
Synthesis and Biological Evaluation of Hydroxylated Monocarbonyl Curcumin Derivatives as Potential Inducers of Neprilysin Activity

 ,
,  , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. General
2.2. Synthesis of Compounds
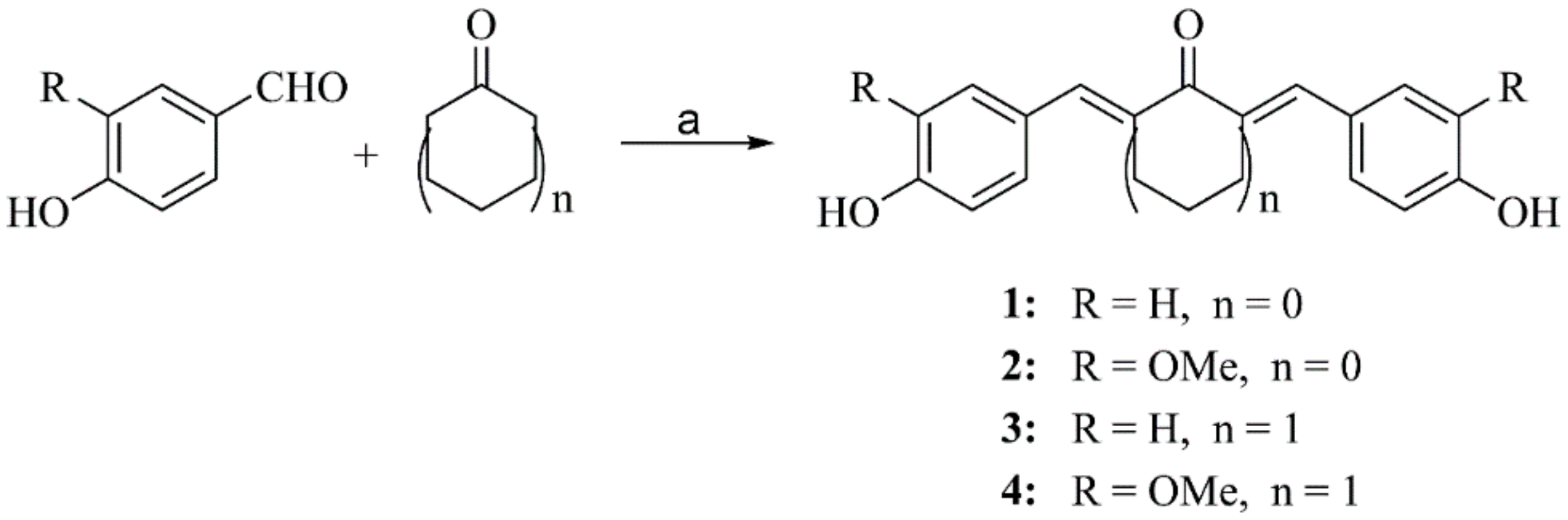
2.2.1. General Procedure for Preparation of Compounds 1–4
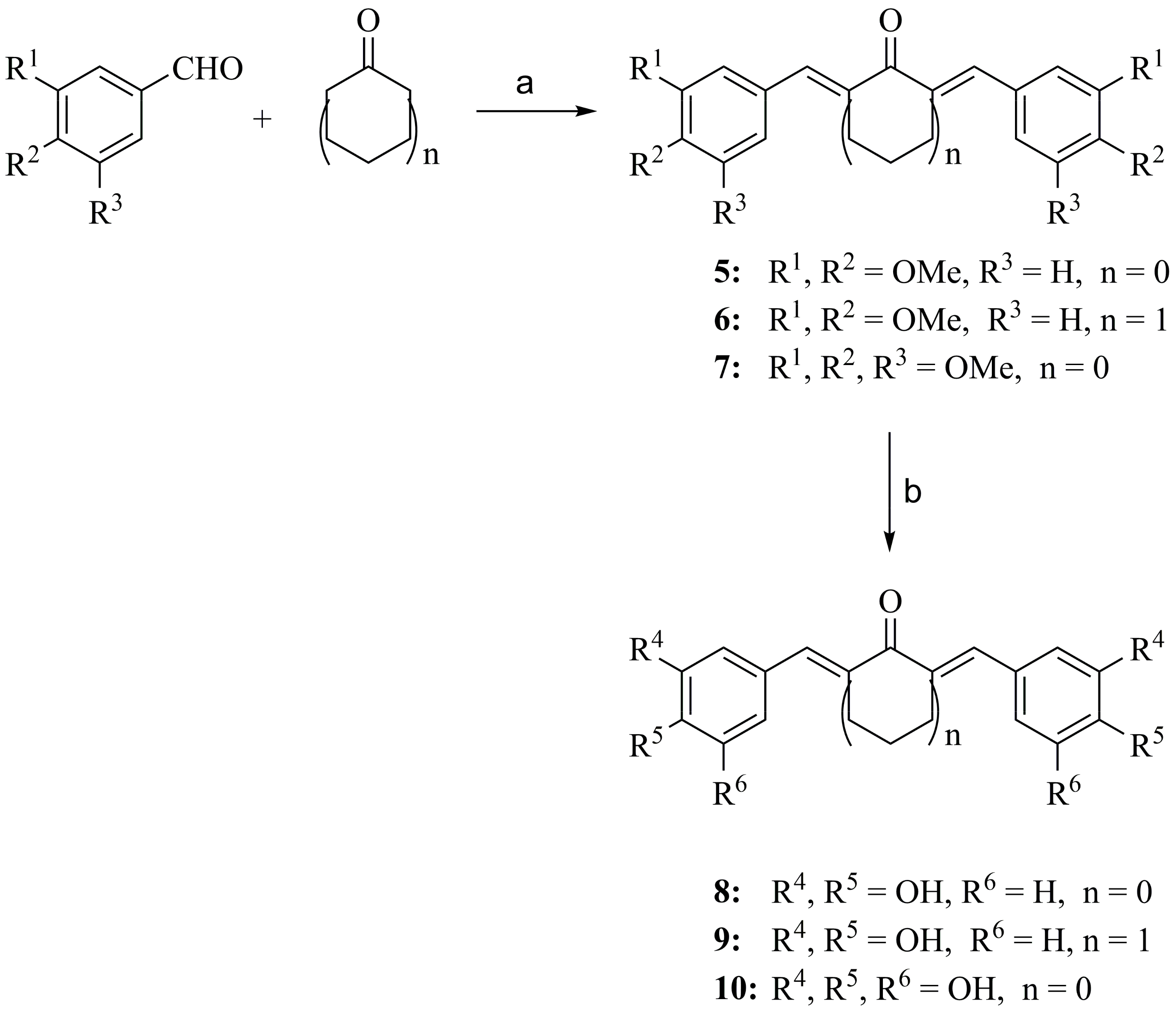
2.2.2. General Procedure for Preparation of Compounds 8–10
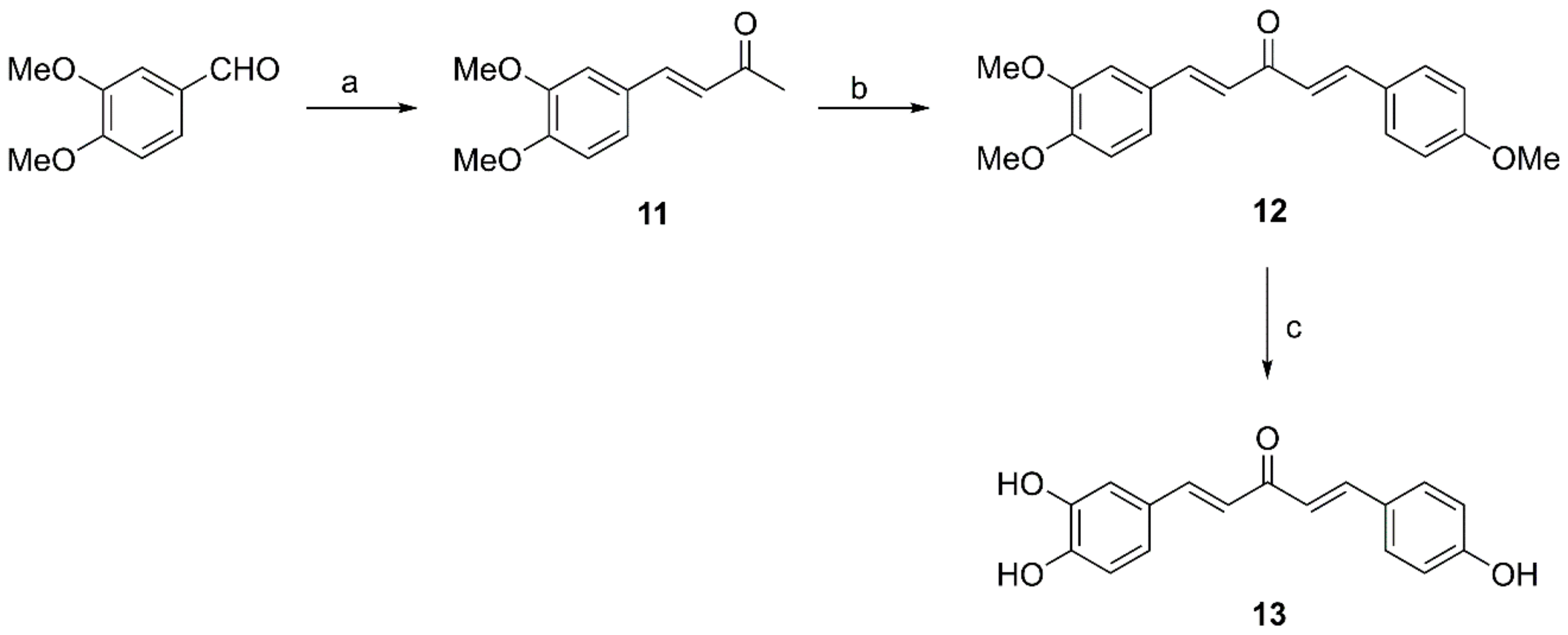
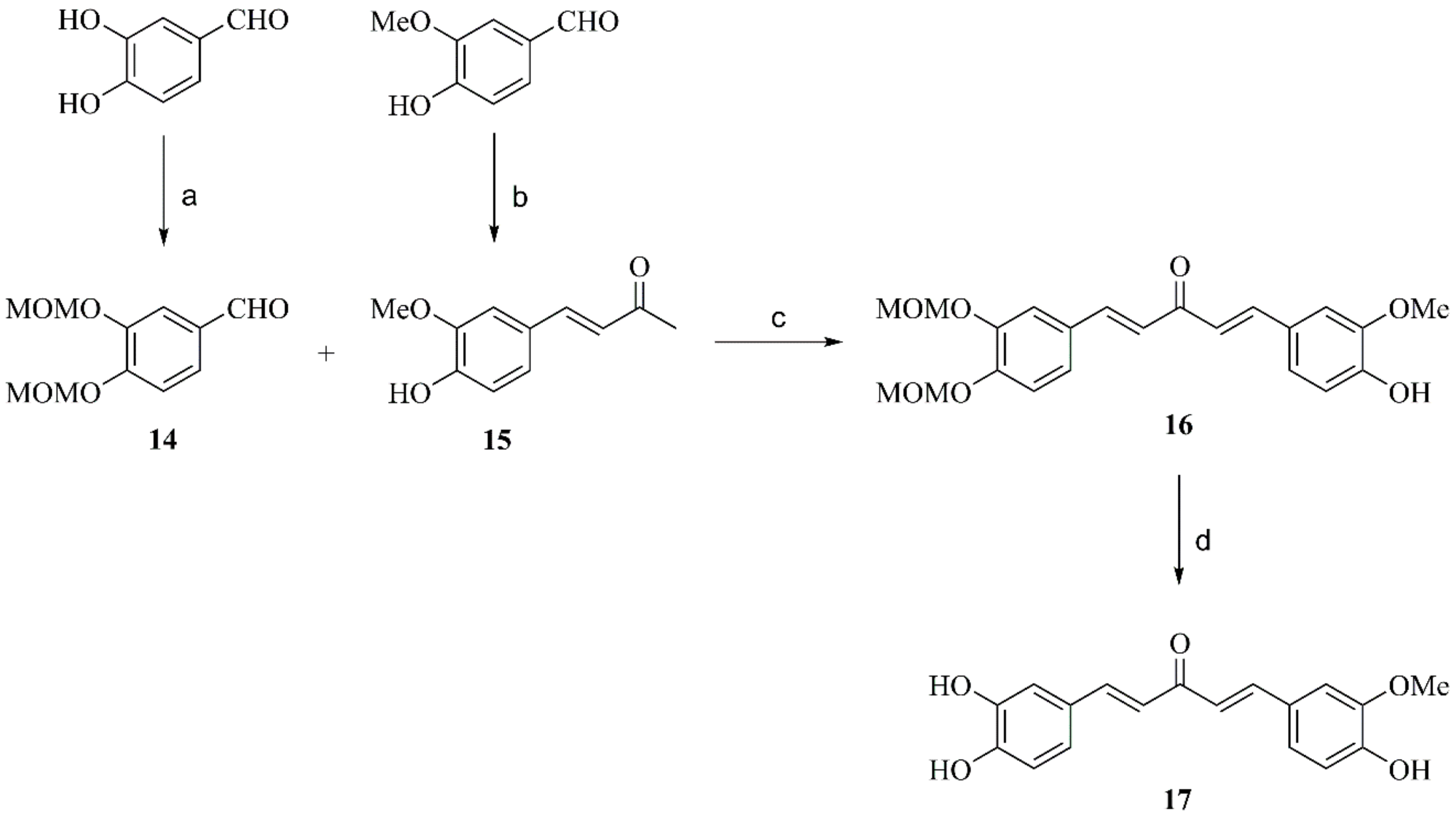
2.2.3. General Procedure for Preparation of Compounds 11, 15 and 18
2.2.4. Preparation of Compound 12
2.2.5. Preparation of Compound 13
2.2.6. Preparation of Compound 14
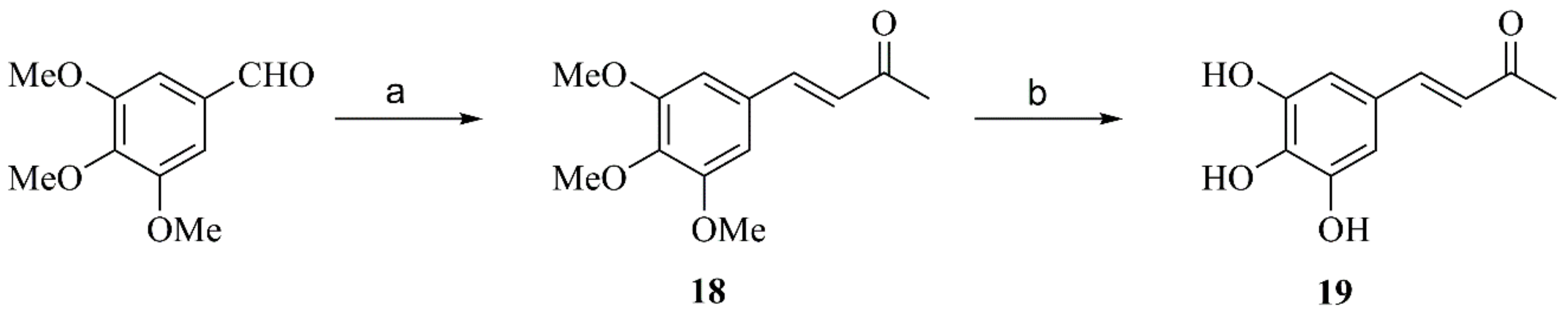
2.2.7. Preparation of Compound 17
2.2.8. Synthesis of Compound 19
2.3. Synthesis of qf-Aβ(1–7)C
2.4. Synthesis of qf-Aβ(12–16)AAC-EDANS
2.5. Biological Evaluation
2.5.1. Cell Culture
2.5.2. Neprilysin Aβ-Degrading Activity Assay Using qf-Aβ(1–7)C or qf-Aβ(12–16)AAC-EDANS as Substrate
2.6. Statistical Analysis
3. Results and Discussion
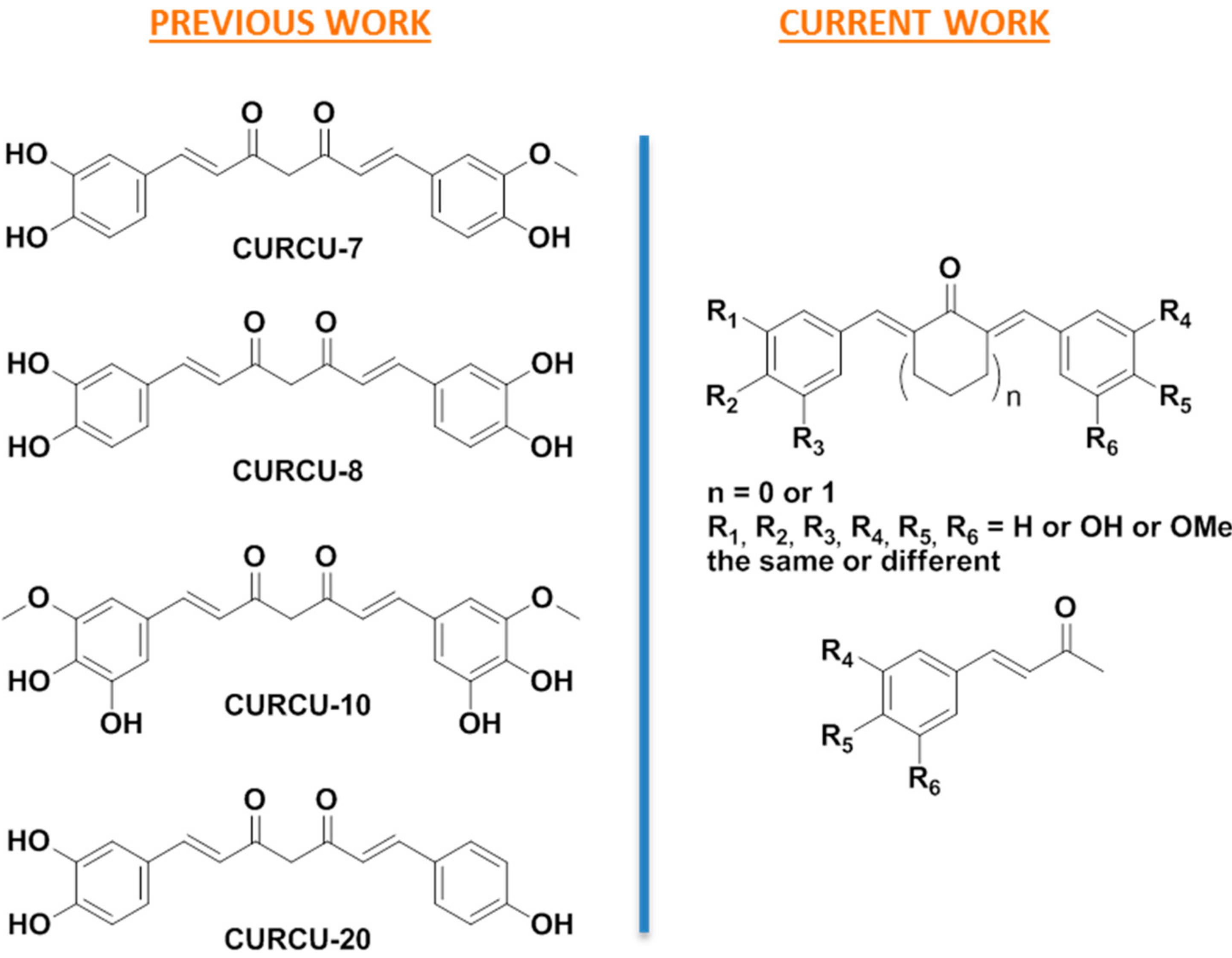
3.1. Design
3.2. Synthesis
3.3. Biological Evaluation
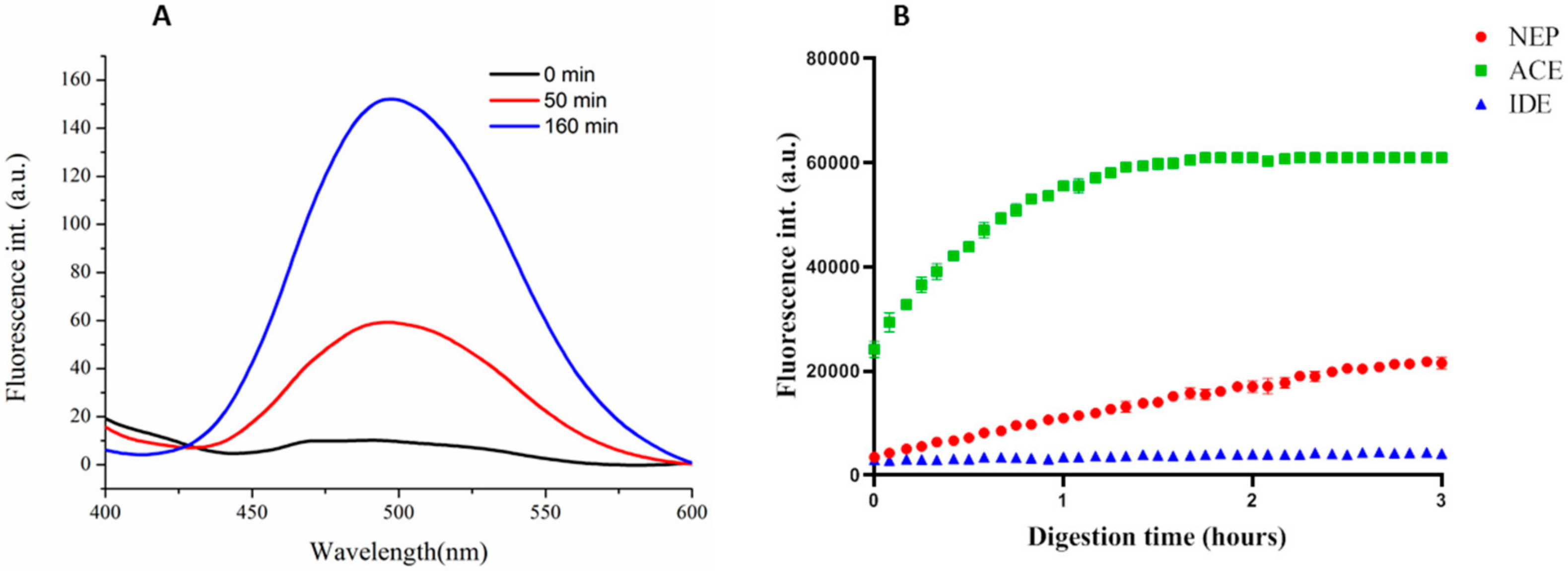
3.3.1. Synthesis and Specificity of Fluorescent Peptide NEP Substrates
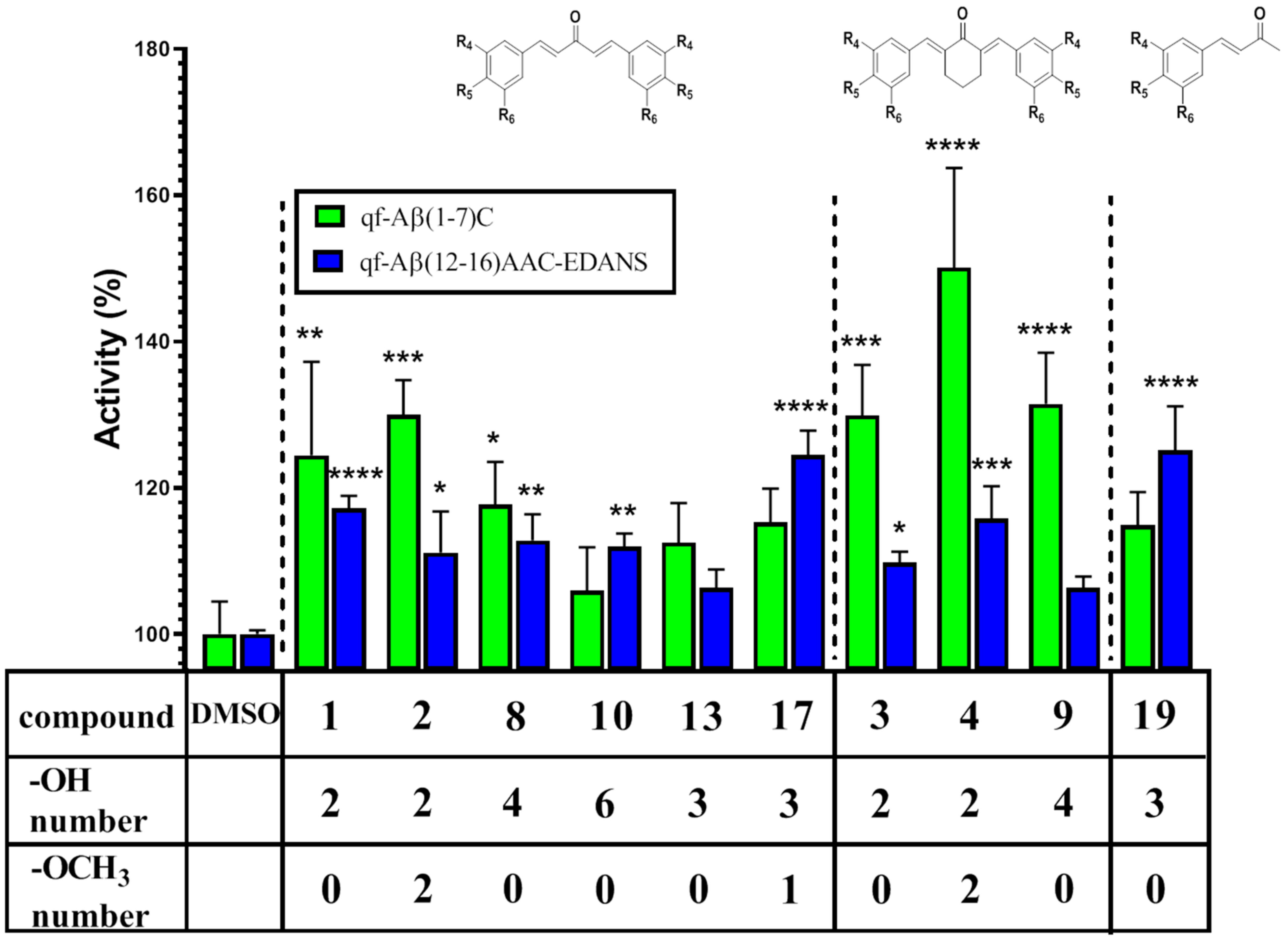
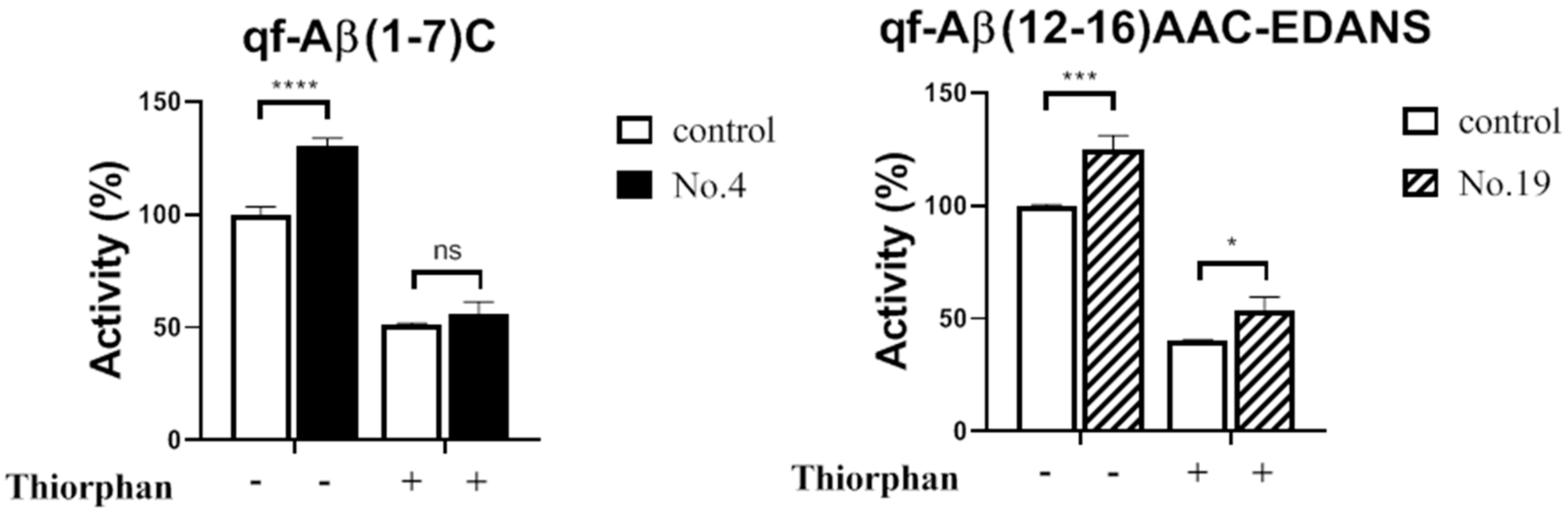
3.3.2. NEP Aβ-Degrading Activity in Cells and Inhibition Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Karran, E.; De Strooper, B. The amyloid cascade hypothesis: Are we poised for success or failure? J. Neurochem. 2016, 139, 237–252. [Google Scholar] [CrossRef]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef]
- Hardy, J. The amyloid hypothesis for Alzheimer’s disease: A critical reappraisal. J. Neurochem. 2009, 110, 1129–1134. [Google Scholar] [CrossRef]
