
The Roles of Post-Translational Modifications in STAT3 Biological Activities and Functions



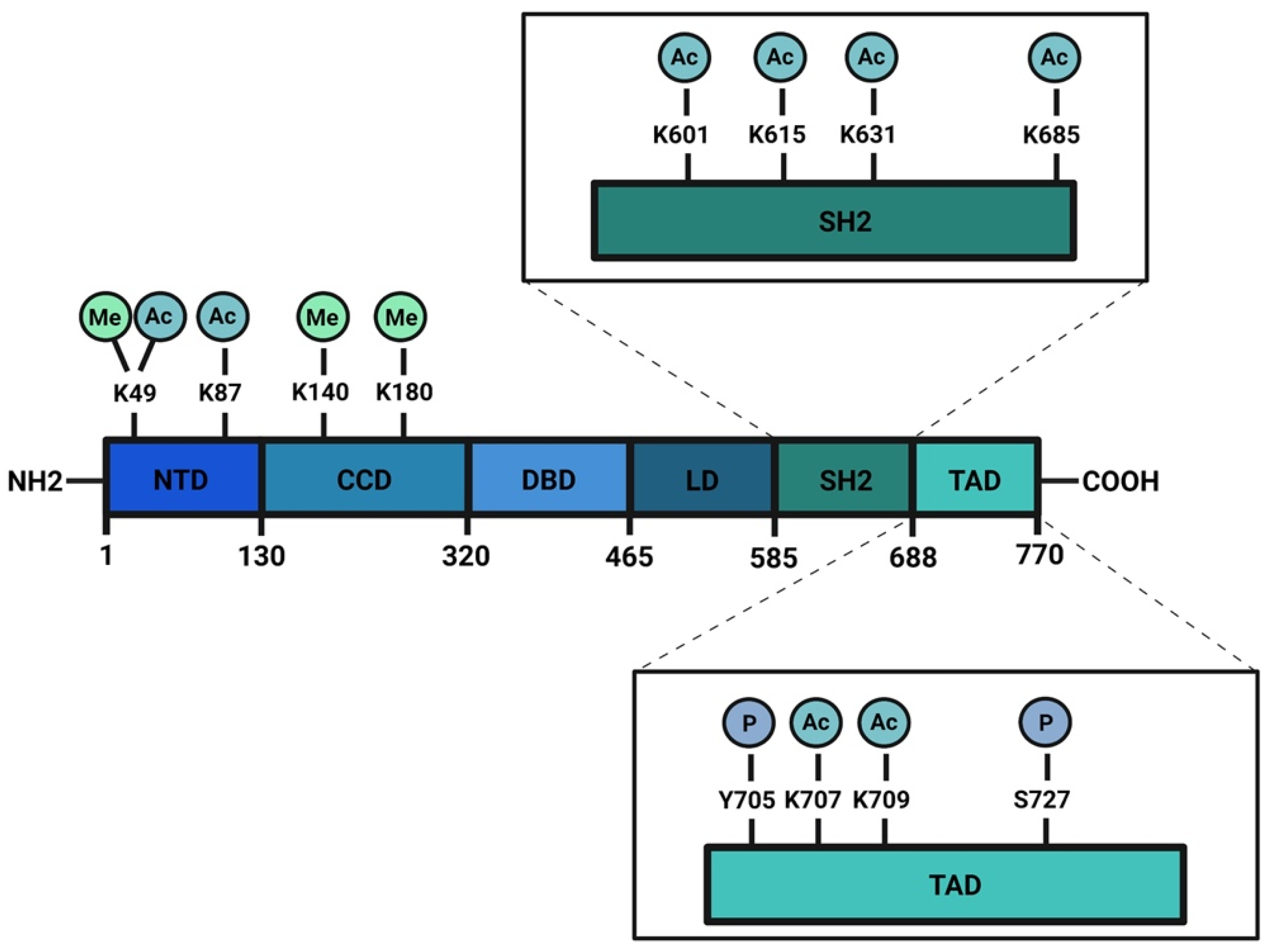{kind=link}
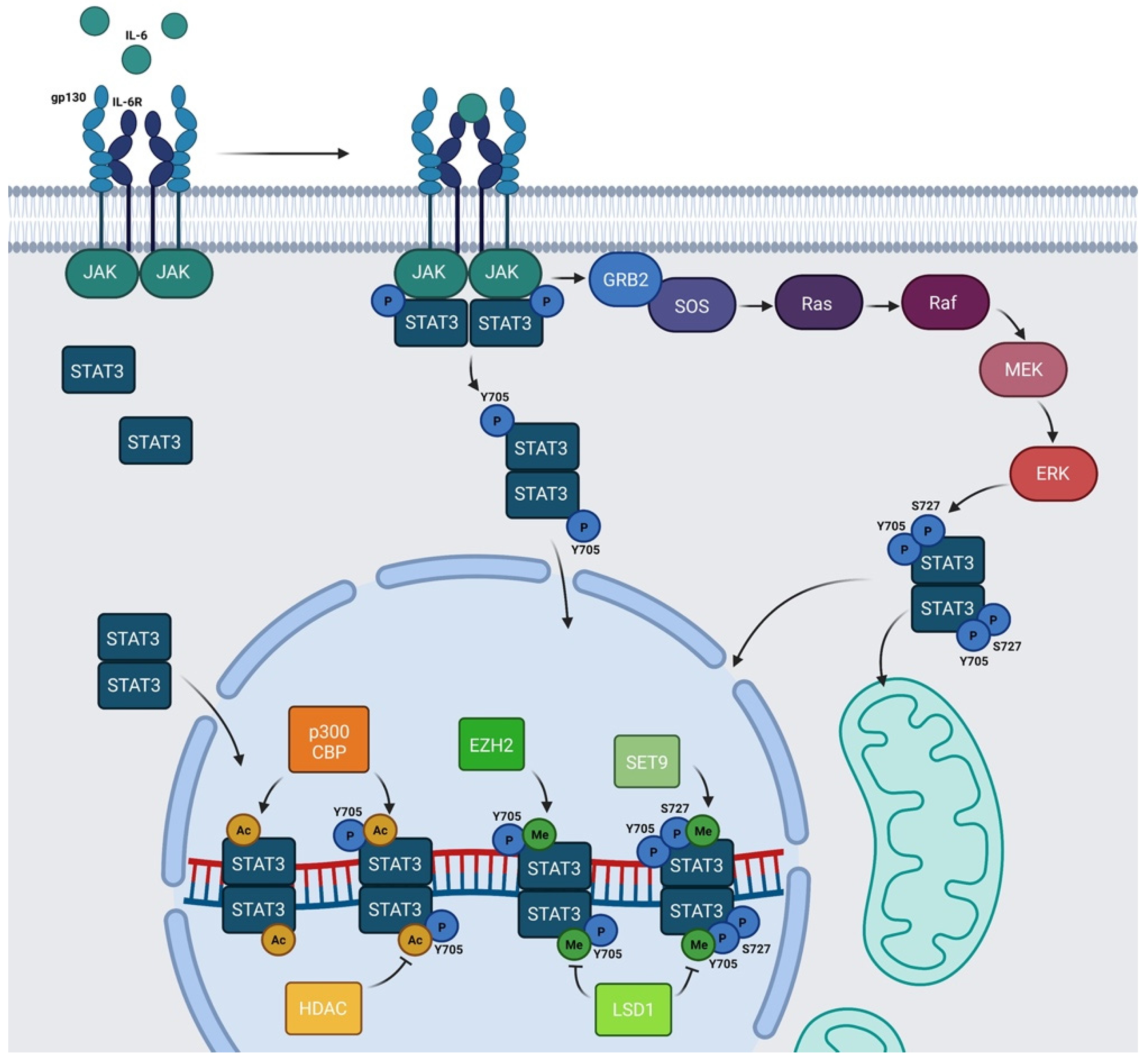{kind=link}
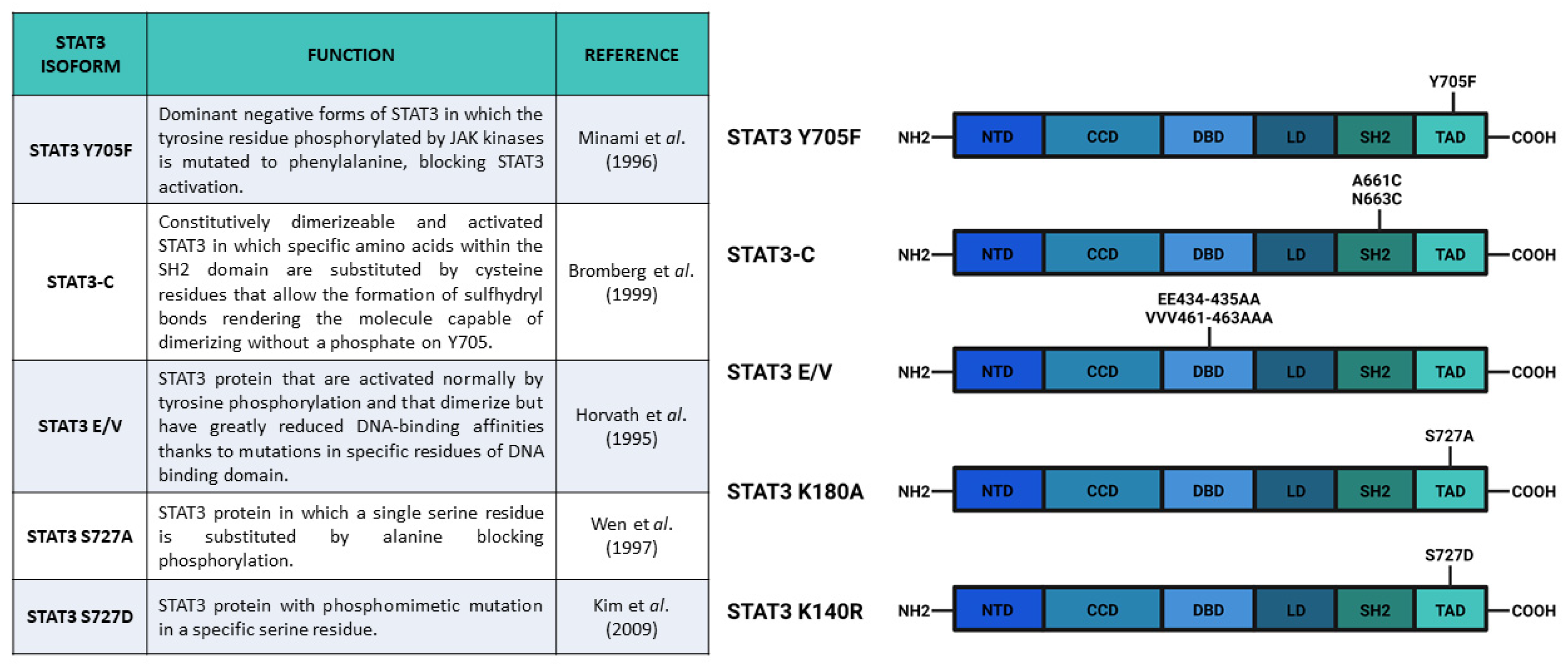{kind=link}
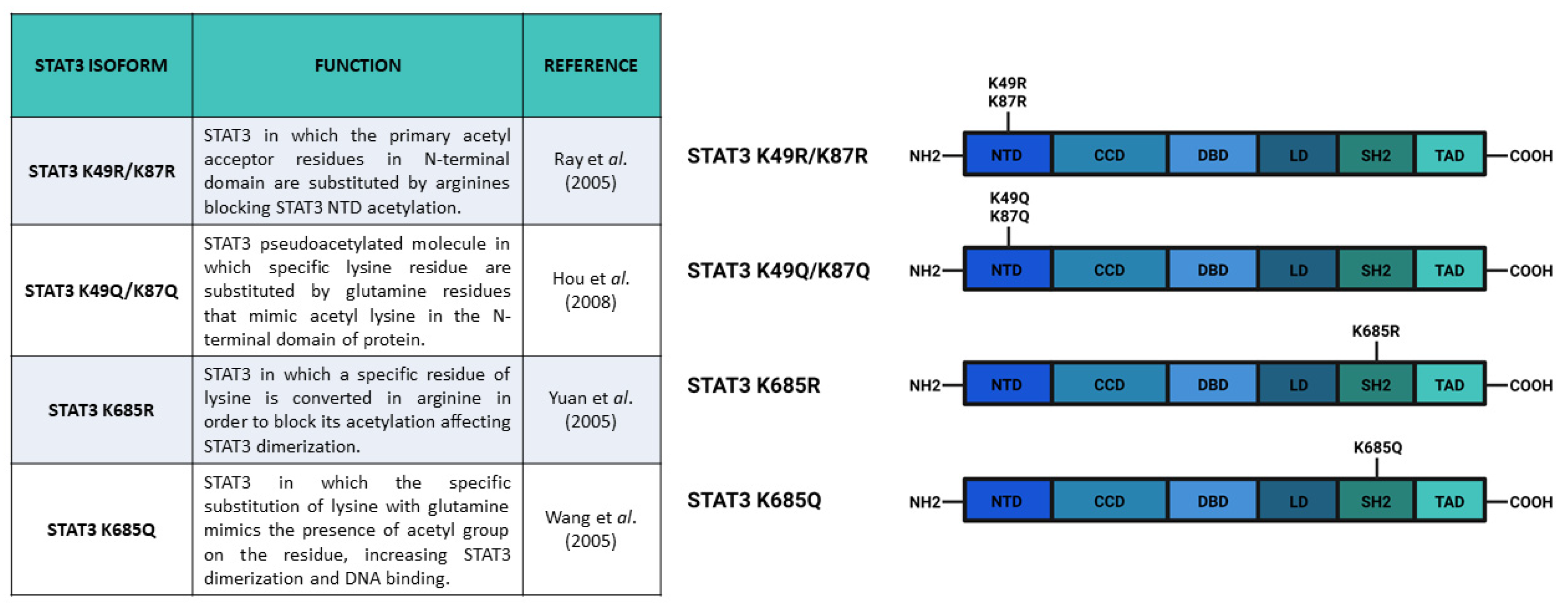{kind=link}
{kind=link}
Abstract
1. Introduction
2. Phosphorylation
3. Acetylation
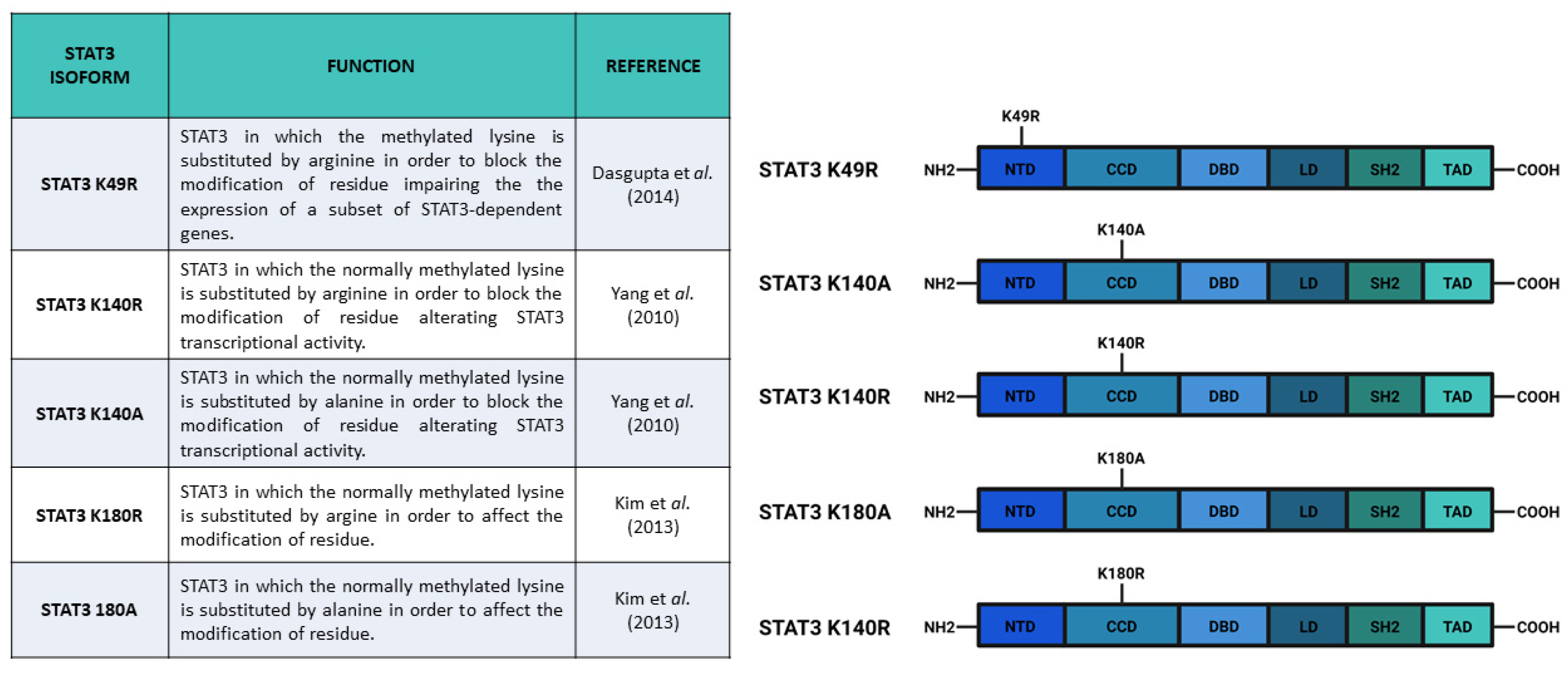
4. Methylation
5. Discussion and Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Guanizo, A.C.; Fernando, C.D.; Garama, D.J.; Gough, D.J. STAT3: A multifaceted oncoprotein. Growth Factors 2018, 36, 1–14. [Google Scholar] [CrossRef]
- Zhang, X.; Darnell, J.E., Jr. Functional importance of Stat3 tetramerization in activation of the alpha 2-macroglobulin gene. J. Biol. Chem. 2001, 276, 33576–33581. [Google Scholar] [CrossRef]
- Wang, Y.; Levy, D.E. Comparative evolutionary genomics of the STAT family of transcription factors. JAKSTAT 2012, 1, 23–33. [Google Scholar] [CrossRef]
- Avalle, L.; Camporeale, A.; Camperi, A.; Poli, V. STAT3 in cancer: A double edged sword. Cytokine 2017, 98, 42–50. [Google Scholar] [CrossRef]
- Johnson, D.E.; O’Keefe, R.A.; Grandis, J.R. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat. Rev. Clin. Oncol. 2018, 15, 234–248. [Google Scholar] [CrossRef]
- Park, O.K.; Schaefer, T.S.; Nathans, D. In vitro activation of Stat3 by epidermal growth factor receptor kinase. Proc. Natl. Acad. Sci. USA 1996, 93, 13704–13708. [Google Scholar] [CrossRef]
- Chan, K.S.; Carbajal, S.; Kiguchi, K.; Clifford, J.; Sano, S.; DiGiovanni, J. Epidermal Growth Factor Receptor-Mediated Activation of Stat3 during Multistage Skin. Carcinog. Cancer Res. 2004, 7, 2382–2389. [Google Scholar] [CrossRef]
- Delgoffe, G.M.; Vignali, D.A.A. STAT heterodimers in immunity. JAKSTAT 2013, 2, e23060. [Google Scholar] [CrossRef]
- Betto, R.M.; Diamante, L.; Perrera, V.; Audano, M.; Rapelli, S.; Lauria, A.; Incarnato, D.; Arboit, M.; Pedretti, S.; Rigoni, G.; et al. Metabolic control of DNA methylation in naive pluripotent cells. Nat. Genet. 2021, 53, 215–229. [Google Scholar] [CrossRef] [PubMed]
- Dauer, D.J.; Ferraro, B.; Song, L.; Yu, B.; Mora, L.; Buettner, R.; Enkemann, S.; Jove, R.; Haura, E.B. Stat3 regulates genes common to both wound healing and cancer. Oncogene 2004, 24, 3397–3408. [Google Scholar] [CrossRef]
- Fan, Y.; Mao, R.; Yang, J. NF-κB and STAT3 signaling pathways collaboratively link inflammation to cancer. Protein Cell 2013, 4, 176–185. [Google Scholar] [CrossRef]
- Hillmer, E.J.; Zhang, H.; Li, H.S.; Watowich, S.S. STAT3 signaling in immunity. Cytokine Growth Factor Rev. 2016, 31, 1–15. [Google Scholar] [CrossRef]
- Xu, Y.S.; Liang, J.J.; Wang, Y.; Zhao, X.J.; Xu, L.; Xu, Y.; Zou, Q.C.; Zhang, J.M.; Tu, C.; Cui, Y.; et al. STAT3 Undergoes Acetylation-dependent Mitochondrial Translocation to Regulate Pyruvate Metabolism. Sci. Rep. 2016, 6, 39517. [Google Scholar] [CrossRef]
- Peron, M.; Dinarello, A.; Meneghetti, G.; Martorano, L.; Facchinello, N.; Vettori, A.; Licciardello, G.; Tiso, N.; Argenton, F. The stem-like Stat3-responsive cells of zebrafish intestine are Wnt/β-catenin dependent. Development 2020, 147, dev188987. [Google Scholar] [CrossRef]
