
Role of Epigenetics in Type 2 Diabetes and Obesity

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. T2D and OB: Risks, Prevalence, and Genetics
3. Epigenetics and OB
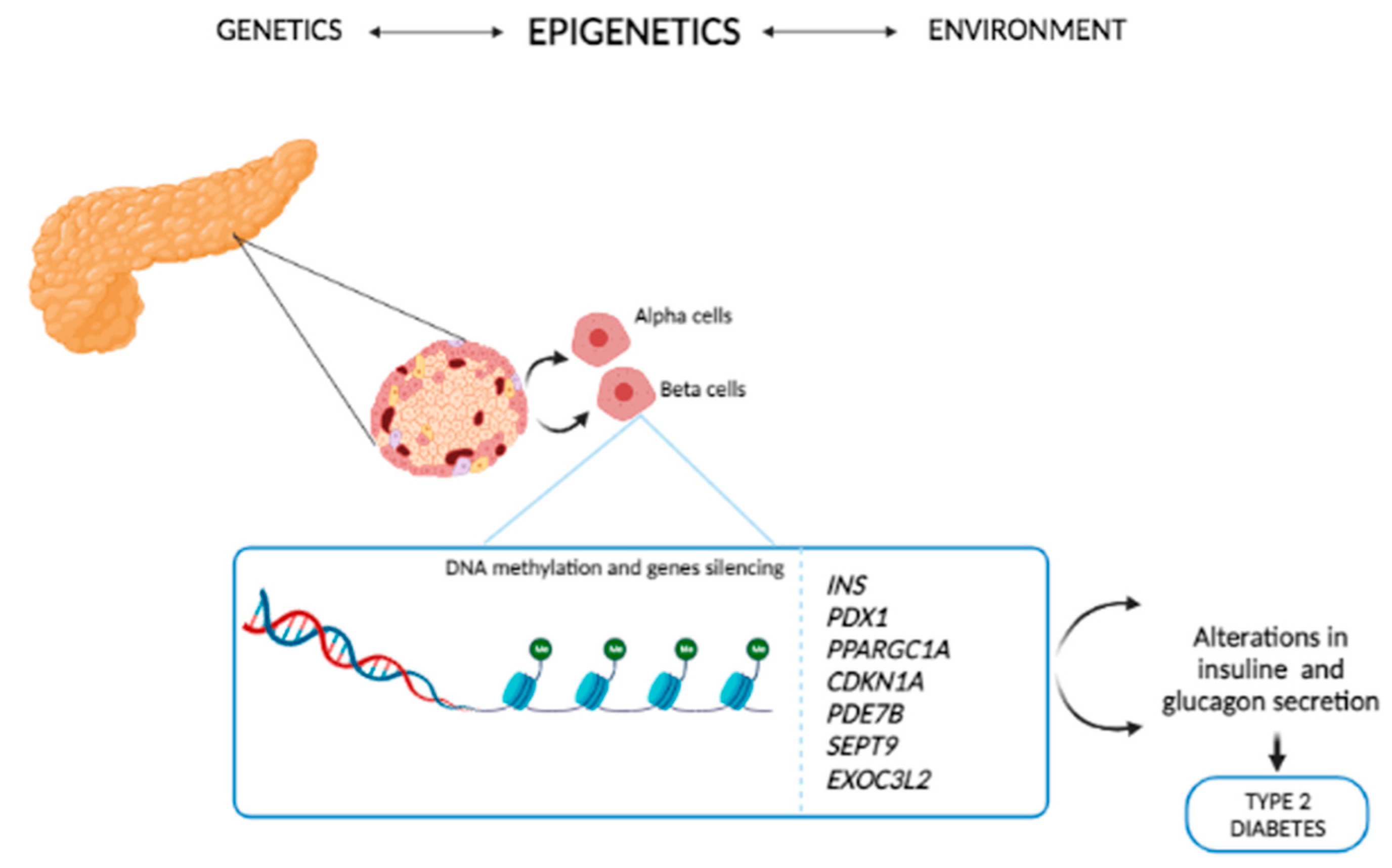
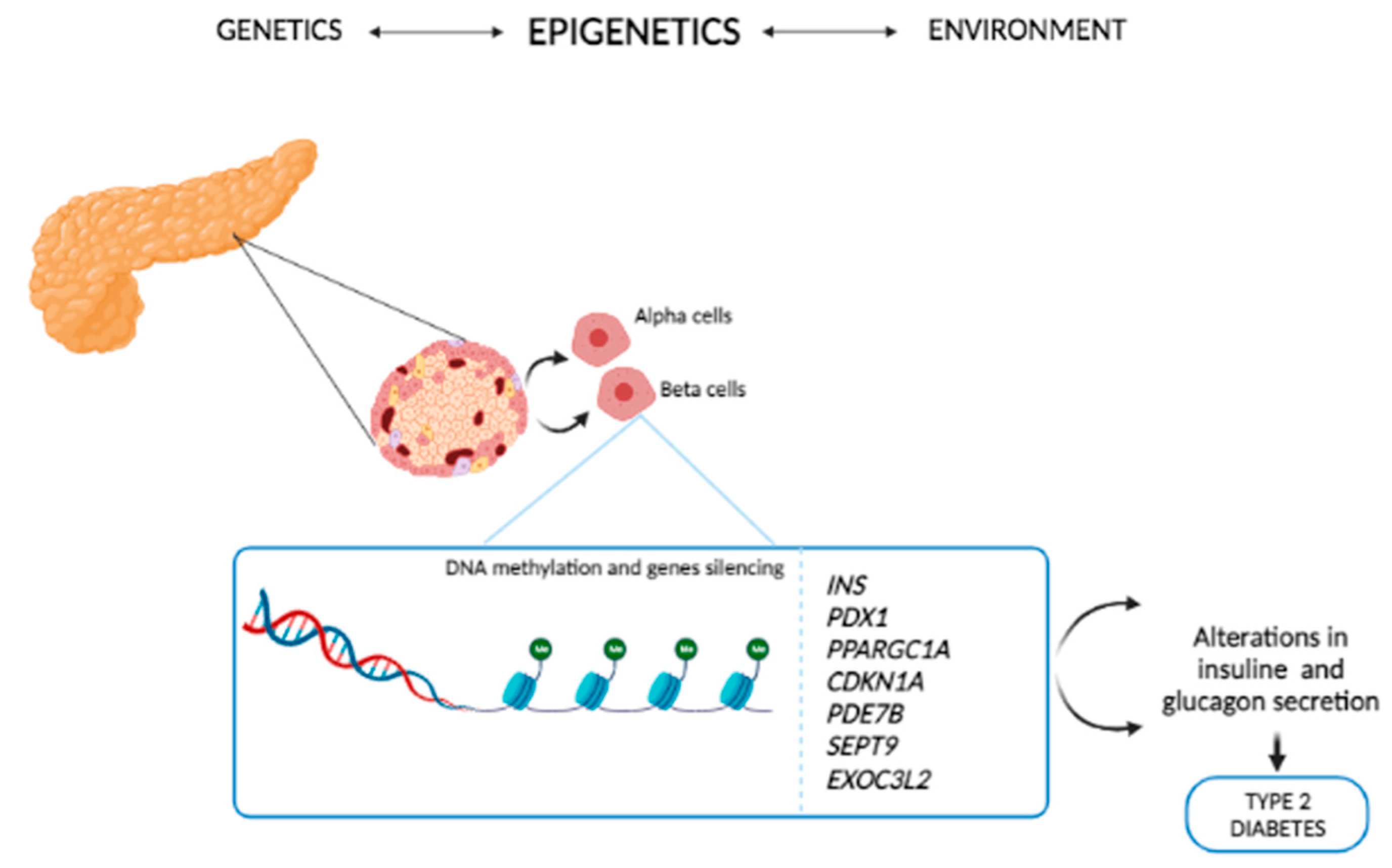
4. Epigenetics of T2D
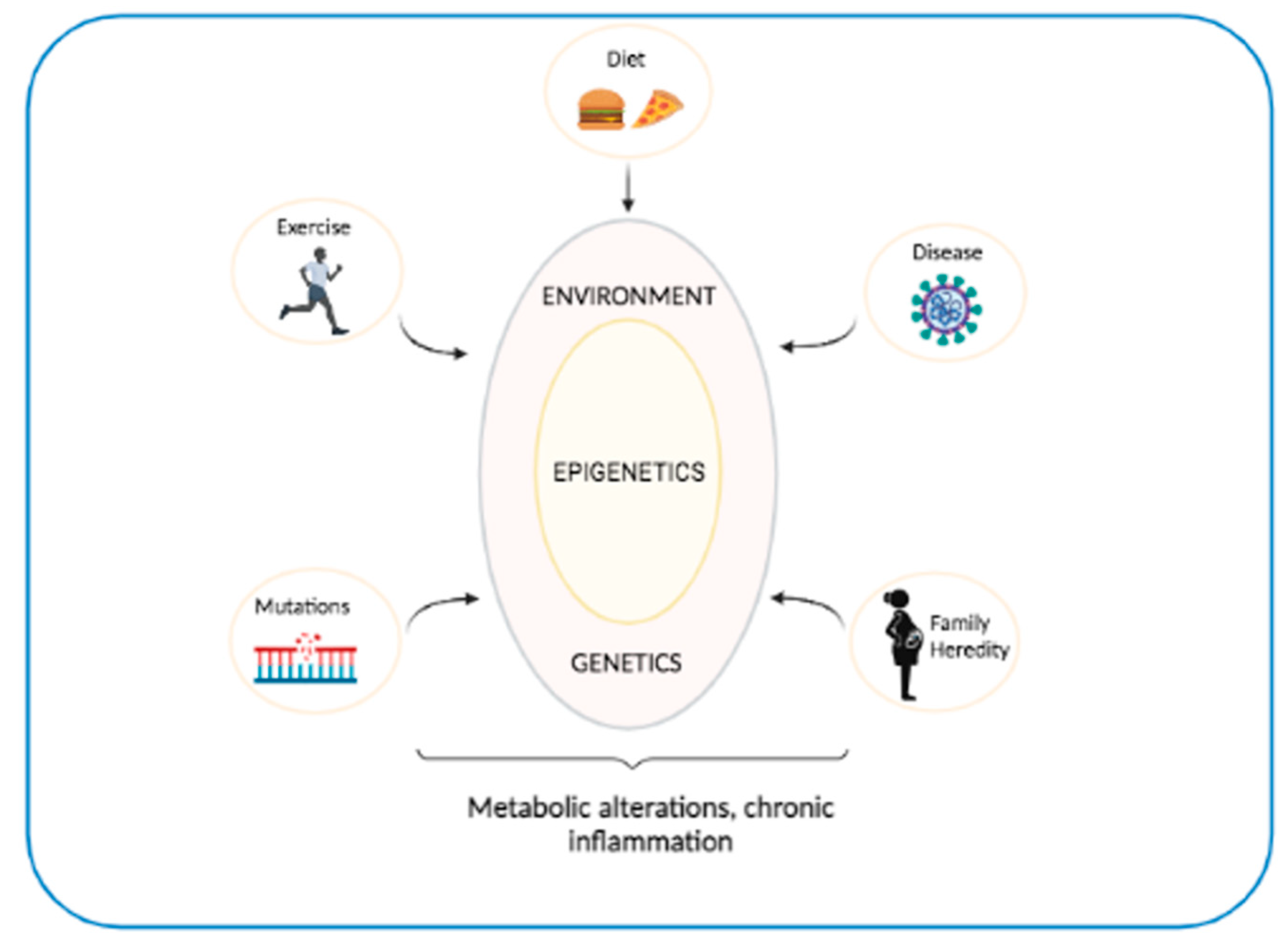
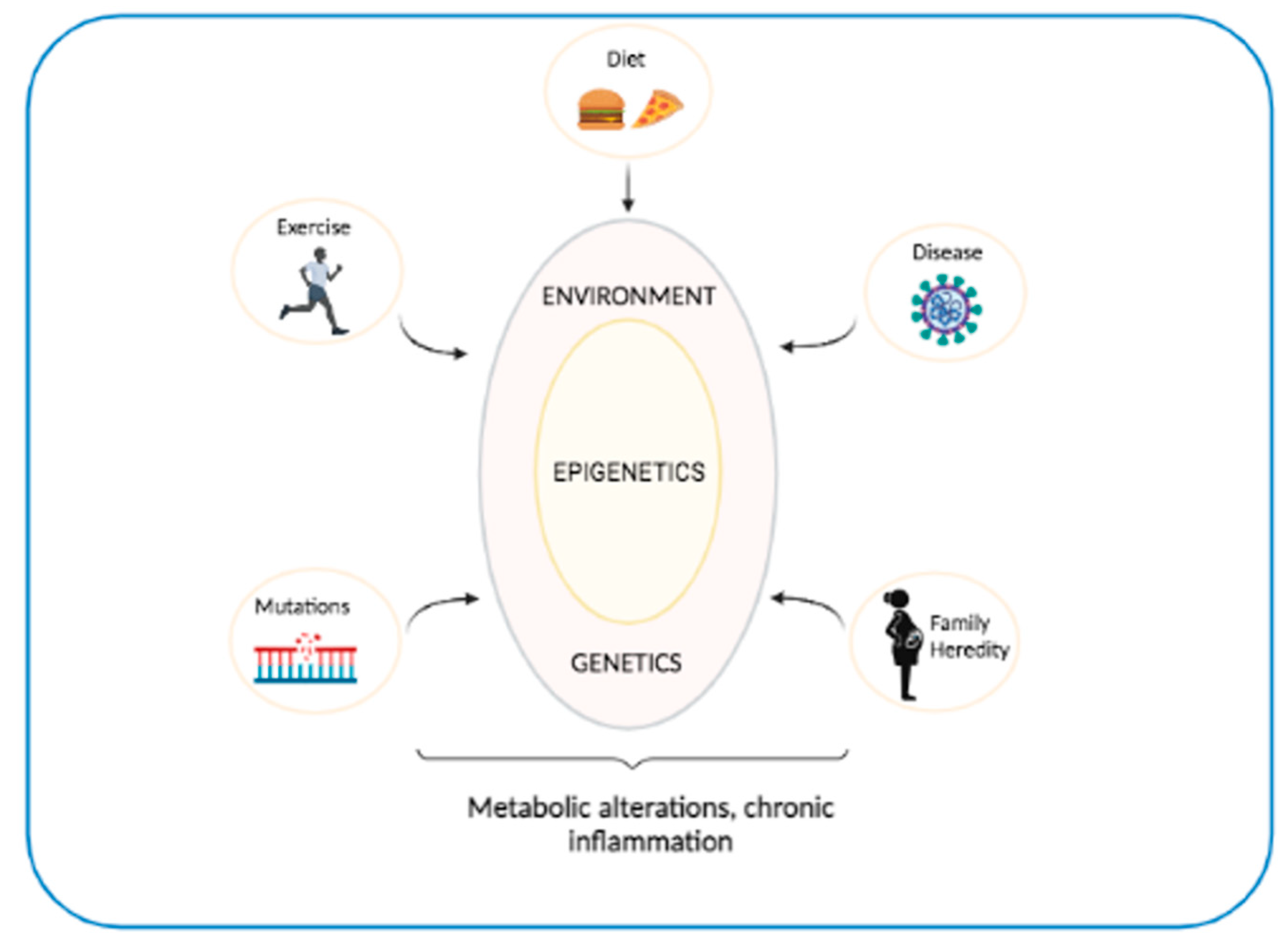
5. Interaction between Genetics and Epigenetics in T2D and OB
6. Role of Aging in T2D and OB
7. Intermittent Fasting, Epigenetics and Aging
8. Discussion
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| T2D | Type 2 Diabetes |
| OB | Obesity |
| CVD | Cardiovascular disease |
| ChIP | Chromatin Immunoprecipitation |
| IGF2 | Insulin-like growth factor 2 |
| BMI | Body Mass Index |
| GWASSs | Genome-wide association studies |
| HIF3A | Hypoxia Inducible Factor 3 Subunit Alpha |
| TNF-α | Tumor necrosis factor-alpha |
| PGC1α | PPARg coactivator 1 alpha |
| PDK4 | Pyruvate dehydrogenase |
| INS | Insulin |
| PDX1 | Pancreatic and Duodenal homeobox 1 |
| PPARGC1A | PPARg coactivator 1 alpha |
| CDKN1A | Cyclin Dependent Kinase Inhibitor 1A |
| PDE7B | Phosphodiesterase 7B |
| SEPT9 | Septin 9 |
| EXOC3L2 | Exocyst Complex Component 3 Like 2 |
| NAFLD | Non-Alcoholic Fatty Liver Disease |
| PDGFA | Platelet-Derived Growth Factor Alpha |
| ped/pea-15 | phosphoprotein enriched in diabetes/phosphoprotein enriched in astrocytes-15 |
| SNP | Single Nucleotide polymorphism |
| TCF7L2 | Transcription Factor 7 Like 2 |
| LPS | Lipopolysaccharide |
| USF1 | Upstream Transcription Factor 1 |
| NEC | Necrotizing Enterocolitis |
| DR | Dietary Restriction |
| CR | Caloric Restriction |
| IF | Intermittent Fasting |
| ATP | Adenosine Triphosphate |
References
- Bird, A. Perceptions of epigenetics. Nature 2007, 447, 396–398. [Google Scholar] [CrossRef] [PubMed]
- Ling, C.; Rönn, T. Epigenetics in Human Obesity and Type 2 Diabetes. Cell Metab. 2019, 29, 1028–1044. [Google Scholar] [CrossRef] [Green Version]
- Horsthemke, B. A critical view on transgenerational epigenetic inheritance in humans. Nat. Commun. 2018, 9, 2973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.; Yan, W. Epigenetic inheritance of acquired traits through sperm RNAs and sperm RNA modifications. Nat. Rev. Genet. 2016, 17, 733–743. [Google Scholar] [CrossRef]