- Rofo, F.; Ugur Yilmaz, C.; Metzendorf, N.; Gustavsson, T.; Beretta, C.; Erlandsson, A.; Sehlin, D.; Syvänen, S.; Nilsson, P.; Hultqvist, G. Enhanced neprilysin-mediated degradation of hippocampal Aβ42 with a somatostatin peptide that enters the brain. Theranostics 2021, 11, 789–804. [Google Scholar] [CrossRef] [PubMed]
- Mawuenyega, K.G.; Sigurdson, W.; Ovod, V.; Munsell, L.; Kasten, T.; Morris, J.C.; Yarasheski, K.E.; Bateman, R.J. Decreased clearance of CNS β-Amyloid in Alzheimer’s Disease. Science 2010, 330, 1774. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Gu, B.J.; Masters, C.L.; Wang, Y.J. A systemic view of Alzheimer disease—Insights from amyloid-β metabolism beyond the brain. Nat. Rev. Neurol. 2017, 13, 612–623. [Google Scholar] [CrossRef]
- Kurochkin, I.V.; Guarnera, E.; Berezovsky, I.N. Insulin degrading enzyme in the fight against Alzheimer’s disease. Trends Pharmacol. Sci. 2018, 39, 49–58. [Google Scholar] [CrossRef]
- Huang, J.Y.; Hafez, D.M.; James, B.D.; Bennett, D.A.; Marr, R.A. Altered NEP2 expression and activity in mild cognitive impairment and Alzheimer’s disease. J. Alzheimer’s Dis. 2012, 28, 433–441. [Google Scholar] [CrossRef]
- Huang, S.M.; Mouri, A.; Kokubo, H.; Nakajima, R.; Suemoto, T.; Higuchi, M.; Staufenbiel, M.; Noda, Y.; Yamaguchi, H.; Nabeshima, T.; et al. Neprilysin-sensitive synapse-associated amyloid-β peptide oligomers impair neuronal plasticity and cognitive function. J. Biol. Chem. 2006, 281, 17941–17951. [Google Scholar] [CrossRef] [PubMed]
- Hüttenrauch, M.; Baches, S.; Gerth, J.; Bayer, T.A.; Weggen, S.; Wirths, O. Neprilysin deficiency alters the neuropathological and behavioral phenotype in the 5XFAD mouse model of Alzheimer’s disease. J. Alzheimer’s Dis. 2015, 44, 1291–1302. [Google Scholar] [CrossRef]
- Farris, W.; Schütz, S.G.; Cirrito, J.R.; Shankar, G.M.; Sun, X.; George, A.; Leissring, A.; Walsh, D.M.; Qiu, W.Q.; Holtzman, D.M.; et al. Loss of neprilysin function promotes amyloid plaque formation and causes cerebral amyloid angiopathy. Am. J. Pathol. 2007, 171, 241–251. [Google Scholar] [CrossRef]