- Huynh, J.; Chand, A.; Gough, D.; Ernst, M. Therapeutically exploiting STAT3 activity in cancer-using tissue repair as a road map. Nat. Rev. Cancer 2019, 19, 82–96. [Google Scholar] [CrossRef]
- Szczepanek, K.; Chen, Q.; Derecka, M.; Salloum, F.N.; Zhang, Q.; Szelag, M.; Cichy, J.; Kukreja, R.C.; Dulak, J.; Lesnefsky, E.J.; et al. Mitochondrial-targeted Signal Transducer and Activator of Transcription 3 (STAT3) Protects against Ischemia-induced Changes in the Electron Transport Chain and the Generation of Reactive Oxygen Species. J. Biol. Chem. 2011, 286, 29610–29620. [Google Scholar] [CrossRef]
- Wegrzyn, J.; Potla, R.; Chwae, Y.J.; Sepuri, N.B.; Zhang, Q.; Koeck, T.; Derecka, M.; Szczepanek, K.; Szelag, M.; Gornicka, A.; et al. Function of mitochondrial Stat3 in cellular respiration. Science 2009, 323, 793–797. [Google Scholar] [CrossRef] [PubMed]
- Peron, M.; Dinarello, A.; Meneghetti, G.; Martorano, L.; Betto, R.M.; Facchinello, N.; Tesoriere, A.; Tiso, N.; Martello, G.; Argenton, F. Y705 and S727 are required for mitochondrial import and transcriptional activities of STAT3 and regulate proliferation of embryonic and tissue stem cells. Development 2021. accepted. [Google Scholar]
- Carbognin, E.; Betto, R.M.; Soriano, M.E.; Smith, A.G.; Martello, G. Stat3 promotes mitochondrial transcription and oxidative respiration during maintenance and induction of naive pluripotency. EMBO J. 2016, 35, 618–634. [Google Scholar] [CrossRef] [PubMed]
- Avalle, L.; Camporeale, A.; Morciano, G.; Caroccia, N.; Ghetti, E.; Orecchia, V.; Viavattene, D.; Giorgi, C.; Pinton, P.; Poli, V. STAT3 localizes to the ER, acting as a gatekeeper for ER-mitochondrion Ca2+ fluxes and apoptotic responses. Cell Death Differ. 2019, 26, 932–942. [Google Scholar] [CrossRef] [PubMed]
- Meier, J.A.; Larner, A.C. Toward a new STATe: The role of STATs in mitochondrial function. Semin. Immunol. 2014, 26, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Do, N.N.; Willenborg, S.; Eckes, B.; Jüngst, C.; Sengle, G.; Zaucke, F.; Eming, S.A. Myeloid Cell-Restricted STAT3 Signaling Controls a Cell-Autonomous Antifibrotic Repair Program. J. Immunol. 2018, 201, 663–674. [Google Scholar] [CrossRef] [PubMed]
- Nguyen-Jackson, H.; Panopoulos, A.D.; Zhang, H.; Li, H.S.; Watowich, S.S. STAT3 controls the neutrophil migratory response to CXCR2 ligands by direct activation of G-CSF-induced CXCR2 expression and via modulation of CXCR2 signal transduction. Blood 2010, 115, 3354–3363. [Google Scholar] [CrossRef] [PubMed]
- Nguyen-Jackson, H.T.; Li, H.S.; Zhang, H.; Ohashi, E.; Watowich, S.S. G-CSF-activated STAT3 enhances production of the chemokine MIP-2 in bone marrow neutrophils. J. Leukoc. Biol. 2012, 92, 1215–1225. [Google Scholar] [CrossRef] [PubMed]
- Rozovski, U.; Harris, D.M.; Li, P.; Liu, Z.; Wu, J.Y.; Grgurevic, S.; Faderl, S.; Ferrajoli, A.; Wierda, W.G.; Martinez, M.; et al. At High Levels, Constitutively Activated STAT3 Induces Apoptosis of Chronic Lymphocytic Leukemia. Cells J. Immunol. 2016, 196, 4400–4409. [Google Scholar] [CrossRef]