- Feinberg, A.P. Epigenomics reveals a functional genome anatomy and a new approach to common disease. Nat. Biotechnol. 2010, 28, 1049–1052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eichler, E.E.; Flint, J. Missing heritability and strategies for finding the underlying causes of complex disease. Nat. Rev. Genet. 2010, 11, 446–450. [Google Scholar] [CrossRef] [Green Version]
- Berdasco, M.; Esteller, M. Clinical epigenetics: Seizing opportunities for translation. Nat. Rev. Genet. 2019, 20, 109–127. [Google Scholar] [CrossRef]
- Ma, F.; Zhang, C.Y. Histone modifying enzymes: Novel disease biomarkers and assay development. Expert Rev. Mol. Diagn. 2016, 16, 297–306. [Google Scholar] [CrossRef]
- Šestáková, Š.; Šálek, C.; Remešová, H. DNA Methylation Validation Methods: A Coherent Review with Practical Comparison. Biol. Proced. Online 2019, 21, 19. [Google Scholar] [CrossRef]
- Mørkve, K.T.; Rezwan, F.I. Transgenerational and intergenerational epigenetic inheritance in allergic diseases. J. Allergy Clin. Immunol. 2018, 142, 765–772. [Google Scholar] [CrossRef]
- Van Dijk, S.J.; Molloy, P.L. Members of EpiSCOPE. Epigenetics and human obesity. Int. J. Obes. 2015, 39, 85–97. [Google Scholar] [CrossRef] [Green Version]
- Martin-Gronert, M.S.; Ozanne, S.E. Mechanisms underlying the developmental origins of disease. Rev. Endocr. Metab. Disord. 2012, 13, 85–92. [Google Scholar] [CrossRef]
- Barker, D.J.; Osmond, C. Growth in utero, blood pressure in childhood and adult life, and mortality from cardiovascular disease. Br. Med. J. 1989, 298, 564–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gluckman, P.D.; Hanson, M.A. Effect of in utero and early-life conditions on adult health and disease. N. Engl. J. Med. 2008, 359, 61–73. [Google Scholar] [CrossRef] [Green Version]
- Bateson, P.; Barker, D. Developmental plasticity and human health. Nature 2004, 430, 419–421. [Google Scholar] [CrossRef]
- Barker, D.J.; Osmond, C. Infant mortality, childhood nutrition, and ischaemic heart disease in England and Wales. Lancet 1986, 1, 1077–1081. [Google Scholar] [CrossRef]
- Osmond, C.; Barker, D.J. Early growth and death from cardiovascular disease in women. BMJ 1993, 307, 1519–1524. [Google Scholar] [CrossRef] [Green Version]