- Apelt, J.; Ach, K.; Schliebs, R. Aging-related downregulation of neprilysin, a putative β-amyloid-degrading enzyme, in transgenic Tg2576 Alzheimer-like mouse brain is accompanied by an astroglial upregulation in the vicinity of β-amyloid plaques. Neurosci. Lett. 2003, 339, 183–186. [Google Scholar] [CrossRef]
- Nalivaeva, N.N.; Turner, A.J. Targeting amyloid clearance in Alzheimer’s disease as a therapeutic strategy. Br. J. Pharmacol. 2018, 176, 3447–3463. [Google Scholar] [CrossRef] [PubMed]
- Iwata, N.; Tsubuki, S.; Takaki, Y.; Watanabe, K.; Sekiguchi, M.; Hosoki, E.; Kawashima-Morishima, M.; Lee, H.-J.; Hama, E.; Sekine-Aizawa, Y.; et al. Identification of the major Aβ1-42-degrading catabolic pathway in brain parenchyma: Suppression leads to biochemical and pathological deposition. Nat. Med. 2000, 6, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Sandoval, K.E.; Farr, S.A.; Banks, W.A.; Crider, A.M.; Morley, J.E.; Witt, K.A. Somatostatin receptor subtype-4 agonist NNC 26-9100 decreases extracellular and intracellular Aβ1-42 trimers. Eur. J. Pharmacol. 2012, 683, 116–124. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sandoval, K.E.; Farr, S.A.; Banks, W.A.; Crider, A.M.; Morley, J.E.; Witt, K.A. Somatostatin receptor subtype-4 agonist NNC 26-9100 mitigates the effect of soluble Aβ42 oligomers via a metalloproteinase-dependent mechanism. Brain Res. 2013, 1520, 145–156. [Google Scholar] [CrossRef][Green Version]
- Belyaev, N.D.; Nalivaeva, N.N.; Makova, N.Z.; Turner, A.J. Neprilysin gene expression requires binding of the amyloid precursor protein intracellular domain to its promoter: Implications for Alzheimer disease. EMBO Rep. 2009, 10, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Nalivaeva, N.N.; Belyaev, N.D.; Lewis, D.I.; Pickles, A.R.; Makova, N.Z.; Bagrova, D.I.; Dubrovskaya, N.M.; Plesneva, S.A.; Zhuravin, I.A.; Turner, A.J. Effect of sodium valproate administration on brain neprilysin expression and memory in rats. J. Mol. Neurosci. 2012, 46, 569–577. [Google Scholar] [CrossRef]