- Tkach, M.; Rosemblit, C.; Rivas, M.A.; Proietti, C.J.; Díaz Flaqué, M.C.; Mercogliano, M.F.; Beguelin, W.; Maronna, E.; Guzmán, P.; Gercovich, F.G.; et al. p42/p44 MAPK-mediated Stat3Ser727 phosphorylation is required for progestin-induced full activation of Stat3 and breast cancer growth. Endocr. Relat. Cancer 2013, 20, 197–212. [Google Scholar] [CrossRef]
- Levy, D.E.; Lee, C.K. What does Stat3 do? J. Clin. Investig. 2002, 109, 1143–1148. [Google Scholar] [CrossRef]
- Giraud, S.; Bienvenu, F.; Avril, S.; Gascan, H.; Heery, D.M.; Coqueret, O. Functional interaction of STAT3 transcription factor with the coactivator NcoA/SRC1a. J. Biol. Chem. 2002, 277, 8004–8011. [Google Scholar] [CrossRef]
- Paulson, M.; Pisharody, S.; Pan, L.; Guadagno, S.; Mui, A.L.; Levy, D.E. Stat protein transactivation domains recruit p300/CBP through widely divergent sequences. J. Biol. Chem. 1999, 274, 25343–25349. [Google Scholar] [CrossRef]
- Boulton, T.G.; Zhong, Z.; Wen, Z.; Darnell, J.E., Jr.; Stahl, N.; Yancopoulos, G.D. STAT3 activation by cytokines utilizing gp130 and related transducers involves a secondary modification requiring an H7-sensitive kinase. Proc. Natl. Acad. Sci. USA 1995, 92, 6915–6919. [Google Scholar] [CrossRef]
- Zhang, X.; Blenis, J.; Li, H.C.; Schindler, C.; Chen-Kiang, S. Requirement of serine phosphorylation for formation of STAT-promoter complexes. Science 1995, 267, 1990–1994. [Google Scholar] [CrossRef]
- Wen, Z.; Zhong, Z.; Darnell, J.E., Jr. Maximal activation of transcription by Stat1 and Stat3 requires both tyrosine and serine phosphorylation. Cell 1995, 82, 241–250. [Google Scholar] [CrossRef]
- Horvath, C.M.; Wen, Z.; Darnell, J.E., Jr. A STAT protein domain that determines DNA sequence recognition suggests a novel DNA-binding domain. Genes Dev. 1995, 9, 984–994. [Google Scholar] [CrossRef]
- Decker, T.; Kovarik, P. Serine phosphorylation of STATs. Oncogene 2000, 19, 2628–2637. [Google Scholar] [CrossRef] [PubMed]
- Schuringa, J.J.; Wierenga, A.T.; Kruijer, W.; Vellenga, E. Constitutive Stat3, Tyr705, and Ser727 phosphorylation in acute myeloid leukemia cells caused by the autocrine secretion of interleukin-6. Blood 2000, 95, 3765–3770. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Baumann, H. The carboxyl-terminal region of STAT3 controls gene induction by the mouse haptoglobin promoter. J. Biol. Chem. 1997, 272, 14571–14579. [Google Scholar] [CrossRef]
- Chung, C.D.; Liao, J.; Liu, B.; Rao, X.; Jay, P.; Berta, P.; Shuai, K. Specific inhibition of Stat3 signal transduction by PIAS3. Science 1997, 278, 1803–1805. [Google Scholar] [CrossRef] [PubMed]
- Bromberg, J.F.; Horvath, C.M.; Besser, D.; Lathem, W.W.; Darnell, J.E., Jr. Stat3 activation is required for cellular transformation by v-src. Mol. Cell Biol 1998, 18, 2553–2558. [Google Scholar] [CrossRef] [PubMed]
- Turkson, J.; Bowman, T.; Garcia, R.; Caldenhoven, E.; De Groot, R.P.; Jove, R. Stat3 activation by Src induces specific gene regulation and is required for cell transformation. Mol. Cell Biol. 1998, 18, 2545–2552. [Google Scholar] [CrossRef]