- Ainge, H.; Thompson, C. A systematic review on animal models of maternal high fat feeding and offspring glycaemic control. Int. J. Obes. 2011, 35, 325–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seki, Y.; Williams, L. Minireview: Epigenetic Programming of Diabetes and Obesity: Animal Models. Endocrinology 2012, 153, 1031–1038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heijmans, B.T.; Tobi, E.W. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc. Natl. Acad. Sci. USA 2008, 105, 17046–17049. [Google Scholar] [CrossRef] [Green Version]
- Lumey, L.; Stein, A.D. Lipid profiles in middle-aged men and women after famine exposure during gestation: The Dutch Hunger Winter Families Study. Am. J. Clin. Nutr. 2009, 89, 1737–1743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Hu, F.B. The global implications of diabetes and cancer. Lancet 2014, 383, 1947–1948. [Google Scholar] [CrossRef]
- Agardh, E.; Lundstig, A. Genome-wide analysis of DNA methylation in subjects with type 1 diabetes identifies epigenetic modifications associated with proliferative diabetic retinopathy. BMC Med. 2015, 13, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, C.G.; Finer, S. Integrated Genetic and Epigenetic Analysis Identifies Haplotype-Specific Methylation in the FTO Type 2 Diabetes and Obesity Susceptibility Locus. PLoS ONE 2010, 5, e14040. [Google Scholar] [CrossRef]
- Nakatochi, M.; Ichihara, S. Epigenome-wide association of myocardial infarction with DNA methylation sites at loci related to cardiovascular disease. Clin. Epigenetics 2017, 9, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pillon, N.J.; Loos, R.J.F. Metabolic consequences of obesity and type 2 diabetes: Balancing genes and environment for personalized care. Cell 2021, 184, 1530–1544. [Google Scholar] [CrossRef]
- Yengo, L.; Sidorenko, J. Meta-analysis of genome-wide association studies for height and body mass index in ∼700000 individuals of European ancestry. Hum. Mol. Genet. 2018, 27, 3641–3649. [Google Scholar] [CrossRef]
- Mahajan, A.; Taliun, D. Fine-mapping type 2 diabetes loci to single-variant resolution using high-density imputation and islet-specific epigenome maps. Nat. Genet. 2018, 50, 1505–1513. [Google Scholar] [CrossRef] [Green Version]
- Locke, A.E.; Kahali, B. Genetic studies of body mass index yield new insights for obesity biology. Nature 2015, 518, 197–206. [Google Scholar] [CrossRef] [Green Version]