- Klein, C.; Roussel, G.; Brun, S.; Rusu, C.; Patte-Mensah, C.; Maitre, M.; Mensah-Nyagan, A.-G. 5-HIAA induces neprilysin to ameliorate pathophysiology and symptoms in a mouse model for Alzheimer’s disease. Acta Neuropathol. Commun. 2018, 6, 136. [Google Scholar] [CrossRef]
- Jha, N.K.; Jha, S.K.; Kumar, D.; Kejriwal, N.; Sharma, R.; Ambasta, R.K.; Kumar, P. Impact of Insulin Degrading Enzyme and Neprilysin in Alzheimer’s Disease Biology: Characterization of Putative Cognates for Therapeutic Applications. J. Alzheimer’s Dis. 2015, 48, 891–917. [Google Scholar] [CrossRef]
- Melzig, M.F.; Janka, M. Enhancement of neutral endopeptidase activity in SK-N-SH cells by green tea extract. Phytomedicine 2003, 10, 494–498. [Google Scholar] [CrossRef]
- El-Sayed, N.S.; Bayan, Y. Possible role of resveratrol targeting estradiol and neprilysin pathways in lipopolysaccharide model of Alzheimer disease. In GeNeDis; Springer: Cham, Switzerland, 2014; Volume 822, pp. 107–118. [Google Scholar] [CrossRef]
- Ayoub, S.; Melzig, M.F. Induction effect of apigenin, luteolin and vinpocetin on neutral endopeptidase (NEP) and angiotensin-converting enzyme (ACE) activity of SK-N-SH cell. Nat. Prod. Commun. 2006, 1, 633–639. [Google Scholar] [CrossRef]
- Ayoub, S.; Melzig, M.F. Influence of selected natural products on neutral endopeptidase activity and β-amyloid production in SKN-SH cells. Pharmaceut. Biol. 2008, 46, 425–432. [Google Scholar] [CrossRef]
- Chen, P.T.; Chen, Z.T.; Hou, W.C.; Yu, L.C.; Chen, R.P. Polyhydroxycurcuminoids but not curcumin upregulate neprilysin and can be applied to the prevention of Alzheimer’s disease. Sci. Rep. 2016, 6, 29760. [Google Scholar] [CrossRef]