- Shen, Y.; Schlessinger, K.; Zhu, X.; Meffre, E.; Quimby, F.; Levy, D.E.; Darnell, J.E., Jr. Essential role of STAT3 in postnatal survival and growth revealed by mice lacking STAT3 serine 727 phosphorylation. Mol. Cell Biol. 2004, 24, 407–419. [Google Scholar] [CrossRef]
- Hazan-Halevy, I.; Harris, D.; Liu, Z.; Liu, J.; Li, P.; Chen, X.; Shanker, S.; Ferrajoli, A.; Keating, M.J.; Estrov, Z. STAT3 is constitutively phosphorylated on serine 727 residues, binds DNA, and activates transcription in CLL cells. Blood 2010, 115, 2852–2863. [Google Scholar] [CrossRef] [PubMed]
- Proietti, C.; Salatino, M.; Rosemblit, C.; Carnevale, R.; Pecci, A.; Kornblihtt, A.R.; Molinolo, A.A.; Frahm, I.; Charreau, E.H.; Schillaci, R.; et al. Progestins induce transcriptional activation of signal transducer and activator of transcription 3 (Stat3) via a Jak- and Src-dependent mechanism in breast cancer cells. Mol. Cell Biol. 2005, 25, 4826–4840. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.; Yan, H.; Ye, S.; Tong, C.; Ying, Q.L. STAT3 phosphorylation at tyrosine 705 and serine 727 differentially regulates mouse ESC fates. Stem Cells 2014, 32, 1149–1160. [Google Scholar] [CrossRef] [PubMed]
- Aziz, M.H.; Hafeez, B.B.; Sand, J.M.; Pierce, D.B.; Aziz, S.W.; Dreckschmidt, N.E.; Verma, A.K. Protein kinase Cvarepsilon mediates Stat3Ser727 phosphorylation, Stat3-regulated gene expression, and cell invasion in various human cancer cell lines through integration with MAPK cascade (RAF-1, MEK1/2, and ERK1/2). Oncogene 2010, 29, 3100–3109. [Google Scholar] [CrossRef] [PubMed]
- Aziz, M.H.; Manoharan, H.T.; Church, D.R.; Dreckschmidt, N.E.; Zhong, W.; Oberley, T.D.; Wilding, G.; Verma, A.K. Protein kinase Cepsilon interacts with signal transducers and activators of transcription 3 (Stat3), phosphorylates Stat3Ser727, and regulates its constitutive activation in prostate cancer. Cancer Res. 2007, 67, 8828–8838. [Google Scholar] [CrossRef] [PubMed]
- Aziz, M.H.; Manoharan, H.T.; Sand, J.M.; Verma, A.K. Protein kinase Cepsilon interacts with Stat3 and regulates its activation that is essential for the development of skin cancer. Mol. Carcinog. 2007, 46, 646–653. [Google Scholar] [CrossRef]
- Aziz, M.H.; Manoharan, H.T.; Verma, A.K. Protein kinase C epsilon, which sensitizes skin to sun’s UV radiation-induced cutaneous damage and development of squamous cell carcinomas, associates with Stat3. Cancer Res. 2007, 67, 1385–13894. [Google Scholar] [CrossRef]
- Kim, M.; Morales, L.D.; Jang, I.S.; Cho, Y.Y.; Kim, D.J. Protein Tyrosine Phosphatases as Potential Regulators of STAT3 Signaling. Int. J. Mol. Sci. 2018, 19, 2708. [Google Scholar] [CrossRef]
- Kuusanmäki, H.; Dufva, O.; Parri, E.; van Adrichem, A.J.; Rajala, H.; Majumder, M.M.; Yadav, B.; Parsons, A.; Chan, W.C.; Wennerberg, K.; et al. Drug sensitivity profiling identifies potential therapies for lymphoproliferative disorders with overactive JAK/STAT3 signaling. Oncotarget 2017, 8, 97516–97527. [Google Scholar] [CrossRef]
- Avalle, L.; Poli, V. Nucleus, Mitochondrion, or Reticulum? STAT3 à La Carte. Int. J. Mol. Sci. 2018, 19, 2820. [Google Scholar] [CrossRef]
- Gough, D.J.; Corlett, A.; Schlessinger, K.; Wegrzyn, J.; Larner, A.C.; Levy, D.E. Mitochondrial STAT3 supports Ras-dependent oncogenic transformation. Science 2009, 324, 1713–1716. [Google Scholar] [CrossRef]