- Barroso, I.; McCarthy, M.I. The Genetic Basis of Metabolic Disease. Cell 2019, 177, 146–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, Y.; Yiorkas, A.M. Genome-Wide and Abdominal MRI Data Provide Evidence That a Genetically Determined Favorable Adiposity Phenotype Is Characterized by Lower Ectopic Liver Fat and Lower Risk of Type 2 Diabetes, Heart Disease, and Hypertension. Diabetes 2019, 68, 207–219. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Z.Z.; Hou, L. Predictors of global methylation levels in blood DNA of healthy subjects: A combined analysis. Int. J. Epidemiol. 2012, 41, 126. [Google Scholar] [CrossRef] [Green Version]
- Cash, H.L.; McGarvey, S.T. Cardiovascular disease risk factors and DNA methylation at the LINE-1 repeat region in peripheral blood from Samoan Islanders. Epigenetics 2011, 6, 1257–1264. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.; Long, T.I. DNA Methylation as a Biomarker for Cardiovascular Disease Risk. PLoS ONE 2010, 5, e9692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Su, S. A genome-wide methylation study on obesity: Differential variability and differential methylation. Epigenetics 2013, 8, 522–533. [Google Scholar] [CrossRef] [Green Version]
- Dick, K.J.; Nelson, C.P. DNA methylation and body-mass index: A genome-wide analysis. Lancet 2014, 383, 1990–1998. [Google Scholar] [CrossRef] [Green Version]
- Wahl, S.; Drong, A. Epigenome-wide association study of body mass index, and the adverse outcomes of adiposity. Nature 2016, 541, 81–86. [Google Scholar] [CrossRef] [Green Version]
- Dahlman, I.; Sinha, I. The fat cell epigenetic signature in post-obese women is characterized by global hypomethylation and differential DNA methylation of adipogenesis genes. Int. J. Obes. 2015, 39, 910–919. [Google Scholar] [CrossRef] [PubMed]
- Hermsdorff, H.H.; Mansego, M.L. TNF-alpha promoter methylation in peripheral white blood cells: Relationship with circulating TNFα, truncal fat and n-6 PUFA intake in young women. Cytokine 2013, 64, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Barres, R.; Kirchner, H. Weight loss after gastric bypass surgery in human obesity remodels promoter methylation. Cell Rep. 2013, 3, 1020–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obermann-Borst, S.A.; Eilers, P.H. Duration of breastfeeding and gender are associated with methylation of the LEPTIN gene in very young children. Pediatr. Res. 2013, 74, 344–349. [Google Scholar] [CrossRef] [Green Version]