- Endo, H.; Nikaido, Y.; Nakadate, M.; Ise, S.; Konno, H. Structure activity relationship study of curcumin analogues toward the amyloid-beta aggregation inhibitor. Bioorg. Med. Chem. Lett. 2014, 24, 5621–5626. [Google Scholar] [CrossRef]
- Matiadis, D.; Nowak, K.E.; Alexandratou, E.; Hatzidimitriou, A.; Sagnou, M.; Papadakis, R. Synthesis and (fluoro)solvatochromism of two 3-styryl-2-pyrazoline derivatives bearing benzoic acid moiety: A spectral, crystallographic and computational study. J. Mol. Liq. 2021, 331, 115737. [Google Scholar] [CrossRef]
- Matiadis, D.; Mavroidi, B.; Panagiotopoulou, A.; Methenitis, C.; Pelecanou, M.; Sagnou, M. (E)-(1-(4-Ethoxycarbonylphenyl)-5-(3,4-dimethoxyphenyl)-3-(3,4-dimethoxystyryl)-2-pyrazoline: Synthesis, characterization, DNA-interaction, and evaluation of activity against drug-resistant cell lines. Molbank 2020, 2020, M1114. [Google Scholar] [CrossRef]
- Matiadis, D.; Karagiaouri, M.; Mavroidi, B.; Nowak, K.E.; Katsipis, G.; Pelecanou, M.; Pantazaki, A.; Sagnou, M. Synthesis and antimicrobial evaluation of a pyrazoline-pyridine silver(I) complex: DNA-interaction and anti-biofilm activity. Biometals 2021, 34, 67–85. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.-Y.; Liu, R.-R.; Shao, W.-Y.; Mao, X.-P.; Ma, L.; Gu, L.-Q.; Huang, Z.-S.; Chan, A.S.C. α-Glucosidase inhibition of natural curcuminoids and curcumin analogs. Eur. J. Med. Chem. 2006, 41, 213–218. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.-Y.; Bao, Y.-D.; Liu, Z.; Qiao, W.; Ma, L.; Huang, Z.-S.; Gu, L.-Q.; Chan, A.S.C. Curcumin analogues as potent aldose reductase inhibitors. Arch. Pharm. Chem. Life Sci. 2006, 339, 123–128. [Google Scholar] [CrossRef]
- Tatsuzaki, J.; Bastow, K.F.; Nakagawa-Goto, K.; Nakamura, S.; Itokawa, H.; Lee, K.-H. Dehydrozingerone, chalcone, and isoeugenol analogues as in vitro anticancer agents. J. Nat. Prod. 2006, 69, 1445–14493. [Google Scholar] [CrossRef]
