
Wilson’s Disease: Facing the Challenge of Diagnosing a Rare Disease

 and
and 
Abstract
:1. Introduction
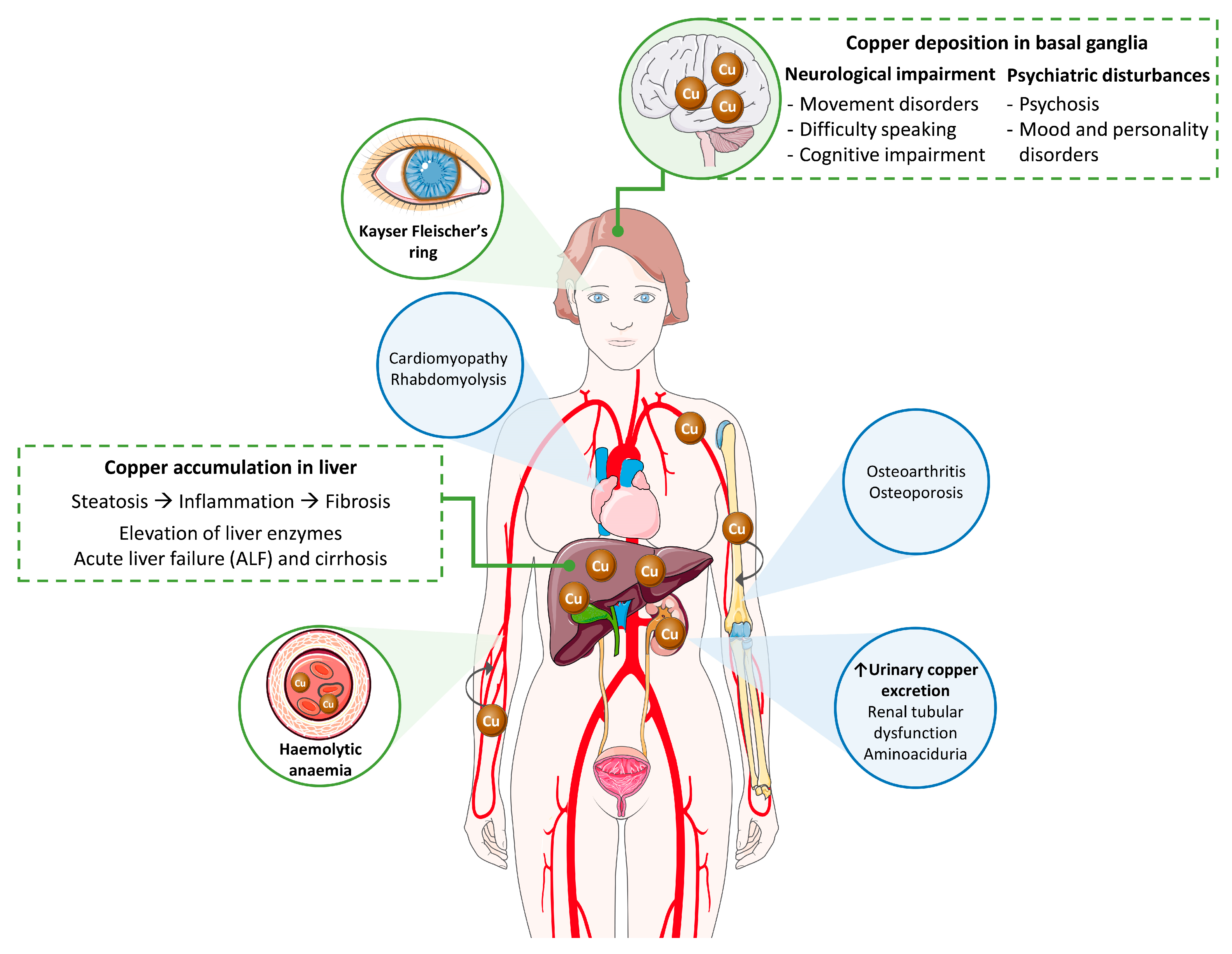
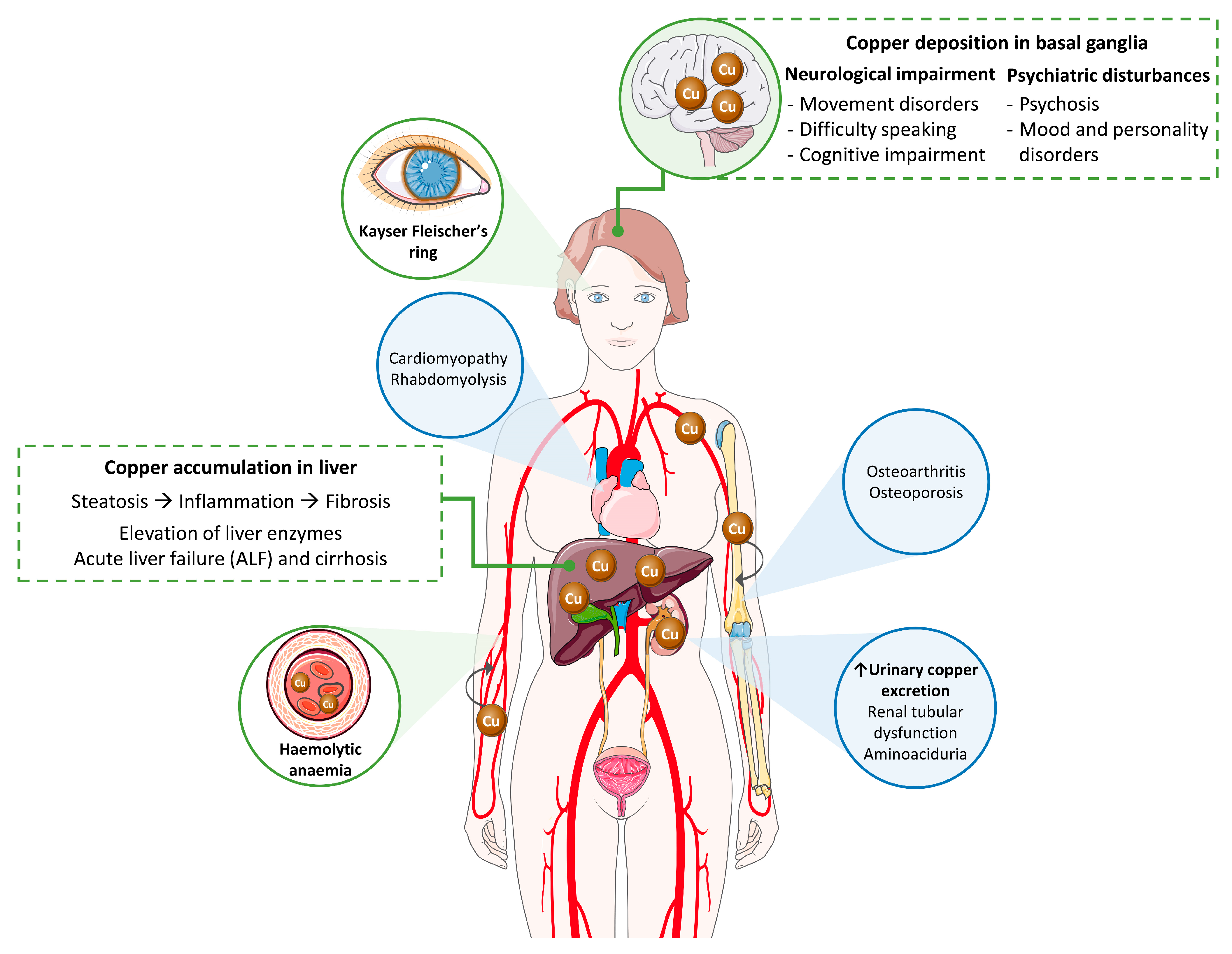
2. Clinical Picture of Wilson’s Disease
2.1. Clinical Signs
2.1.1. Liver Disease
2.1.2. Neurological and Psychiatric Disease
2.1.3. Additional Clinical Manifestations
2.2. Diagnosis: Just a Matter of the Leipzig Scale?
2.2.1. Ceruloplasmin
2.2.2. Serum Copper
2.2.3. Urinary Copper
2.2.4. Additional Blood Tests
2.2.5. Liver Biopsy
2.2.6. Imaging Evaluations: Liver and Brain Assessment
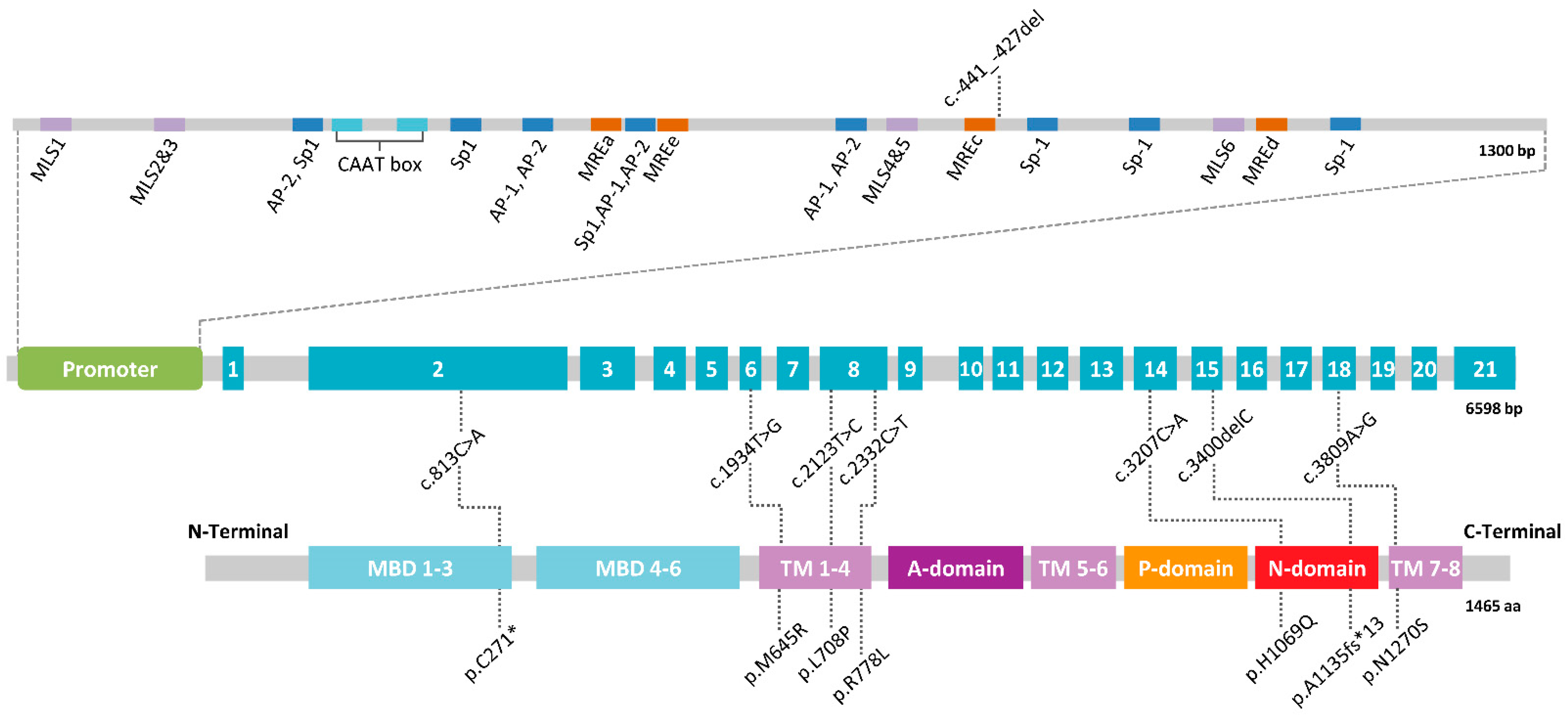
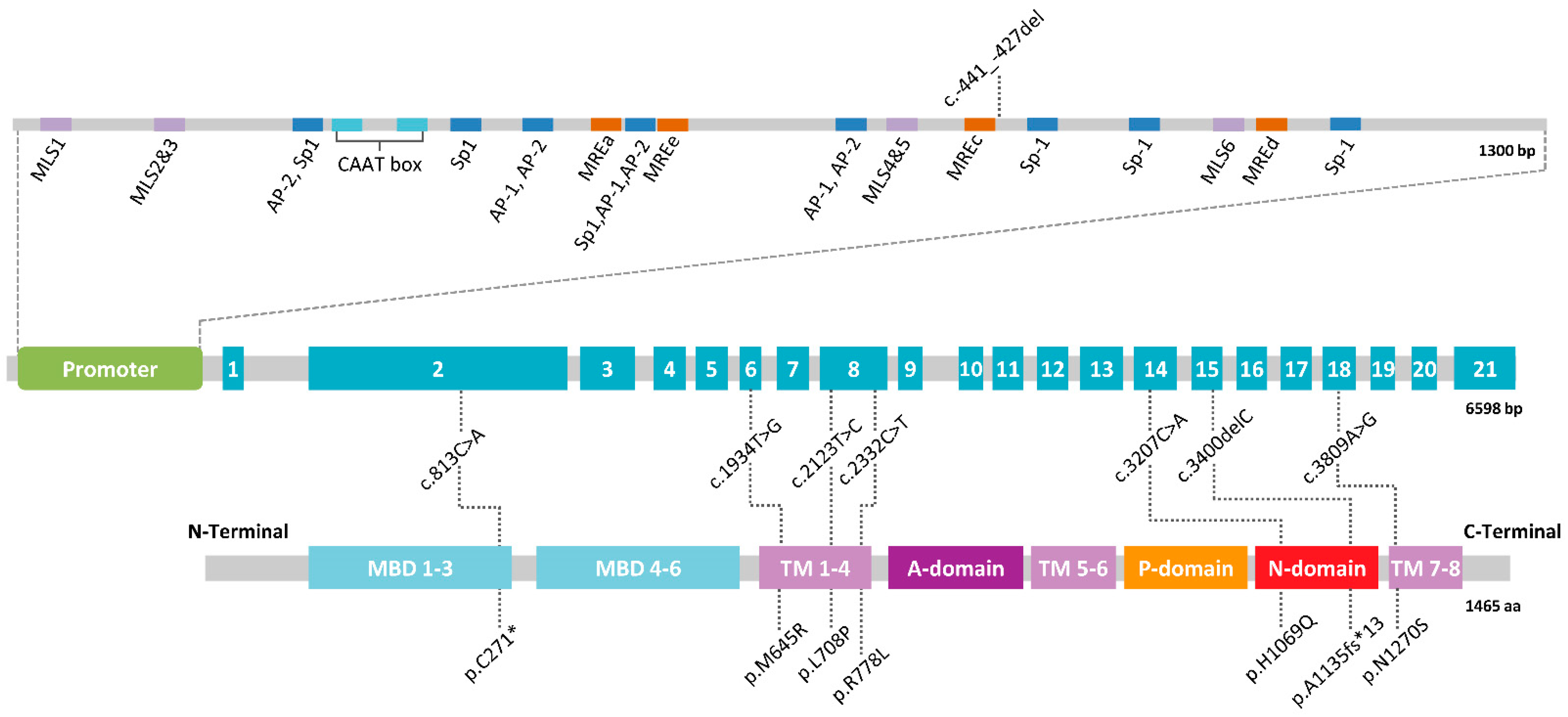
3. A Mendelian Disease Caused by Mutations in ATP7B
4. Wilson’s Disease Is Not That Rare
5. Genetic Modifiers
6. Wilson-Like: Genetic Diseases That Mimic the Wilson Phenotype
6.1. Congenital Disorders of Glycosylation
6.2. Progressive Familial Intrahepatic Cholestasis
6.3. Aceruloplasminaemia
6.4. Menkes Disease
6.5. MEDNIK Syndrome
6.6. Alagille Syndrome
6.7. Idiopathic Copper Toxicosis
7. The Need for Biomarkers for a Better Diagnosis
7.1. Biomarkers in WD and Related Clinical Phenotypes
7.2. Potential of Circulating Micrornas as Biomarkers in WD
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wilson, S.A.K. Progressive lenticular degeneration: A familial nervous disease asociated with cirrhosis of the liver. Brain 1912, 34, 295–507. [Google Scholar] [CrossRef]
- Rumpel, A. Über das Wesen und die Bedeutung der Leberveränderungen und der Pigmentierungen bei den damit verbundenen Fällen von Pseudosklerose, zugleich ein Beitrag zur Lehre von der Pseudosklerose (Westphal-Strümpell). Dtsch. Z. Nervenheilkd. 1913, 49, 54–73. [Google Scholar] [CrossRef]
- Cumings, J.N. The copper and iron content of brain and liver in the normal and in hepato-lenticular degeneration. Brain 1948, 71, 410–415. [Google Scholar] [CrossRef] [PubMed]
- Lalioti, V.; Tsubota, A.; Sandoval, I.V. Disorders in Hepatic Copper Secretion: Wilson’s Disease and Pleomorphic Syndromes. Semin. Liver Dis. 2017, 37, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Czlonkowska, A.; Litwin, T.; Dusek, P.; Ferenci, P.; Lutsenko, S.; Medici, V.; Rybakowski, J.K.; Weiss, K.H.; Schilsky, M.L. Wilson disease. Nat. Rev. Dis. Primers 2018, 4, 21. [Google Scholar] [CrossRef]
- Pak, K.; Ordway, S.; Sadowski, B.; Canevari, M.; Torres, D. Wilson’s Disease and Iron Overload: Pathophysiology and Therapeutic Implications. Clin. Liver Dis. 2021, 17, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Litwin, T.; Gromadzka, G.; Czlonkowska, A. Gender differences in Wilson’s disease. J. Neurol. Sci. 2012, 312, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Litwin, T.; Gromadzka, G.; Czlonkowska, A.; Golebiowski, M.; Poniatowska, R. The effect of gender on brain MRI pathology in Wilson’s disease. Metab. Brain Dis. 2013, 28, 69–75. [Google Scholar] [CrossRef] [Green Version]
- Gromadzka, G.; Wierzbicka, D.; Litwin, T.; Przybylkowski, A. Difference in iron metabolism may partly explain sex-related variability in the manifestation of Wilson’s disease. J. Trace Elem. Med. Biol. 2020, 62, 126637. [Google Scholar] [CrossRef]
- Patil, M.; Sheth, K.A.; Krishnamurthy, A.C.; Devarbhavi, H. A review and current perspective on Wilson disease. J. Clin. Exp. Hepatol. 2013, 3, 321–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shribman, S.; Warner, T.T.; Dooley, J.S. Clinical presentations of Wilson disease. Ann. Transl. Med. 2019, 7 (Suppl. 2), S60. [Google Scholar] [CrossRef] [PubMed]
- Dusek, P.; Smolinski, L.; Redzia-Ogrodnik, B.; Golebiowski, M.; Skowronska, M.; Poujois, A.; Laurencin, C.; Jastrzebska-Kurkowska, I.; Litwin, T.; Czlonkowska, A. Semiquantitative Scale for Assessing Brain MRI Abnormalities in Wilson Disease: A Validation Study. Mov. Disord. 2020, 35, 994–1001. [Google Scholar] [CrossRef]
- Rosencrantz, R.; Schilsky, M. Wilson disease: Pathogenesis and clinical considerations in diagnosis and treatment. Semin. Liver Dis. 2011, 31, 245–259. [Google Scholar] [CrossRef] [PubMed]
- Leinweber, B.; Moller, J.C.; Scherag, A.; Reuner, U.; Gunther, P.; Lang, C.J.; Schmidt, H.H.; Schrader, C.; Bandmann, O.; Czlonkowska, A.; et al. Evaluation of the Unified Wilson’s Disease Rating Scale (UWDRS) in German patients with treated Wilson’s disease. Mov. Disord. 2008, 23, 54–62. [Google Scholar] [CrossRef]
- Aggarwal, A.; Aggarwal, N.; Nagral, A.; Jankharia, G.; Bhatt, M. A novel Global Assessment Scale for Wilson’s Disease (GAS for WD). Mov. Disord. 2009, 24, 509–518. [Google Scholar] [CrossRef] [PubMed]
- Roberts, E.A.; Schilsky, M.L.; American Association for Study of Liver, D. Diagnosis and treatment of Wilson disease: An update. Hepatology 2008, 47, 2089–2111. [Google Scholar] [CrossRef]
- Broniek-Kowalik, K.; Dziezyc, K.; Litwin, T.; Czlonkowska, A.; Szaflik, J.P. Anterior segment optical coherence tomography (AS-OCT) as a new method of detecting copper deposits forming the Kayser-Fleischer ring in patients with Wilson disease. Acta Ophthalmol. 2019, 97, e757–e760. [Google Scholar] [CrossRef]
- Langwinska-Wosko, E.; Litwin, T.; Dziezyc, K.; Czlonkowska, A. The sunflower cataract in Wilson’s disease: Pathognomonic sign or rare finding? Acta Neurol. Belg. 2016, 116, 325–328. [Google Scholar] [CrossRef] [Green Version]