- Drake, A.J.; McPherson, R.C. An unbalanced maternal diet in pregnancy associates with offspring epigenetic changes in genes controlling glucocorticoid action and foetal growth. Clin. Endocrinol. 2012, 77, 808–815. [Google Scholar] [CrossRef]
- Ling, C.; Guerra, S. Epigenetic regulation of PPARGC1A in human type 2 diabetic islets and effect on insulin secretion. Diabetologia 2008, 51, 615–622. [Google Scholar] [CrossRef] [Green Version]
- Yang, B.T.; Dayeh, T.A. Insulin promoter DNA methylation correlates negatively with insulin gene expression and positively with HbA1c levels in human pancreatic islets. Diabetologia 2011, 54, 360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dayeh, T.; Volkov, P. Genome-Wide DNA Methylation Analysis of Human Pancreatic Islets from Type 2 Diabetic and Non-Diabetic Donors Identifies Candidate Genes That Influence Insulin Secretion. PLoS Genet. 2014, 10, e1004160. [Google Scholar] [CrossRef]
- Abderrahmani, A.; Yengo, L. Increased Hepatic PDGF-AA Signaling Mediates Liver Insulin Resistance in Obesity-Associated Type 2 Diabetes. Diabetes 2018, 67, 1310–1321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raciti, G.A.; Longo, M. Understanding type 2 diabetes: From genetics to epigenetics. Acta Diabetol. 2015, 52, 821–827. [Google Scholar] [CrossRef]
- Ungaro, P.; Teperino, R. Molecular cloning and characterization of the human PED/PEA-15 gene promoter reveal antagonistic regulation by hepatocyte nuclear factor 4alpha and chicken ovalbumin upstream promoter transcription factor II. J. Biol Chem. 2008, 283, 30970–30979. [Google Scholar] [CrossRef] [Green Version]
- Ungaro, P.; Mirra, P. Peroxisome proliferator-activated receptor-γ activation enhances insulin-stimulated glucose disposal by reducing ped/pea-15 gene expression in skeletal muscle cells: Evidence for involvement of activator protein-1. J. Biol Chem. 2012, 287, 42951–42961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosen, E.D.; Kaestner, K.H. Epigenetics and Epigenomics: Implications for Diabetes and Obesity. Diabetes 2018, 67, 1923–1931. [Google Scholar] [CrossRef] [Green Version]
- Canani, R.B.; Costanzo, M. The epigenetic effects of butyrate: Potential therapeutic implications for clinical practice. Clin. Epigenetics 2012, 4, 1–7. [Google Scholar]