- Stern, T.; Rückbrod, S.; Czekelius, C.; Donner, C.; Brunner, H. A Selective and Benign Synthesis of Functionalized Benzalacetones via Mizoroki–Heck Reaction Using Aryldiazonium Salts. Adv. Synth. Catal. 2010, 352, 1983–1992. [Google Scholar] [CrossRef]
- Yamakoshi, H.; Ohori, H.; Kudo, C.; Sato, A.; Kanoh, N.; Ishioka, C.; Shibata, H.; Iwabuchi, Y. Structure-activity relationship of C5-curcuminoids and synthesis of their molecular probes thereof. Bioorg. Med. Chem. 2010, 18, 1083–1092. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Du, Z.; Xue, G.; Chen, Q.; Lu, Y.; Zheng, X.; Conney, A.H.; Zhang, K. Synthesis and biological evaluation of unsymmetrical curcumin analogues as tyrosinase inhibitors. Molecules 2013, 18, 3948–3961. [Google Scholar] [CrossRef]
- Nalivaeva, N.N.; Belyaev, N.D.; Zhuravin, I.A.; Turner, A.J. The Alzheimer’s amyloid-degrading peptidase, neprilysin: Can we control it? Int. J. Alzheimer’s Dis. 2012, 2012, 383796. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.-T.; Chen, C.-L.; Lin, L.T.-W.; Lo, C.-H.; Hu, C.-J.; Chen, R.P.-Y.; Wang, S.S.-S. Design of Peptide Substrate for Sensitively and Specifically Detecting Two Aβ-Degrading Enzymes: Neprilysin and Angiotensin-Converting Enzyme. PLoS ONE 2016, 11, e0153360. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.-T.; Liao, T.-Y.; Hu, C.-J.; Wu, S.-T.; Wang, S.S.-S.; Chen, R.P.-Y. A highly sensitive peptide substrate for detecting two Ab-degrading enzymes: Neprilysin and insulin-degrading enzyme. J. Neurosci. Methods 2010, 190, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Shetty, D.; Kim, Y.J.; Shim, H.; Snyder, J.P. Eliminating the heart from the curcumin molecule: Monocarbonyl curcumin mimics (MACs). Molecules 2015, 20, 249–292. [Google Scholar] [CrossRef]
- Hotsumi, M.; Tajiri, M.; Nikaido, Y.; Sato, T.; Makabe, K.; Konno, H. Design, synthesis, and evaluation of a water soluble C5-monoketone type curcumin analogue as a potent amyloid β aggregation inhibitor, Bioorg. Med. Chem. Lett. 2019, 29, 2157–2161. [Google Scholar] [CrossRef] [PubMed]