- Jang, H.J.; Kim, J.M.; Choi, C.Y. Elemental analysis of sunflower cataract in Wilson’s disease: A study using scanning transmission electron microscopy and energy dispersive spectroscopy. Exp. Eye Res. 2014, 121, 58–65. [Google Scholar] [CrossRef]
- Litwin, T.; Langwinska-Wosko, E.; Dziezyc, K.; Czlonkowska, A. Sunflower cataract: Do not forget Wilson’s disease. Pract. Neurol. 2015, 15, 385–386. [Google Scholar] [CrossRef] [PubMed]
- Langwinska-Wosko, E.; Litwin, T.; Dziezyc, K.; Karlinski, M.; Czlonkowska, A. Optical coherence tomography as a marker of neurodegeneration in patients with Wilson’s disease. Acta Neurol. Belg. 2017, 117, 867–871. [Google Scholar] [CrossRef] [PubMed]
- Svetel, M.; Bozic, M.; Vitkovic, J.; Jovanovic, C.; Dragasevic, N.; Pekmezovic, T.; Svetel, M.; Tomic, A.; Kresojevic, N.; Kostic, V. Optical coherence tomography in patients with Wilson’s disease. Acta Neurol. Scand. 2021, 144, 149–154. [Google Scholar] [CrossRef] [PubMed]
- Grandis, D.J.; Nah, G.; Whitman, I.R.; Vittinghoff, E.; Dewland, T.A.; Olgin, J.E.; Marcus, G.M. Wilson’s Disease and Cardiac Myopathy. Am. J. Cardiol. 2017, 120, 2056–2060. [Google Scholar] [CrossRef] [PubMed]
- Kuan, P. Fatal cardiac complications of Wilson’s disease. Am. Heart J. 1982, 104, 314–316. [Google Scholar] [CrossRef]
- Hlubocka, Z.; Marecek, Z.; Linhart, A.; Kejkova, E.; Pospisilova, L.; Martasek, P.; Aschermann, M. Cardiac involvement in Wilson disease. J. Inherit. Metab. Dis. 2002, 25, 269–277. [Google Scholar] [CrossRef]
- Bobbio, E.; Forsgard, N.; Oldfors, A.; Szamlewski, P.; Bollano, E.; Andersson, B.; Lingbrant, M.; Bergh, N.; Karason, K.; Polte, C.L. Cardiac arrest in Wilson’s disease after curative liver transplantation: A life-threatening complication of myocardial copper excess? ESC Heart Fail. 2019, 6, 228–231. [Google Scholar] [CrossRef]
- Chevalier, K.; Benyounes, N.; Obadia, M.A.; Van Der Vynckt, C.; Morvan, E.; Tibi, T.; Poujois, A. Cardiac involvement in Wilson disease: Review of the literature and description of three cases of sudden death. J. Inherit. Metab. Dis. 2021. [Google Scholar] [CrossRef]
- Zhang, K.; Reuner, U.; Hempel, C.; Speiser, U.; Ibrahim, K.; Heinzel, F.R.; Pieske, B.; Christoph, M.; Heidrich, F.M.; Quick, S. Evaluation of Myocardial Strain Using Cardiac Magnetic Resonance in Patients with Wilson’s Disease. J. Clin. Med. 2021, 10, 335. [Google Scholar] [CrossRef]
- Zhuang, X.H.; Mo, Y.; Jiang, X.Y.; Chen, S.M. Analysis of renal impairment in children with Wilson’s disease. World J. Pediatr. 2008, 4, 102–105. [Google Scholar] [CrossRef]
- Dziezyc-Jaworska, K.; Litwin, T.; Czlonkowska, A. Clinical manifestations of Wilson disease in organs other than the liver and brain. Ann. Transl. Med. 2019, 7 (Suppl. 2), S62. [Google Scholar] [CrossRef]
- European Association For The Study Of The Liver. EASL Clinical Practice Guidelines: Wilson’s disease. J. Hepatol. 2012, 56, 671–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mak, C.M. Biochemical diagnosis of Wilson disease. In Clinical and Translational Perspectives on Wilson Disease; Kerkar, N., Roberts, E.A., Eds.; Elsevier Academic Press: London, UK, 2019; pp. 237–248. [Google Scholar]
- Poujois, A.; Poupon, J.; Woimant, F. Direct determination of non-ceruloplasmin-bound copper in plasma. In Clinical and Translational Perspectives on Wilson Disease; Kerkar, N., Roberts, E.A., Eds.; Elsevier Academic Press: London, UK, 2019; pp. 249–255. [Google Scholar]
- Merle, U.; Eisenbach, C.; Weiss, K.H.; Tuma, S.; Stremmel, W. Serum ceruloplasmin oxidase activity is a sensitive and highly specific diagnostic marker for Wilson’s disease. J. Hepatol. 2009, 51, 925–930. [Google Scholar] [CrossRef] [PubMed]
- Ryan, A.; Nevitt, S.J.; Tuohy, O.; Cook, P. Biomarkers for diagnosis of Wilson’s disease. Cochrane Database Syst. Rev. 2019, 11. [Google Scholar] [CrossRef]
- Mohr, I.; Weiss, K.H. Biochemical Markers for the Diagnosis and Monitoring of Wilson Disease. Clin. Biochem. Rev. 2019, 40, 59–77. [Google Scholar] [PubMed]
- El Balkhi, S.; Trocello, J.M.; Poupon, J.; Chappuis, P.; Massicot, F.; Girardot-Tinant, N.; Woimant, F. Relative exchangeable copper: A new highly sensitive and highly specific biomarker for Wilson’s disease diagnosis. Clin. Chim. Acta 2011, 412, 2254–2260. [Google Scholar] [CrossRef] [PubMed]
- Trocello, J.M.; El Balkhi, S.; Woimant, F.; Girardot-Tinant, N.; Chappuis, P.; Lloyd, C.; Poupon, J. Relative exchangeable copper: A promising tool for family screening in Wilson disease. Mov. Disord. 2014, 29, 558–562. [Google Scholar] [CrossRef] [PubMed]
- Poujois, A.; Trocello, J.M.; Djebrani-Oussedik, N.; Poupon, J.; Collet, C.; Girardot-Tinant, N.; Sobesky, R.; Habes, D.; Debray, D.; Vanlemmens, C.; et al. Exchangeable copper: A reflection of the neurological severity in Wilson’s disease. Eur. J. Neurol. 2017, 24, 154–160. [Google Scholar] [CrossRef] [Green Version]
- Guillaud, O.; Brunet, A.S.; Mallet, I.; Dumortier, J.; Pelosse, M.; Heissat, S.; Rivet, C.; Lachaux, A.; Bost, M. Relative exchangeable copper: A valuable tool for the diagnosis of Wilson disease. Liver Int. 2018, 38, 350–357. [Google Scholar] [CrossRef]
- Woimant, F.; Djebrani-Oussedik, N.; Poujois, A. New tools for Wilson’s disease diagnosis: Exchangeable copper fraction. Ann. Transl. Med. 2019, 7 (Suppl. 2), S70. [Google Scholar] [CrossRef]