- JP, D. Abdominal obesity and cardiovascular disease: Is inflammation the missing link? Can. J. Cardiol. 2012, 28, 642–652. [Google Scholar]
- Kumar, H.; Lund, R. Gut microbiota as an epigenetic regulator: Pilot study based on whole-genome methylation analysis. mBio 2014, 5, e02113-14. [Google Scholar] [CrossRef] [Green Version]
- Ley, R.E.; Turnbaugh, P.J. Microbial ecology: Human gut microbes associated with obesity. Nature 2006, 444, 1022–1023. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, M.; Ogasawara, N. Transglucosidase improves the gut microbiota profile of type 2 diabetes mellitus patients: A randomized double-blind, placebo-controlled study. BMC Gastroenterol. 2013, 13, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manco, M.; Putignani, L. Gut microbiota, lipopolysaccharides, and innate immunity in the pathogenesis of obesity and cardiovascular risk. Endocr. Rev. 2010, 31, 817–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Hoenig, J.D. 16S rRNA gene-based analysis of fecal microbiota from preterm infants with and without necrotizing enterocolitis. ISME J. 2009, 3, 944–954. [Google Scholar] [CrossRef] [Green Version]
- Cortese, R.; Lu, L. Epigenome-Microbiome crosstalk: A potential new paradigm influencing neonatal susceptibility to disease. Epigenetics 2016, 11, 205–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradley, D.; Hsueh, W. Type 2 Diabetes in the Elderly: Challenges in a Unique Patient Population. J. Geriatr. Med. Gerontol. 2016, 2, 14. [Google Scholar] [CrossRef] [PubMed]
- DeFronzo, R.A. Pathogenesis of type 2 diabetes mellitus. Med. Clin. N. Am. 2004, 88, 787–835. [Google Scholar] [CrossRef]
- Harman, D. The biologic clock: The mitochondria? J. Am. Geriatr. Soc. 1972, 20, 145–147. [Google Scholar] [CrossRef]
- Ng, G.Y.-Q.; Fann, D.Y.-W. Dietary Restriction and Epigenetics: Part, I. Cond. Med. 2019, 2, 284–299. [Google Scholar]
- Calvanese, V.; Lara, E. The role of epigenetics in aging and age-related diseases. Ageing Res. Rev. 2009, 8, 268–276. [Google Scholar] [CrossRef]
- Anton, S.D.; Moehl, K. Flipping the Metabolic Switch: Understanding and Applying the Health Benefits of Fasting. Obesity 2018, 26, 254–268. [Google Scholar] [CrossRef]
