova, V.M.; Vronskaia, A.A.; Knyazev, N.A.; Shamova, O.V. Targeting ErbB3 Receptor in Cancer with Inhibitory Antibodies from Llama. Biomedicines 2021, 9, 1106. https://doi.org/10.3390/biomedicines9091106
Eliseev IE, Ukrainskaya VM, Yudenko AN, Mikushina AD, Shmakov SV, Afremova AI, Ekimova VM, Vronskaia AA, Knyazev NA, Shamova OV. Targeting ErbB3 Receptor in Cancer with Inhibitory Antibodies from Llama. Biomedicines. 2021; 9(9):1106. https://doi.org/10.3390/biomedicines9091106
Chicago/Turabian StyleEliseev, Igor E., Valeria M. Ukrainskaya, Anna N. Yudenko, Anna D. Mikushina, Stanislav V. Shmakov, Anastasiya I. Afremova, Viktoria M. Ekimova, Anna A. Vronskaia, Nickolay A. Knyazev, and Olga V. Shamova. 2021. "Targeting ErbB3 Receptor in Cancer with Inhibitory Antibodies from Llama" Biomedicines 9, no. 9: 1106. https://doi.org/10.3390/biomedicines9091106
APA StyleEliseev, I. E., Ukrainskaya, V. M., Yudenko, A. N., Mikushina, A. D., Shmakov, S. V., Afremova, A. I., Ekimova, V. M., Vronskaia, A. A., Knyazev, N. A., & Shamova, O. V. (2021). Targeting ErbB3 Receptor in Cancer with Inhibitory Antibodies from Llama. Biomedicines, 9(9), 1106. https://doi.org/10.3390/biomedicines9091106