- Nicastro, E.; Ranucci, G.; Vajro, P.; Vegnente, A.; Iorio, R. Re-evaluation of the diagnostic criteria for Wilson disease in children with mild liver disease. Hepatology 2010, 52, 1948–1956. [Google Scholar] [CrossRef] [Green Version]
- Pfeiffenberger, J.; Lohse, C.M.; Gotthardt, D.; Rupp, C.; Weiler, M.; Teufel, U.; Weiss, K.H.; Gauss, A. Long-term evaluation of urinary copper excretion and non-caeruloplasmin associated copper in Wilson disease patients under medical treatment. J. Inherit. Metab. Dis. 2019, 42, 371–380. [Google Scholar] [CrossRef]
- Dziezyc, K.; Litwin, T.; Chabik, G.; Czlonkowska, A. Measurement of urinary copper excretion after 48-h d-penicillamine cessation as a compliance assessment in Wilson’s disease. Funct. Neurol. 2015, 30, 264–268. [Google Scholar]
- Korman, J.D.; Volenberg, I.; Balko, J.; Webster, J.; Schiodt, F.V.; Squires, R.H., Jr.; Fontana, R.J.; Lee, W.M.; Schilsky, M.L.; Pediatric and Adult Acute Liver Failure Study Group. Screening for Wilson disease in acute liver failure: A comparison of currently available diagnostic tests. Hepatology 2008, 48, 1167–1174. [Google Scholar] [CrossRef] [PubMed]
- Weiss, K.H.; Gotthardt, D.N.; Klemm, D.; Merle, U.; Ferenci-Foerster, D.; Schaefer, M.; Ferenci, P.; Stremmel, W. Zinc monotherapy is not as effective as chelating agents in treatment of Wilson disease. Gastroenterology 2011, 140, 1189–1198.e1. [Google Scholar] [CrossRef] [PubMed]
- Czlonkowska, A.; Rodo, M.; Wierzchowska-Ciok, A.; Smolinski, L.; Litwin, T. Accuracy of the radioactive copper incorporation test in the diagnosis of Wilson disease. Liver Int. 2018, 38, 1860–1866. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.; Whiteaker, J.R.; Zhao, L.; Yoo, H.W.; Paulovich, A.G.; Hahn, S.H. Quantification of ATP7B Protein in Dried Blood Spots by Peptide Immuno-SRM as a Potential Screen for Wilson’s Disease. J. Proteome Res. 2017, 16, 862–871. [Google Scholar] [CrossRef] [PubMed]
- Collins, C.J.; Yi, F.; Dayuha, R.; Duong, P.; Horslen, S.; Camarata, M.; Coskun, A.K.; Houwen, R.H.J.; Pop, T.L.; Zoller, H.; et al. Direct Measurement of ATP7B Peptides Is Highly Effective in the Diagnosis of Wilson Disease. Gastroenterology 2021, 160, 2367–2382.e1. [Google Scholar] [CrossRef]
- Sini, M.; Sorbello, O.; Sanna, F.; Battolu, F.; Civolani, A.; Fanni, D.; Faa, G.; Demelia, L. Histologic evolution and long-term outcome of Wilson’s disease: Results of a single-center experience. Eur. J. Gastroenterol. Hepatol. 2013, 25, 111–117. [Google Scholar] [CrossRef]
- Cope-Yokoyama, S.; Finegold, M.J.; Sturniolo, G.C.; Kim, K.; Mescoli, C.; Rugge, M.; Medici, V. Wilson disease: Histopathological correlations with treatment on follow-up liver biopsies. World J. Gastroenterol. 2010, 16, 1487–1494. [Google Scholar] [CrossRef]
- Calvopina, D.A.; Coleman, M.A.; Lewindon, P.J.; Ramm, G.A. Function and Regulation of MicroRNAs and Their Potential as Biomarkers in Paediatric Liver Disease. Int. J. Mol. Sci. 2016, 17, 1795. [Google Scholar] [CrossRef] [Green Version]
- Sini, M.; Sorbello, O.; Civolani, A.; Liggi, M.; Demelia, L. Non-invasive assessment of hepatic fibrosis in a series of patients with Wilson’s Disease. Dig. Liver Dis. 2012, 44, 487–491. [Google Scholar] [CrossRef]
- Hwang, J.; Yoon, H.M.; Jung, A.Y.; Lee, J.S.; Kim, K.M.; Oh, S.H.; Cho, Y.A. Diagnostic Performance of Ultrasound Elastography and Serologic Fibrosis Indices for Evaluation of Hepatic Involvement in Wilson Disease. J. Ultrasound Med. 2020, 39, 2231–2242. [Google Scholar] [CrossRef] [PubMed]
- Paternostro, R.; Pfeiffenberger, J.; Ferenci, P.; Stattermayer, A.F.; Stauber, R.E.; Wrba, F.; Longerich, T.; Lackner, K.; Trauner, M.; Ferlitsch, A.; et al. Non-invasive diagnosis of cirrhosis and long-term disease monitoring by transient elastography in patients with Wilson disease. Liver Int. 2020, 40, 894–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castera, L.; Forns, X.; Alberti, A. Non-invasive evaluation of liver fibrosis using transient elastography. J. Hepatol. 2008, 48, 835–847. [Google Scholar] [CrossRef]
- Przybylkowski, A.; Szeligowska, J.; Januszewicz, M.; Raszeja-Wyszomirska, J.; Szczepankiewicz, B.; Nehring, P.; Gornicka, B.; Litwin, T.; Czlonkowska, A. Evaluation of liver fibrosis in patients with Wilson’s disease. Eur. J. Gastroenterol. Hepatol. 2021, 33, 535–540. [Google Scholar] [CrossRef] [PubMed]
- Vargas, O.; Faraoun, S.A.; Dautry, R.; Guerrache, Y.; Woimant, F.; Hamzi, L.; Boudiaf, M.; Poujois, A.; Soyer, P.; Dohan, A. MR imaging features of liver involvement by Wilson disease in adult patients. Radiol. Med. 2016, 121, 546–556. [Google Scholar] [CrossRef] [PubMed]
- Czlonkowska, A.; Litwin, T.; Chabik, G. Wilson disease: Neurologic features. Handb. Clin. Neurol. 2017, 142, 101–119. [Google Scholar] [PubMed]
- Dusek, P.; Skoloudik, D.; Maskova, J.; Huelnhagen, T.; Bruha, R.; Zahorakova, D.; Niendorf, T.; Ruzicka, E.; Schneider, S.A.; Wuerfel, J. Brain iron accumulation in Wilson’s disease: A longitudinal imaging case study during anticopper treatment using 7.0T MRI and transcranial sonography. J. Magn. Reson. Imaging 2018, 47, 282–285. [Google Scholar] [CrossRef]