- Kudo, C.; Yamakoshi, H.; Sato, A.; Nanjo, H.; Ohori, H.; Ishioka, C.; Iwabuchi, Y.; Shibata, H. Synthesis of 86 species of 1,5-diaryl-3-oxo-1,4-pentadienes analogs of curcumin can yield a good lead in vivo. BMC Pharmacol. 2011, 11, 4. [Google Scholar] [CrossRef]
- Mandrekar-Colucci, S.; Karlo, J.C.; Landreth, G.E. Mechanisms underlying the rapid peroxisome proliferator-activated receptor-γ-mediated amyloid clearance and reversal of cognitive deficits in a murine model of Alzheimer’s disease. J. Neurosci. 2012, 32, 10117–10128. [Google Scholar] [CrossRef]
- Jiang, Q.; Lee, C.Y.D.; Mandrekar, S.; Wilkinson, B.; Cramer, P.; Zelcer, N.; Mann, K.; Lamb, B.; Willson, T.M.; Collins, J.L.; et al. ApoE promotes the proteolytic degradation of Aβ. Neuron 2008, 58, 681–693, 2008. [Google Scholar] [CrossRef]
- Wang, H.M.; Zhao, Y.X.; Zhang, S.; Liu, G.D.; Kang, W.Y.; Tang, H.D.; Ding, J.Q.; Chen, S.D. PPARγ agonist curcumin reduces the amyloid-β-stimulated inflammatory responses in primary astrocytes. J. Alzheimer’s Dis. 2010, 20, 1189–1199. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Wang, S.; Malter, J.S.; Wang, D.S. Effects of HNE-modification induced by Aβ on neprilysin expression and activity in SH-SY5Y cells. J. Neurochem. 2008, 108, 1072–1082. [Google Scholar] [CrossRef]
- Zheng, Q.-T.; Yang, Z.-H.; Yu, L.-Y.; Ren, Y.-Y.; Huang, Q.-X.; Liu, Q.; Ma, X.-Y.; Chen, Z.-K.; Wang, Z.-B.; Zheng, X. Synthesis and antioxidant activity of curcumin analogs. J. Asian Nat. Prod. Res. 2017, 19, 489–503. [Google Scholar] [CrossRef] [PubMed]
- Kumboonma, P.; Senawong, T.; Saenglee, S.; Senawong, G.; Somsakeesit, L.-O.; Yenjai, C.; Phaosiri, C. New histone deacetylase inhibitors and anticancer agents from Curcuma Longa. Med. Chem. Res. 2019, 28, 1773–1782. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | % Activity | |