- Paoli, A.; Tinsley, G. The Influence of Meal Frequency and Timing on Health in Humans: The Role of Fasting. Nutrients 2019, 11, 719. [Google Scholar] [CrossRef] [Green Version]
- Kroeger, C.M.; Klempel, M.C. Improvement in coronary heart disease risk factors during an intermittent fasting/calorie restriction regimen: Relationship to adipokine modulations. Nutr. Metab. 2012, 9, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zampieri, M.; Ciccarone, F. Reconfiguration of DNA methylation in aging. Mech. Ageing Dev. 2015, 151, 60–70. [Google Scholar] [CrossRef]
- Lopetuso, L.R.; Scaldaferri, F. Commensal Clostridia: Leading players in the maintenance of gut homeostasis. Gut Pathog. 2013, 5, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aron-Wisnewsky, J.; Julia, Z. Effect of bariatric surgery-induced weight loss on SR-BI-, ABCG1-, and ABCA1-mediated cellular cholesterol efflux in obese women. J. Clin. Endocrinol. Metab. 2011, 96, 1151–1159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duh, E.J.; Sun, J.K. Diabetic retinopathy: Current understanding, mechanisms, and treatment strategies. JCI Insight. 2017, 2, e93751. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.; Barnes, A.C. Can type 2 diabetes be reversed and how can this best be achieved? James Lind Alliance research priority number one. Diabet. Med. 2019, 36, 308–315. [Google Scholar] [CrossRef]
- Nesse, R.M.; Williams, G.C. Why We Get Sick: The New Science of Darwinian Medicine; Vintage Books: New York, NY, USA, 1996; p. 290. [Google Scholar]
- Atlasi, Y.; Stunnenberg, H.G. The interplay of epigenetic marks during stem cell differentiation and development. Nat. Rev. Genet. 2017, 18, 643–658. [Google Scholar] [CrossRef]
- Kiefer, J.C. Epigenetics in development. Dev. Dyn. 2007, 236, 1144–1156. [Google Scholar] [CrossRef]
- Wolf, R. Stability and flexibility of epigenetic gene regulation in mammalian development. Nature 2007, 447, 425–432. [Google Scholar]
- Patel, C.J.; Chen, R. Systematic identification of interaction effects between genome- and environment-wide associations in type 2 diabetes mellitus. Hum. Genet. 2013, 132, 495–508. [Google Scholar] [CrossRef] [Green Version]
- Campión, J.; Milagro, F.I. Individuality and epigenetics in obesity. Obes. Rev. 2009, 10, 383–392. [Google Scholar] [CrossRef]