- Sudmeyer, M.; Saleh, A.; Wojtecki, L.; Cohnen, M.; Gross, J.; Ploner, M.; Hefter, H.; Timmermann, L.; Schnitzler, A. Wilson’s disease tremor is associated with magnetic resonance imaging lesions in basal ganglia structures. Mov. Disord. 2006, 21, 2134–2139. [Google Scholar] [CrossRef]
- Dusek, P.; Bahn, E.; Litwin, T.; Jablonka-Salach, K.; Luciuk, A.; Huelnhagen, T.; Madai, V.I.; Dieringer, M.A.; Bulska, E.; Knauth, M.; et al. Brain iron accumulation in Wilson disease: A post mortem 7 Tesla MRI—Histopathological study. Neuropathol. Appl. Neurobiol. 2017, 43, 514–532. [Google Scholar] [CrossRef] [Green Version]
- Czlonkowska, A.; Tarnacka, B.; Moller, J.C.; Leinweber, B.; Bandmann, O.; Woimant, F.; Oertel, W.H. Unified Wilson’s Disease Rating Scale—A proposal for the neurological scoring of Wilson’s disease patients. Neurol. Neurochir. Pol. 2007, 41, 1–12. [Google Scholar] [CrossRef]
- Dezortova, M.; Lescinskij, A.; Dusek, P.; Herynek, V.; Acosta-Cabronero, J.; Bruha, R.; Jiru, F.; Robinson, S.D.; Hajek, M. Multiparametric Quantitative Brain MRI in Neurological and Hepatic Forms of Wilson’s Disease. J. Magn. Reson. Imaging 2020, 51, 1829–1835. [Google Scholar] [CrossRef] [PubMed]
- Dusek, P.; Lescinskij, A.; Ruzicka, F.; Acosta-Cabronero, J.; Bruha, R.; Sieger, T.; Hajek, M.; Dezortova, M. Associations of Brain Atrophy and Cerebral Iron Accumulation at MRI with Clinical Severity in Wilson Disease. Radiology 2021, 299, 662–672. [Google Scholar] [CrossRef] [PubMed]
- Bull, P.C.; Thomas, G.R.; Rommens, J.M.; Forbes, J.R.; Cox, D.W. The Wilson disease gene is a putative copper transporting P-type ATPase similar to the Menkes gene. Nat. Genet. 1993, 5, 327–337. [Google Scholar] [CrossRef]
- Tanzi, R.E.; Petrukhin, K.; Chernov, I.; Pellequer, J.L.; Wasco, W.; Ross, B.; Romano, D.M.; Parano, E.; Pavone, L.; Brzustowicz, L.M.; et al. The Wilson disease gene is a copper transporting ATPase with homology to the Menkes disease gene. Nat. Genet. 1993, 5, 344–350. [Google Scholar] [CrossRef]
- Petrukhin, K.; Fischer, S.G.; Pirastu, M.; Tanzi, R.E.; Chernov, I.; Devoto, M.; Brzustowicz, L.M.; Cayanis, E.; Vitale, E.; Russo, J.J.; et al. Mapping, cloning and genetic characterization of the region containing the Wilson disease gene. Nat. Genet. 1993, 5, 338–343. [Google Scholar] [CrossRef]
- Oh, W.J.; Kim, E.K.; Park, K.D.; Hahn, S.H.; Yoo, O.J. Cloning and characterization of the promoter region of the Wilson disease gene. Biochem. Biophys. Res. Commun. 1999, 259, 206–211. [Google Scholar] [CrossRef]
- Sanchez-Monteagudo, A.; Alvarez-Sauco, M.; Sastre, I.; Martinez-Torres, I.; Lupo, V.; Berenguer, M.; Espinos, C. Genetics of Wilson disease and Wilson-like phenotype in a clinical series from eastern Spain. Clin. Genet. 2020, 97, 758–763. [Google Scholar] [CrossRef]
- Espinos, C.; Ferenci, P. Are the new genetic tools for diagnosis of Wilson disease helpful in clinical practice? JHEP Rep. 2020, 2, 100114. [Google Scholar] [CrossRef]
- Garcia-Villarreal, L.; Daniels, S.; Shaw, S.H.; Cotton, D.; Galvin, M.; Geskes, J.; Bauer, P.; Sierra-Hernandez, A.; Buckler, A.; Tugores, A. High prevalence of the very rare Wilson disease gene mutation Leu708Pro in the Island of Gran Canaria (Canary Islands, Spain): A genetic and clinical study. Hepatology 2000, 32, 1329–1336. [Google Scholar] [CrossRef] [PubMed]
- Gojova, L.; Jansova, E.; Kulm, M.; Pouchla, S.; Kozak, L. Genotyping microarray as a novel approach for the detection of ATP7B gene mutations in patients with Wilson disease. Clin. Genet. 2008, 73, 441–452. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.H.; Lee, T.; Bang, S.; Kim, Y.E.; Cho, E.H. Carrier frequency of Wilson’s disease in the Korean population: A DNA-based approach. J. Hum. Genet. 2017, 62, 815–818. [Google Scholar] [CrossRef] [PubMed]
- Xu, A.; Lv, T.; Zhang, B.; Zhang, W.; Ou, X.; Huang, J. Development and evaluation of an unlabeled probe high-resolution melting assay for detection of ATP7B mutations in Wilson’s disease. J. Clin. Lab. Anal. 2017, 31, e22064. [Google Scholar] [CrossRef] [PubMed]
- Poon, K.S.; Teo, Z.H.; Yap, J.H.; Koay, E.S.; Tan, K. Challenges in molecular diagnosis of Wilson disease: Viewpoint from the clinical laboratory. J. Clin. Pathol. 2020, 73, 231–234. [Google Scholar] [CrossRef]
- Chen, H.I.; Jagadeesh, K.A.; Birgmeier, J.; Wenger, A.M.; Guturu, H.; Schelley, S.; Bernstein, J.A.; Bejerano, G. An MTF1 binding site disrupted by a homozygous variant in the promoter of ATP7B likely causes Wilson Disease. Eur. J. Hum. Genet. 2018, 26, 1810–1818. [Google Scholar] [CrossRef]
- Todorov, T.; Balakrishnan, P.; Savov, A.; Socha, P.; Schmidt, H.H. Intragenic Deletions in ATP7B as an Unusual Molecular Genetics Mechanism of Wilson’s Disease Pathogenesis. PLoS ONE 2016, 11, e0168372. [Google Scholar] [CrossRef] [Green Version]
- Woimant, F.; Poujois, A.; Bloch, A.; Jordi, T.; Laplanche, J.L.; Morel, H.; Collet, C. A novel deep intronic variant in ATP7B in five unrelated families affected by Wilson disease. Mol. Genet. Genomic. Med. 2020, 8, e1428. [Google Scholar] [CrossRef]
- Scheinberg, I.H.; Sternlieb, I. Wilson’s Disease (A Volume in the Major Problems in Internal Medicine Series); Elsevier: Philadelphia, PA, USA, 1984; p. 192. [Google Scholar]
- Sandahl, T.D.; Laursen, T.L.; Munk, D.E.; Vilstrup, H.; Weiss, K.H.; Ott, P. The Prevalence of Wilson disease. An Update. Hepatology 2019, 12, 333–363. [Google Scholar]