- Gough, D.J.; Koetz, L.; Levy, D.E. The MEK-ERK pathway is necessary for serine phosphorylation of mitochondrial STAT3 and Ras-mediated transformation. PLoS ONE 2013, 8, e83395. [Google Scholar] [CrossRef]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R., 3rd; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007, 316, 1160–1166. [Google Scholar] [CrossRef] [PubMed]
- Daub, H.; Olsen, J.V.; Bairlein, M.; Gnad, F.; Oppermann, F.S.; Körner, R.; Greff, Z.; Kéri, G.; Stemmann, O.; Mann, M. Kinase-selective enrichment enables quantitative phosphoproteomics of the kinome across the cell cycle. Mol. Cell 2008, 31, 438–448. [Google Scholar] [CrossRef] [PubMed]
- Dephoure, N.; Zhou, C.; Villén, J.; Beausoleil, S.A.; Bakalarski, C.E.; Elledge, S.J.; Gygi, S.P. A quantitative atlas of mitotic phosphorylation. Proc. Natl. Acad. Sci. USA 2008, 105, 10762–10767. [Google Scholar] [CrossRef] [PubMed]
- Old, W.M.; Shabb, J.B.; Houel, S.; Wang, H.; Couts, K.L.; Yen, C.Y.; Litman, E.S.; Croy, C.H.; Meyer-Arendt, K.; Miranda, J.G.; et al. Functional proteomics identifies targets of phosphorylation by B-Raf signaling in melanoma. Mol. Cell. 2009, 34, 115–131. [Google Scholar] [CrossRef] [PubMed]
- Voss, A.K.; Thomas, T. Histone Lysine and Genomic Targets of Histone Acetyltransferases in Mammals. Bioessays 2018, 40, e1800078. [Google Scholar] [CrossRef]
- Thiagarajan, D.; Vedantham, S.; Ananthakrishnan, R.; Schmidt, A.M.; Ramasamy, R. Mechanisms of transcription factor acetylation and consequences in hearts. Biochim. Biophys. Acta 2016, 1862, 2221–2231. [Google Scholar] [CrossRef]
- Glozak, M.A.; Sengupta, N.; Zhang, X.; Seto, E. Acetylation and deacetylation of non-histone proteins. Gene 2005, 363, 15–23. [Google Scholar] [CrossRef]
- Zhuang, S. Regulation of STAT signaling by acetylation. Cell Signal. 2013, 25, 1924–1931. [Google Scholar] [CrossRef] [PubMed]
- Wieczorek, M.; Ginter, T.; Brand, P.; Heinzel, T.; Krämer, O.H. Acetylation modulates the STAT signaling code. Cytokine Growth Factor Rev. 2012, 23, 293–305. [Google Scholar] [CrossRef]
- Ray, S.; Boldogh, I.; Brasier, A.R. STAT3 NH2-terminal acetylation is activated by the hepatic acute-phase response and required for IL-6 induction of angiotensinogen. Gastroenterology 2005, 129, 1616–1632. [Google Scholar] [CrossRef]
- Ray, S.; Lee, C.; Hou, T.; Boldogh, I.; Brasier, A.R. Requirement of histone deacetylase1 (HDAC1) in signal transducer and activator of transcription 3 (STAT3) nucleocytoplasmic distribution. Nucleic Acids Res. 2008, 36, 4510–4520. [Google Scholar] [CrossRef]
- Ma, L.; Huang, C.; Wang, X.J.; Xin, D.E.; Wang, L.S.; Zou, Q.C.; Zhang, Y.S.; Tan, M.D.; Wang, Y.M.; Zhao, T.C.; et al. Lysyl Oxidase 3 Is a Dual-Specificity Enzyme Involved in STAT3 Deacetylation and Deacetylimination Modulation. Mol. Cell 2017, 65, 296–309. [Google Scholar] [CrossRef]
- Wang, R.; Cherukuri, P.; Luo, J. Activation of Stat3 sequence-specific DNA binding and transcription by p300/CREB-binding protein-mediated acetylation. J. Biol. Chem. 2005, 280, 11528–11534. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.L.; Guan, Y.J.; Chatterjee, D.; Chin, Y.E. Stat3 dimerization regulated by reversible acetylation of a single lysine residue. Science 2005, 307, 269–273. [Google Scholar] [CrossRef]