|---|---|---|
| qf-Aβ(1–7)C | qf-Aβ(12–16)AAC-EDANS | |
| Control | 100 ± 4.47 | 100 ± 0.5 |
| 1 | 124.45 ± 12.78 | 117.17 ± 1.73 |
| 2 | 129.96 ± 4.76 | 111.13 ± 5.61 |
| 3 | 129.88 ± 6.92 | 109.78 ± 1.49 |
| 4 | 150.08 ± 13.63 | 115.78 ± 4.43 |
| 8 | 117.70 ± 5.85 | 112.78 ± 3.57 |
| 9 | 131.46 ± 7.02 | 106.35 ± 1.53 |
| 10 | 105.95 ± 5.91 | 111.92 ± 1.81 |
| 13 | 112.49 ± 5.41 | 106.30 ± 2.54 |
| 17 | 115.29 ± 4.60 | 124.48 ± 3.34 |
| 19 | 114.86 ± 4.57 | 125.10 ± 6.02 |
| CURCU-8 | 126.10 ± 5.39 a | 125.134 ± 8.22 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matiadis, D.; Ng, S.-T.; Chen, E.H.-L.; Nigianni, G.; Vidali, V.P.; Canko, A.; Chen, R.P.-Y.; Sagnou, M. Synthesis and Biological Evaluation of Hydroxylated Monocarbonyl Curcumin Derivatives as Potential Inducers of Neprilysin Activity. Biomedicines 2021, 9, 955. https://doi.org/10.3390/biomedicines9080955
Matiadis D, Ng S-T, Chen EH-L, Nigianni G, Vidali VP, Canko A, Chen RP-Y, Sagnou M. Synthesis and Biological Evaluation of Hydroxylated Monocarbonyl Curcumin Derivatives as Potential Inducers of Neprilysin Activity. Biomedicines. 2021; 9(8):955. https://doi.org/10.3390/biomedicines9080955
Chicago/Turabian StyleMatiadis, Dimitris, See-Ting Ng, Eric H.-L. Chen, Georgia Nigianni, Veroniki P. Vidali, Aleksander Canko, Rita P.-Y. Chen, and Marina Sagnou. 2021. "Synthesis and Biological Evaluation of Hydroxylated Monocarbonyl Curcumin Derivatives as Potential Inducers of Neprilysin Activity" Biomedicines 9, no. 8: 955. https://doi.org/10.3390/biomedicines9080955
APA StyleMatiadis, D., Ng, S.-T., Chen, E. H.-L., Nigianni, G., Vidali, V. P., Canko, A., Chen, R. P.-Y., & Sagnou, M. (2021). Synthesis and Biological Evaluation of Hydroxylated Monocarbonyl Curcumin Derivatives as Potential Inducers of Neprilysin Activity. Biomedicines, 9(8), 955. https://doi.org/10.3390/biomedicines9080955