- Gialluisi, A.; Incollu, S.; Pippucci, T.; Lepori, M.B.; Zappu, A.; Loudianos, G.; Romeo, G. The homozygosity index (HI) approach reveals high allele frequency for Wilson disease in the Sardinian population. Eur. J. Hum. Genet. 2013, 21, 1308–1311. [Google Scholar] [CrossRef] [Green Version]
- Zappu, A.; Magli, O.; Lepori, M.B.; Dessi, V.; Diana, S.; Incollu, S.; Kanavakis, E.; Nicolaidou, P.; Manolaki, N.; Fretzayas, A.; et al. High incidence and allelic homogeneity of Wilson disease in 2 isolated populations: A prerequisite for efficient disease prevention programs. J. Pediatr. Gastroenterol. Nutr. 2008, 47, 334–338. [Google Scholar]
- Leung, M.; Aronowitz, P.B.; Medici, V. The Present and Future Challenges of Wilson’s Disease Diagnosis and Treatment. Clin. Liver Dis. 2021, 17, 267–270. [Google Scholar] [CrossRef]
- Gao, J.; Brackley, S.; Mann, J.P. The global prevalence of Wilson disease from next-generation sequencing data. Genet. Med. 2019, 21, 1155–1163. [Google Scholar] [CrossRef] [PubMed]
- Tai, C.S.; Wu, J.F.; Chen, H.L.; Hsu, H.Y.; Chang, M.H.; Ni, Y.H. Modality of treatment and potential outcome of Wilson disease in Taiwan: A population-based longitudinal study. J. Formos. Med. Assoc. 2018, 117, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Poujois, A.; Woimant, F. Wilson’s disease: A 2017 update. Clin. Res. Hepatol. Gastroenterol. 2018, 46, 512–520. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Marro, S.; Barrachina-Bonet, L.; Paramo-Rodriguez, L.; Alonso-Ferreira, V.; Guardiola-Vilarroig, S.; Vicente, E.; Garcia-Lopez, M.; Palomar-Rodriguez, J.; Zoni, A.C.; Zurriaga, O.; et al. Wilson’s disease in Spain: Validation of sources of information used by the Rare Diseases Registries. Gac. Sanit. 2020. [Google Scholar] [CrossRef]
- Coffey, A.J.; Durkie, M.; Hague, S.; McLay, K.; Emmerson, J.; Lo, C.; Klaffke, S.; Joyce, C.J.; Dhawan, A.; Hadzic, N.; et al. A genetic study of Wilson’s disease in the United Kingdom. Brain 2013, 136, 1476–1487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collet, C.; Laplanche, J.L.; Page, J.; Morel, H.; Woimant, F.; Poujois, A. High genetic carrier frequency of Wilson’s disease in France: Discrepancies with clinical prevalence. BMC Med. Genet. 2018, 19, 143. [Google Scholar] [CrossRef]
- Panagiotakaki, E.; Tzetis, M.; Manolaki, N.; Loudianos, G.; Papatheodorou, A.; Manesis, E.; Nousia-Arvanitakis, S.; Syriopoulou, V.; Kanavakis, E. Genotype-phenotype correlations for a wide spectrum of mutations in the Wilson disease gene (ATP7B). Am. J. Med. Genet. A 2004, 131, 168–173. [Google Scholar] [CrossRef]
- Gupta, A.; Aikath, D.; Neogi, R.; Datta, S.; Basu, K.; Maity, B.; Trivedi, R.; Ray, J.; Das, S.K.; Gangopadhyay, P.K.; et al. Molecular pathogenesis of Wilson disease: Haplotype analysis, detection of prevalent mutations and genotype-phenotype correlation in Indian patients. Hum. Genet. 2005, 118, 49–57. [Google Scholar] [CrossRef]
- Denoyer, Y.; Woimant, F.; Bost, M.; Edan, G.; Drapier, S. Neurological Wilson’s disease lethal for the son, asymptomatic in the father. Mov. Disord. 2013, 28, 402–403. [Google Scholar] [CrossRef]
- Dufernez, F.; Lachaux, A.; Chappuis, P.; De Lumley, L.; Bost, M.; Woimant, F.; Misrahi, M.; Debray, D. Wilson disease in offspring of affected patients: Report of four French families. Clin. Res. Hepatol. Gastroenterol. 2013, 37, 240–245. [Google Scholar] [CrossRef]
- Stattermayer, A.F.; Entenmann, A.; Gschwantler, M.; Zoller, H.; Hofer, H.; Ferenci, P. The dilemma to diagnose Wilson disease by genetic testing alone. Eur. J. Clin. Investig. 2019, 49, e13147. [Google Scholar] [CrossRef]
- Maher, B. Personal genomes: The case of the missing heritability. Nature 2008, 456, 18–21. [Google Scholar] [CrossRef]
- Medici, V.; LaSalle, J.M. Genetics and epigenetic factors of Wilson disease. Ann. Transl. Med. 2019, 7 (Suppl. 2), S58. [Google Scholar] [CrossRef] [PubMed]
- Stattermayer, A.F.; Traussnigg, S.; Dienes, H.P.; Aigner, E.; Stauber, R.; Lackner, K.; Hofer, H.; Stift, J.; Wrba, F.; Stadlmayr, A.; et al. Hepatic steatosis in Wilson disease--Role of copper and PNPLA3 mutations. J. Hepatol. 2015, 63, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Schiefermeier, M.; Kollegger, H.; Madl, C.; Polli, C.; Oder, W.; Kuhn, H.; Berr, F.; Ferenci, P. The impact of apolipoprotein E genotypes on age at onset of symptoms and phenotypic expression in Wilson’s disease. Brain 2000, 123, 585–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medici, V.; Weiss, K.H. Genetic and environmental modifiers of Wilson disease. Handb. Clin. Neurol. 2017, 142, 35–41. [Google Scholar] [PubMed]
- Sibani, S.; Christensen, B.; O’Ferrall, E.; Saadi, I.; Hiou-Tim, F.; Rosenblatt, D.S.; Rozen, R. Characterization of six novel mutations in the methylenetetrahydrofolate reductase (MTHFR) gene in patients with homocystinuria. Hum. Mutat. 2000, 15, 280–287. [Google Scholar] [CrossRef]
- Gromadzka, G.; Rudnicka, M.; Chabik, G.; Przybylkowski, A.; Czlonkowska, A. Genetic variability in the methylenetetrahydrofolate reductase gene (MTHFR) affects clinical expression of Wilson’s disease. J. Hepatol. 2011, 55, 913–919. [Google Scholar] [CrossRef] [PubMed]