- O’Shea, J.J.; Kanno, Y.; Chen, X.; Levy, D.E. Cell signaling. Stat acetylation--a key facet of cytokine signaling? Science 2005, 307, 217–218. [Google Scholar] [CrossRef] [PubMed]
- Génin, P.; Morin, P.; Civas, A. Impairment of interferon-induced IRF-7 gene expression due to inhibition of ISGF3 formation by trichostatin A. J. Virol. 2003, 77, 7113–7119. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Shankaranarayanan, P.; Chaitidis, P.; Kühn, H.; Nigam, S. Acetylation by histone acetyltransferase CREB-binding protein/p300 of STAT6 is required for transcriptional activation of the 15-lipoxygenase-1 gene. J. Biol. Chem. 2001, 276, 42753–42760. [Google Scholar] [CrossRef]
- McDonald, C.; Reich, N.C. Cooperation of the transcriptional coactivators CBP and p300 with Stat6. J. Interferon Cytokine Res. 1999, 19, 711–722. [Google Scholar] [CrossRef]
- Belo, Y.; Mielko, Z.; Nudelman, H.; Afek, A.; Ben-David, O.; Shahar, A.; Zarivach, R.; Gordan, R.; Arbely, E. Unexpected implications of STAT3 acetylation revealed by genetic encoding of acetyl-lysine. Biochim. Biophys. Acta Gen. Subj. 2019, 1863, 1343–1350. [Google Scholar] [CrossRef] [PubMed]
- Garama, D.J.; Harris, T.J.; White, C.L.; Rossello, F.J.; Abdul-Hay, M.; Gough, D.J.; Levy, D.E. A Synthetic Lethal Interaction between Glutathione Synthesis and Mitochondrial Reactive Oxygen Species Provides a Tumor-Specific Vulnerability Dependent on STAT3. Mol. Cell Biol. 2015, 35, 3646–3656. [Google Scholar] [CrossRef]
- Yang, J.; Liao, X.; Agarwal, M.K.; Barnes, L.; Auron, P.E.; Stark, G.R. Unphosphorylated STAT3 accumulates in response to IL-6 and activates transcription by binding to NFkappaB. Genes Dev. 2007, 21, 1396–1408. [Google Scholar] [CrossRef] [PubMed]
- Yue, H.; Li, W.; Desnoyer, R.; Karnik, S.S. Role of nuclear unphosphorylated STAT3 in angiotensin II type 1 receptor-induced cardiac hypertrophy. Cardiovasc. Res. 2010, 85, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, M.; Unal, H.; Willard, B.; Yang, J.; Karnik, S.S.; Stark, G.R. Critical role for lysine 685 in gene expression mediated by transcription factor unphosphorylated STAT3. J. Biol. Chem. 2014, 289, 30763–30771. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Zhang, P.; Herrmann, A.; Yang, C.; Xin, H.; Wang, Z.; Hoon, D.S.; Forman, S.J.; Jove, R.; Riggs, A.D.; et al. Acetylated STAT3 is crucial for methylation of tumor-suppressor gene promoters and inhibition by resveratrol results in demethylation. Proc. Natl. Acad. Sci. USA 2012, 109, 7765–7769. [Google Scholar] [CrossRef]
- Gupta, M.; Han, J.J.; Stenson, M.; Wellik, L.; Witzig, T.E. Regulation of STAT3 by histone deacetylase-3 in diffuse large B-cell lymphoma: Implications for therapy. Leukemia 2012, 26, 1356–1364. [Google Scholar] [CrossRef]
- Sun, Y.; Chin, Y.E.; Weisiger, E.; Malter, C.; Tawara, I.; Toubai, T.; Gatza, E.; Mascagni, P.; Dinarello, C.A.; Reddy, P. Cutting edge: Negative regulation of dendritic cells through acetylation of the nonhistone protein STAT-3. J. Immunol. 2009, 182, 5899–5903. [Google Scholar] [CrossRef]