- Baylin, S.B.; Jones, P.A. Epigenetic Determinants of Cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuomo, P.; Papaianni, M. An In Vitro Model to Investigate the Role of Helicobacter pylori in Type 2 Diabetes, Obesity, Alzheimer’s Disease and Cardiometabolic Disease. Int. J. Mol. Sci. 2020, 21, 8369. [Google Scholar] [CrossRef] [PubMed]
- Felice, C.; Federico, C. The hypothesis that helicobacter pylori predisposes to Alzheimer’s disease is biologically plausible. Sci. Rep. 2017, 7, 1–14. [Google Scholar]
- Fulgione, A.; Papaianni, M. Interaction between MyD88, TIRAP and IL1RL1 against Helicobacter pylori infection. Sci. Rep. 2020, 10, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Streata, I.; Weiner, J.R., 3rd. The CARD9 Polymorphisms rs4077515, rs10870077 and rs10781499 Are Uncoupled from Susceptibility to and Severity of Pulmonary Tuberculosis. PLoS ONE 2016, 11, e0165853. [Google Scholar]
- Laland, K.; Uller, T. Does evolutionary theory need a rethink? Nature 2014, 514, 161–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bürger, R.; Willensdorfer, M. Why Are Phenotypic Mutation Rates Much Higher Than Genotypic Mutation Rates? Genetics 2006, 172, 197–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, Q.; Christiansen, L. Twin methodology in epigenetic studies. J. Exp. Biol. 2015, 218 Pt 1, 134–139. [Google Scholar] [CrossRef] [Green Version]
- Skinner, M.K. Endocrine disruptor induction of epigenetic transgenerational inheritance of disease. Mol. Cell. Endocrinol. 2014, 398, 4–12. [Google Scholar] [CrossRef] [Green Version]
- Skinner, M.K.; Gurerrero-Bosagna, C. Epigenetics and the evolution of Darwin’s Finches. Genome Biol. Evol. 2014, 6, 1972–1978. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Capparelli, R.; Iannelli, D. Role of Epigenetics in Type 2 Diabetes and Obesity. Biomedicines 2021, 9, 977. https://doi.org/10.3390/biomedicines9080977
Capparelli R, Iannelli D. Role of Epigenetics in Type 2 Diabetes and Obesity. Biomedicines. 2021; 9(8):977. https://doi.org/10.3390/biomedicines9080977
Chicago/Turabian StyleCapparelli, Rosanna, and Domenico Iannelli. 2021. "Role of Epigenetics in Type 2 Diabetes and Obesity" Biomedicines 9, no. 8: 977. https://doi.org/10.3390/biomedicines9080977
APA StyleCapparelli, R., & Iannelli, D. (2021). Role of Epigenetics in Type 2 Diabetes and Obesity. Biomedicines, 9(8), 977. https://doi.org/10.3390/biomedicines9080977