- Hafkemeyer, P.; Schupp, M.; Storch, M.; Gerok, W.; Haussinger, D. Excessive iron storage in a patient with Wilson’s disease. Clin. Investig. 1994, 72, 134–136. [Google Scholar] [CrossRef]
- Walshe, J.M.; Cox, D.W. Effect of treatment of Wilson’s disease on natural history of haemochromatosis. Lancet 1998, 352, 112–113. [Google Scholar] [CrossRef]
- Sorbello, O.; Sini, M.; Civolani, A.; Demelia, L. HFE gene mutations and Wilson’s disease in Sardinia. Dig. Liver Dis. 2010, 42, 216–219. [Google Scholar] [CrossRef] [PubMed]
- Erhardt, A.; Hoffmann, A.; Hefter, H.; Haussinger, D. HFE gene mutations and iron metabolism in Wilson’s disease. Liver 2002, 22, 474–478. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffenberger, J.; Gotthardt, D.N.; Herrmann, T.; Seessle, J.; Merle, U.; Schirmacher, P.; Stremmel, W.; Weiss, K.H. Iron metabolism and the role of HFE gene polymorphisms in Wilson disease. Liver Int. 2012, 32, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Przybylkowski, A.; Gromadzka, G.; Czlonkowska, A. Polymorphisms of metal transporter genes DMT1 and ATP7A in Wilson’s disease. J. Trace Elem. Med. Biol. 2014, 28, 8–12. [Google Scholar] [CrossRef]
- Stuehler, B.; Reichert, J.; Stremmel, W.; Schaefer, M. Analysis of the human homologue of the canine copper toxicosis gene MURR1 in Wilson disease patients. J. Mol. Med. 2004, 82, 629–634. [Google Scholar] [CrossRef]
- Weiss, K.H.; Merle, U.; Schaefer, M.; Ferenci, P.; Fullekrug, J.; Stremmel, W. Copper toxicosis gene MURR1 is not changed in Wilson disease patients with normal blood ceruloplasmin levels. World J. Gastroenterol. 2006, 12, 2239–2242. [Google Scholar] [CrossRef]
- Simon, I.; Schaefer, M.; Reichert, J.; Stremmel, W. Analysis of the human Atox 1 homologue in Wilson patients. World J. Gastroenterol. 2008, 14, 2383–2387. [Google Scholar] [CrossRef]
- Gupta, A.; Chattopadhyay, I.; Mukherjee, S.; Sengupta, M.; Das, S.K.; Ray, K. A novel COMMD1 mutation Thr174Met associated with elevated urinary copper and signs of enhanced apoptotic cell death in a Wilson Disease patient. Behav. Brain Funct. 2010, 6, 33. [Google Scholar] [CrossRef] [Green Version]
- Bost, M.; Piguet-Lacroix, G.; Parant, F.; Wilson, C.M. Molecular analysis of Wilson patients: Direct sequencing and MLPA analysis in the ATP7B gene and Atox1 and COMMD1 gene analysis. J. Trace Elem. Med. Biol. 2012, 26, 97–101. [Google Scholar] [CrossRef]
- Kumari, N.; Kumar, A.; Pal, A.; Thapa, B.R.; Modi, M.; Prasad, R. In-silico analysis of novel p.(Gly14Ser) variant of ATOX1 gene: Plausible role in modulating ATOX1-ATP7B interaction. Mol. Biol. Rep. 2019, 46, 3307–3313. [Google Scholar] [CrossRef]
- Mufti, A.R.; Burstein, E.; Csomos, R.A.; Graf, P.C.; Wilkinson, J.C.; Dick, R.D.; Challa, M.; Son, J.K.; Bratton, S.B.; Su, G.L.; et al. XIAP Is a copper binding protein deregulated in Wilson’s disease and other copper toxicosis disorders. Mol. Cell. 2006, 21, 775–785. [Google Scholar] [CrossRef]
- Burstein, E.; Ganesh, L.; Dick, R.D.; van De Sluis, B.; Wilkinson, J.C.; Klomp, L.W.; Wijmenga, C.; Brewer, G.J.; Nabel, G.J.; Duckett, C.S. A novel role for XIAP in copper homeostasis through regulation of MURR1. EMBO J. 2004, 23, 244–254. [Google Scholar] [CrossRef] [Green Version]
- Maine, G.N.; Mao, X.; Muller, P.A.; Komarck, C.M.; Klomp, L.W.; Burstein, E. COMMD1 expression is controlled by critical residues that determine XIAP binding. Biochem. J. 2009, 417, 601–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, K.H.; Runz, H.; Noe, B.; Gotthardt, D.N.; Merle, U.; Ferenci, P.; Stremmel, W.; Fullekrug, J. Genetic analysis of BIRC4/XIAP as a putative modifier gene of Wilson disease. J. Inherit. Metab. Dis. 2010, 33 (Suppl. 3), S233–S240. [Google Scholar] [CrossRef] [PubMed]
- Girard, M.; Poujois, A.; Fabre, M.; Lacaille, F.; Debray, D.; Rio, M.; Fenaille, F.; Cholet, S.; Ruel, C.; Causse, E.; et al. CCDC115-CDG: A new rare and misleading inherited cause of liver disease. Mol. Genet. Metab. 2018, 124, 228–235. [Google Scholar] [CrossRef]
- Vajro, P.; Zielinska, K.; Ng, B.G.; Maccarana, M.; Bengtson, P.; Poeta, M.; Mandato, C.; D’Acunto, E.; Freeze, H.H.; Eklund, E.A. Three unreported cases of TMEM199-CDG, a rare genetic liver disease with abnormal glycosylation. Orphanet J. Rare Dis. 2018, 13, 4. [Google Scholar] [CrossRef]
- Jansen, J.C.; Timal, S.; van Scherpenzeel, M.; Michelakakis, H.; Vicogne, D.; Ashikov, A.; Moraitou, M.; Hoischen, A.; Huijben, K.; Steenbergen, G.; et al. TMEM199 Deficiency Is a Disorder of Golgi Homeostasis Characterized by Elevated Aminotransferases, Alkaline Phosphatase, and Cholesterol and Abnormal Glycosylation. Am. J. Hum. Genet. 2016, 98, 322–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jansen, J.C.; Cirak, S.; van Scherpenzeel, M.; Timal, S.; Reunert, J.; Rust, S.; Perez, B.; Vicogne, D.; Krawitz, P.; Wada, Y.; et al. CCDC115 Deficiency Causes a Disorder of Golgi Homeostasis with Abnormal Protein Glycosylation. Am. J. Hum. Genet. 2016, 98, 310–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Comstra, H.S.; McArthy, J.; Rudin-Rush, S.; Hartwig, C.; Gokhale, A.; Zlatic, S.A.; Blackburn, J.B.; Werner, E.; Petris, M.; D’Souza, P.; et al. The interactome of the copper transporter ATP7A belongs to a network of neurodevelopmental and neurodegeneration factors. Elife 2017, 6, e24722. [Google Scholar] [CrossRef]