- Blaskovich, M.A.; Sun, J.; Cantor, A.; Turkson, J.; Jove, R.; Sebti, S.M. Discovery of JSI-124 (cucurbitacin I), a selective Janus kinase/signal transducer and activator of transcription 3 signaling pathway inhibitor with potent antitumor activity against human and murine cancer cells in mice. Cancer Res. 2003, 63, 1270–1279. [Google Scholar] [PubMed]
- Sethi, G.; Chatterjee, S.; Rajendran, P.; Li, F.; Shanmugam, M.K.; Wong, K.F.; Kumar, A.P.; Senapati, P.; Behera, A.K.; Hui, K.M.; et al. Inhibition of STAT3 dimerization and acetylation by garcinol suppresses the growth of human hepatocellular carcinoma in vitro and in vivo. Mol. Cancer 2014, 13, 66. [Google Scholar] [CrossRef]
- Cocchiola, R.; Rubini, E.; Altieri, F.; Chichiarelli, S.; Paglia, G.; Romaniello, D.; Carissimi, S.; Giorgi, A.; Giamogante, F.; Macone, A.; et al. STAT3 Post-Translational Modifications Drive Cellular Signaling Pathways in Prostate Cancer Cells. Int. J. Mol. Sci. 2019, 20, 1815. [Google Scholar] [CrossRef]
- Dasgupta, M.; Dermawan, J.K.; Willard, B.; Stark, G.R. STAT3-driven transcription depends upon the dimethylation of K49 by EZH2. Proc. Natl. Acad. Sci. USA 2015, 112, 3985–3990. [Google Scholar] [CrossRef]
- Kim, E.; Kim, M.; Woo, D.H.; Shin, Y.; Shin, J.; Chang, N.; Oh, Y.T.; Kim, H.; Rheey, J.; Nakano, I.; et al. Phosphorylation of EZH2 activates STAT3 signaling via STAT3 methylation and promotes tumorigenicity of glioblastoma stem-like cells. Cancer Cell 2013, 23, 839–852. [Google Scholar] [CrossRef] [PubMed]
- Knutson, S.K.; Wigle, T.J.; Warholic, N.M.; Sneeringer, C.J.; Allain, C.J.; Klaus, C.R.; Sacks, J.D.; Raimondi, A.; Majer, C.R.; Song, J.; et al. A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells. Nat. Chem. Biol. 2012, 8, 890–896. [Google Scholar] [CrossRef] [PubMed]
- McCabe, M.T.; Ott, H.M.; Ganji, G.; Korenchuk, S.; Thompson, C.; Van Aller, G.S.; Liu, Y.; Graves, A.P.; Della Pietra, A., 3rd; Diaz, E.; et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 2012, 492, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Qi, W.; Chan, H.; Teng, L.; Li, L.; Chuai, S.; Zhang, R.; Zeng, J.; Li, M.; Fan, H.; Lin, Y.; et al. Selective inhibition of Ezh2 by a small molecule inhibitor blocks tumor cells proliferation. Proc. Natl. Acad. Sci. USA 2012, 109, 21360–21365. [Google Scholar] [CrossRef] [PubMed]
- Loreti, M.; Shi, D.L.; Carron, C. The regulatory proteins DSCR6 and Ezh2 oppositely regulate Stat3 transcriptional activity in mesoderm patterning during Xenopus development. J. Biol. Chem. 2020, 295, 2724–2735. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Huang, J.; Dasgupta, M.; Sears, N.; Miyagi, M.; Wang, B.; Chance, M.R.; Chen, X.; Du, Y.; Wang, Y.; et al. Reversible methylation of promoter-bound STAT3 by histone-modifying enzymes. Proc. Natl. Acad. Sci. USA 2010, 107, 21499–21504. [Google Scholar] [CrossRef]
- Yang, J.; Chatterjee-Kishore, M.; Staugaitis, S.M.; Nguyen, H.; Schlessinger, K.; Levy, D.E.; Stark, G.R. Novel roles of unphosphorylated STAT3 in oncogenesis and transcriptional regulation. Cancer Res. 2005, 65, 939–947. [Google Scholar]