- Rujano, M.A.; Cannata Serio, M.; Panasyuk, G.; Peanne, R.; Reunert, J.; Rymen, D.; Hauser, V.; Park, J.H.; Freisinger, P.; Souche, E.; et al. Mutations in the X-linked ATP6AP2 cause a glycosylation disorder with autophagic defects. J. Exp. Med. 2017, 214, 3707–3729. [Google Scholar] [CrossRef] [PubMed]
- Ke, P.Y. Diverse Functions of Autophagy in Liver Physiology and Liver Diseases. Int. J. Mol. Sci. 2019, 20, 300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amirneni, S.; Haep, N.; Gad, M.A.; Soto-Gutierrez, A.; Squires, J.E.; Florentino, R.M. Molecular overview of progressive familial intrahepatic cholestasis. World J. Gastroenterol. 2020, 26, 7470–7484. [Google Scholar] [CrossRef] [PubMed]
- Ramraj, R.; Finegold, M.J.; Karpen, S.J. Progressive familial intrahepatic cholestasis type 3: Overlapping presentation with Wilson disease. Clin. Pediatr. 2012, 51, 689–691. [Google Scholar] [CrossRef] [PubMed]
- Boga, S.; Jain, D.; Schilsky, M.L. Presentation of Progressive Familial Intrahepatic Cholestasis Type 3 Mimicking Wilson Disease: Molecular Genetic Diagnosis and Response to Treatment. Pediatr. Gastroenterol. Hepatol. Nutr. 2015, 18, 202–208. [Google Scholar] [CrossRef] [Green Version]
- Ondrejkovicova, M.; Drazilova, S.; Drakulova, M.; Siles, J.L.; Zemjarova Mezenska, R.; Jungova, P.; Fabian, M.; Rychly, B.; Zigrai, M. New mutation of the ceruloplasmin gene in the case of a neurologically asymptomatic patient with microcytic anaemia, obesity and supposed Wilson’s disease. BMC Gastroenterol. 2020, 20, 95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anugwom, C.M.; Moscoso, C.G.; Lim, N.; Hassan, M. Aceruloplasminemia: A Case Report and Review of a Rare and Misunderstood Disorder of Iron Accumulation. Cureus 2020, 12, e11648. [Google Scholar]
- Horn, N.; Wittung-Stafshede, P. ATP7A-Regulated Enzyme Metalation and Trafficking in the Menkes Disease Puzzle. Biomedicines 2021, 9, 391. [Google Scholar] [CrossRef]
- Bansagi, B.; Lewis-Smith, D.; Pal, E.; Duff, J.; Griffin, H.; Pyle, A.; Muller, J.S.; Rudas, G.; Aranyi, Z.; Lochmuller, H.; et al. Phenotypic convergence of Menkes and Wilson disease. Neurol. Genet. 2016, 2, e119. [Google Scholar] [CrossRef] [Green Version]
- Martinelli, D.; Travaglini, L.; Drouin, C.A.; Ceballos-Picot, I.; Rizza, T.; Bertini, E.; Carrozzo, R.; Petrini, S.; de Lonlay, P.; El Hachem, M.; et al. MEDNIK syndrome: A novel defect of copper metabolism treatable by zinc acetate therapy. Brain 2013, 136, 872–881. [Google Scholar] [CrossRef] [Green Version]
- Martinelli, D.; Dionisi-Vici, C. AP1S1 defect causing MEDNIK syndrome: A new adaptinopathy associated with defective copper metabolism. Ann. N. Y. Acad. Sci. 2014, 1314, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, M.A.; Loomes, K.M. Alagille syndrome and non-syndromic paucity of the intrahepatic bile ducts. Transl. Gastroenterol. Hepatol. 2021, 6, 22. [Google Scholar] [CrossRef]
- Guegan, K.; Stals, K.; Day, M.; Turnpenny, P.; Ellard, S. JAG1 mutations are found in approximately one third of patients presenting with only one or two clinical features of Alagille syndrome. Clin. Genet. 2012, 82, 33–40. [Google Scholar] [CrossRef]
- Amson, M.; Lamoureux, E.; Hilzenrat, N.; Tischkowitz, M. Alagille syndrome and Wilson disease in siblings: A diagnostic conundrum. Can. J. Gastroenterol. 2012, 26, 330–332. [Google Scholar] [CrossRef] [PubMed]
- Muller, T.; Muller, W.; Feichtinger, H. Idiopathic copper toxicosis. Am. J. Clin. Nutr. 1998, 67 (Suppl. 5), 1082S–1086S. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.A.; Tsuji, J.S.; Garry, M.R.; McArdle, M.E.; Goodfellow, W.L., Jr.; Adams, W.J.; Menzie, C.A. Critical Review of Exposure and Effects: Implications for Setting Regulatory Health Criteria for Ingested Copper. Environ. Manag. 2020, 65, 131–159. [Google Scholar] [CrossRef] [Green Version]
- Harada, M.; Honma, Y.; Yoshizumi, T.; Kumamoto, K.; Oe, S.; Harada, N.; Tanimoto, A.; Yabuki, K.; Karasuyama, T.; Yoneda, A.; et al. Idiopathic copper toxicosis: Is abnormal copper metabolism a primary cause of this disease? Med. Mol. Morphol. 2019, 53, 50–55. [Google Scholar] [CrossRef]
- Kalita, J.; Kumar, V.; Misra, U.K.; Ranjan, A.; Khan, H.; Konwar, R. A study of oxidative stress, cytokines and glutamate in Wilson disease and their asymptomatic siblings. J. Neuroimmunol. 2014, 274, 141–148. [Google Scholar] [CrossRef]
- Azbukina, N.V.; Lopachev, A.V.; Chistyakov, D.V.; Goriainov, S.V.; Astakhova, A.A.; Poleshuk, V.V.; Kazanskaya, R.B.; Fedorova, T.N.; Sergeeva, M.G. Oxylipin Profiles in Plasma of Patients with Wilson’s Disease. Metabolites 2020, 10, 222. [Google Scholar] [CrossRef]
- Glavind, E.; Gotthardt, D.N.; Pfeiffenberger, J.; Sandahl, T.D.; Bashlekova, T.; Willemoe, G.L.; Hasselby, J.P.; Weiss, K.H.; Moller, H.J.; Vilstrup, H.; et al. The macrophage activation marker soluble CD163 is elevated and associated with liver disease phenotype in patients with Wilson’s disease. Orphanet. J. Rare Dis. 2020, 15, 173. [Google Scholar] [CrossRef]
- Mazagova, M.; Wang, L.; Anfora, A.T.; Wissmueller, M.; Lesley, S.A.; Miyamoto, Y.; Eckmann, L.; Dhungana, S.; Pathmasiri, W.; Sumner, S.; et al. Commensal microbiota is hepatoprotective and prevents liver fibrosis in mice. FASEB J. 2015, 29, 1043–1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, M.; Li, X.; Zhang, X.; Shi, H.; Vos, M.B.; Wei, X.; Wang, Y.; Gao, H.; Rouchka, E.C.; Yin, X.; et al. Dietary copper-fructose interactions alter gut microbial activity in male rats. Am. J. Physiol. Gastrointest. Liver Physiol. 2018, 314, G119–G130. [Google Scholar] [CrossRef]
- Baquero, F.; Nombela, C. The microbiome as a human organ. Clin. Microbiol. Infect. 2012, 18 (Suppl. 4), 2–4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohtani, N.; Kawada, N. Role of the Gut-Liver Axis in Liver Inflammation, Fibrosis, and Cancer: A Special Focus on the Gut Microbiota Relationship. Hepatol. Commun. 2019, 3, 456–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geng, H.; Shu, S.; Dong, J.; Li, H.; Xu, C.; Han, Y.; Hu, J.; Han, Y.; Yang, R.; Cheng, N. Association study of gut flora in Wilson’s disease through high-throughput sequencing. Medicine 2018, 97, e11743. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Dong, J.; Cheng, N.; Yang, R.; Han, Y.; Han, Y. Inflammatory cytokines expression in Wilson’s disease. Neurol. Sci. 2019, 40, 1059–1066. [Google Scholar] [CrossRef] [PubMed]
- Park, J.Y.; Mun, J.H.; Lee, B.H.; Heo, S.H.; Kim, G.H.; Yoo, H.W. Proteomic analysis of sera of asymptomatic, early-stage patients with Wilson’s disease. Proteom. Clin. Appl. 2009, 3, 1185–1190. [Google Scholar] [CrossRef] [Green Version]
- Cai, X.; Deng, L.; Ma, X.; Guo, Y.; Feng, Z.; Liu, M.; Guan, Y.; Huang, Y.; Deng, J.; Li, H.; et al. Altered diversity and composition of gut microbiota in Wilson’s disease. Sci. Rep. 2020, 10, 21825. [Google Scholar] [CrossRef]
- Mazi, T.A.; Shibata, N.M.; Medici, V. Lipid and energy metabolism in Wilson disease. Liver Res. 2020, 4, 5–14. [Google Scholar] [CrossRef]
- Condrat, C.E.; Thompson, D.C.; Barbu, M.G.; Bugnar, O.L.; Boboc, A.; Cretoiu, D.; Suciu, N.; Cretoiu, S.M.; Voinea, S.C. miRNAs as Biomarkers in Disease: Latest Findings Regarding Their Role in Diagnosis and Prognosis. Cells 2020, 9, 276. [Google Scholar] [CrossRef] [Green Version]
- Szelenberger, R.; Kacprzak, M.; Saluk-Bijak, J.; Zielinska, M.; Bijak, M. Plasma MicroRNA as a novel diagnostic. Clin. Chim. Acta 2019, 499, 98–107. [Google Scholar] [CrossRef]
- Bandiera, S.; Pfeffer, S.; Baumert, T.F.; Zeisel, M.B. miR-122—A key factor and therapeutic target in liver disease. J. Hepatol. 2015, 62, 448–457. [Google Scholar] [CrossRef] [Green Version]
- Siaj, R.; Sauer, V.; Stoppeler, S.; Gerss, J.; Spiegel, H.U.; Kohler, G.; Zibert, A.; Schmidt, H.H. Longitudinal analysis of serum miR-122 in a rat model of Wilson’s disease. Hepatol. Int. 2012, 6, 770–777. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
| Test | Parameter | Score |
|---|---|---|
| Kayser-Fleischer ring | Present | 2 |
| Absent | 0 | |
| Neurological symptoms | Severe | 2 |
| Mild | 1 | |
| Absent | 0 | |
| Serum ceruloplasmin | Normal (>0.2 g/L) | 0 |
| 0.1–0.2 g/L | 1 | |
| <0.1 g/L | 2 | |
| Coombs-negative hemolytic anemia | Present | 1 |
| Absent | 0 | |
| Liver copper (in the absence of cholestasis) | >250 µg (>4 µmol) g−1 dry weight | 2 |
| 50–249 µg (0.8–4 µmol) g−1 | 1 | |
| Normal: <50 µg (<0.8 µmol) g−1 | −1 | |
| Rhodanine-positive granules | 1 | |
| Urinary copper (in the absence of acute hepatitis) | Normal | 0 |
| 1–2 × ULN | 1 | |
| >2 × ULN | 2 | |
| Normal but >5 × ULN after D-penicillamine | 2 | |
| Mutation analysis of ATP7B | Biallelic deleterious variants | 4 |
| One deleterious variant | 1 | |
| No mutation detected | 0 | |
| Total score | Evaluation | |
| ≥4 | Diagnosis established | |
| 3 | Diagnosis possible; more tests needed | |
| ≤2 | Diagnosis very unlikely |
| Type of Variant | No. Mutations | Frequency (%) |
|---|---|---|
| Regulatory sequences | 12 | 1.28 |
| Splicing site | 77 | 8.21 |
| Missense and nonsense mutations | 572 | 60.98 |
| Small deletions | 158 | 16.84 |
| Small insertions | 80 | 8.53 |
| Indels | 12 | 1.28 |
| Gross deletions | 26 | 2.77 |
| Deep intronic | 1 | 0.11 |
| Source | Prevalence | Country | Reference |
|---|---|---|---|
| Public health registries | 1.81/100,000 | Taiwan | [86] |
| 1.50/100,000 | France | [87] | |
| 1.64/100,000 | Spain | [88] | |
| Mutational screening in a population | 14.28/100,000 | United Kingdom | [89] |
| 13.22/100,000 | South Korea | [74] | |
| 25/100,000 | France | [90] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sánchez-Monteagudo, A.; Ripollés, E.; Berenguer, M.; Espinós, C. Wilson’s Disease: Facing the Challenge of Diagnosing a Rare Disease. Biomedicines 2021, 9, 1100. https://doi.org/10.3390/biomedicines9091100
Sánchez-Monteagudo A, Ripollés E, Berenguer M, Espinós C. Wilson’s Disease: Facing the Challenge of Diagnosing a Rare Disease. Biomedicines. 2021; 9(9):1100. https://doi.org/10.3390/biomedicines9091100
Chicago/Turabian StyleSánchez-Monteagudo, Ana, Edna Ripollés, Marina Berenguer, and Carmen Espinós. 2021. "Wilson’s Disease: Facing the Challenge of Diagnosing a Rare Disease" Biomedicines 9, no. 9: 1100. https://doi.org/10.3390/biomedicines9091100
APA StyleSánchez-Monteagudo, A., Ripollés, E., Berenguer, M., & Espinós, C. (2021). Wilson’s Disease: Facing the Challenge of Diagnosing a Rare Disease. Biomedicines, 9(9), 1100. https://doi.org/10.3390/biomedicines9091100