- Langlais, D.; Couture, C.; Balsalobre, A.; Drouin, J. The Stat3/GR interaction code: Predictive value of direct/indirect DNA recruitment for transcription outcome. Mol. Cell 2012, 47, 38–49. [Google Scholar] [CrossRef]
- Thaper, D.; Vahid, S.; Kaur, R.; Kumar, S.; Nouruzi, S.; Bishop, J.L.; Johansson, M.; Zoubeidi, A. Galiellalactone inhibits the STAT3/AR signaling axis and suppresses Enzalutamide-resistant Prostate Cancer. Sci. Rep. 2018, 8, 17307. [Google Scholar] [CrossRef] [PubMed]
- Pawlus, M.R.; Wang, L.; Hu, C.J. STAT3 and HIF1α cooperatively activate HIF1 target genes in MDA-MB-231 and RCC4 cells. Oncogene 2014, 33, 1670–1679. [Google Scholar] [CrossRef] [PubMed]
- Ji, Z.; He, L.; Regev, A.; Struhl, K. Inflammatory regulatory network mediated by the joint action of NF-kB, STAT3, and AP-1 factors is involved in many human cancers. Proc. Natl. Acad. Sci. USA 2019, 116, 9453–9462. [Google Scholar] [CrossRef] [PubMed]
- Laudisi, F.; Cherubini, F.; Monteleone, G.; Stolfi, C. STAT3 Interactors as Potential Therapeutic Targets for Cancer Treatment. Int. J. Mol. Sci. 2018, 19, 1787. [Google Scholar] [CrossRef]
- Liu, K.; Petree, C.; Requena, T.; Varshney, P.; Varshney, G.K. Expanding the CRISPR Toolbox in Zebrafish for Studying Development and Disease. Front. Cell Dev. Biol. 2019, 7, 13. [Google Scholar] [CrossRef]
- Grillo, M.; Palmer, C.; Holmes, N.; Sang, F.; Larner, A.C.; Bhosale, R.; Shaw, P.E. Stat3 oxidation-dependent regulation of gene expression impacts on developmental processes and involves cooperation with Hif-1α. PLoS ONE 2020, 12, e0244255. [Google Scholar]
- Meier, J.A.; Hyun, M.; Cantwell, M.; Raza, A.; Mertens, C.; Raje, V.; Sisler, J.; Tracy, E.; Torres-Odio, S.; Gispert, S.; et al. Stress-induced dynamic regulation of mitochondrial STAT3 and its association with cyclophilin D reduce mitochondrial ROS production. Sci. Signal. 2017, 10, eaag2588. [Google Scholar] [CrossRef]
- Aggarwal, V.; Tuli, H.S.; Varol, A.; Thakral, F.; Yerer, M.B.; Sak, K.; Varol, M.; Jain, A.; Khan, M.A.; Sethi, G. Role of Reactive Oxygen Species in Cancer Progression: Molecular Mechanisms and Recent Advancements. Biomolecules 2019, 9, 735. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tesoriere, A.; Dinarello, A.; Argenton, F. The Roles of Post-Translational Modifications in STAT3 Biological Activities and Functions. Biomedicines 2021, 9, 956. https://doi.org/10.3390/biomedicines9080956
Tesoriere A, Dinarello A, Argenton F. The Roles of Post-Translational Modifications in STAT3 Biological Activities and Functions. Biomedicines. 2021; 9(8):956. https://doi.org/10.3390/biomedicines9080956
Chicago/Turabian StyleTesoriere, Annachiara, Alberto Dinarello, and Francesco Argenton. 2021. "The Roles of Post-Translational Modifications in STAT3 Biological Activities and Functions" Biomedicines 9, no. 8: 956. https://doi.org/10.3390/biomedicines9080956
APA StyleTesoriere, A., Dinarello, A., & Argenton, F. (2021). The Roles of Post-Translational Modifications in STAT3 Biological Activities and Functions. Biomedicines, 9(8), 956. https://doi.org/10.3390/biomedicines9080956