
Enzymes in the Cholesterol Synthesis Pathway: Interactomics in the Cancer Context

 , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. TCGA Datasets Analysis
2.2. Interactomics Data Acquisition and Processing
2.3. Annotation and Enrichment Analysis
3. Results
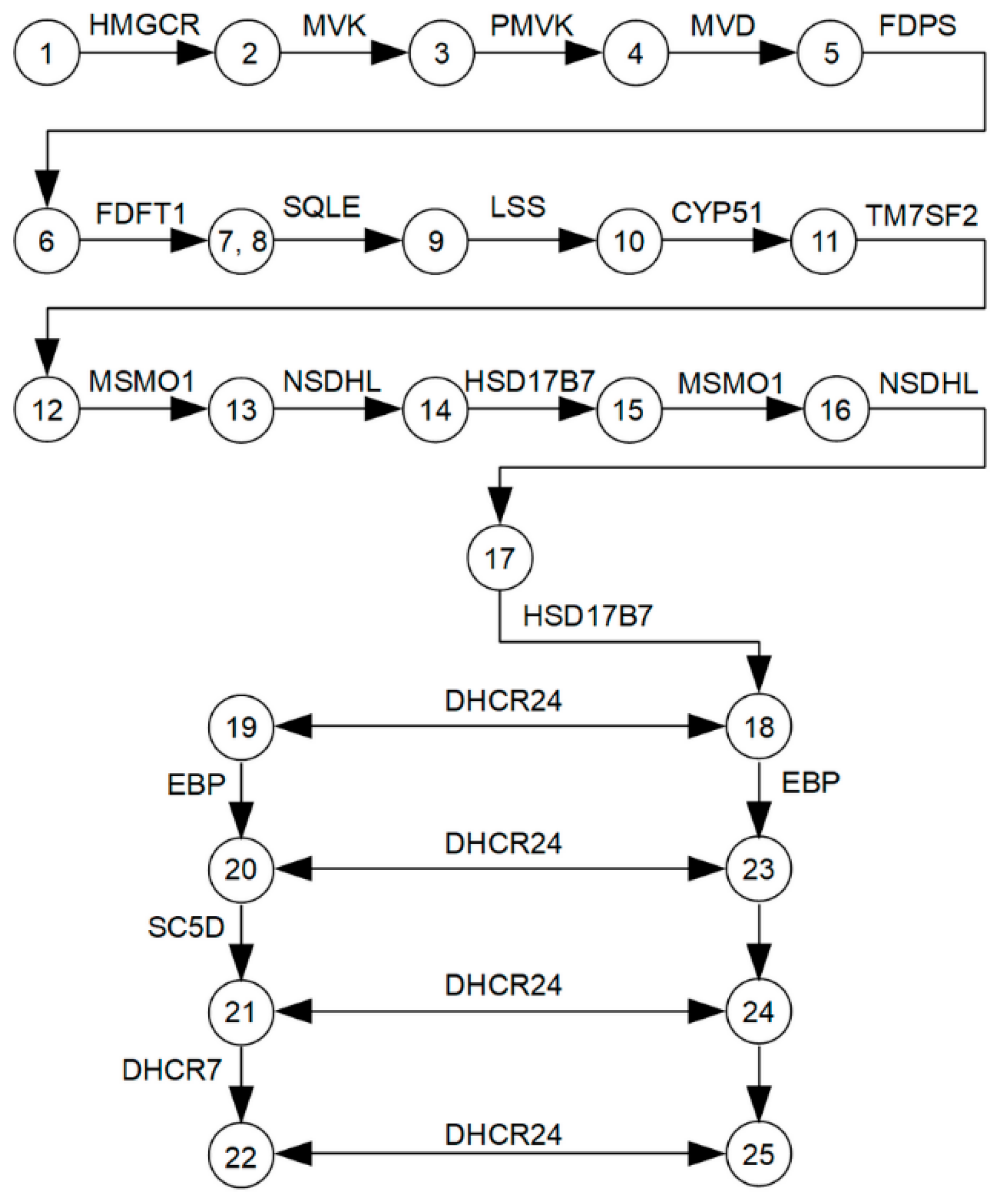
3.1. The Main Characteristics of Cholesterol Synthesis Enzymes
3.2. Transcriptomic Landscape of Genes, Encoding Cholesterol Synthesis Enzymes
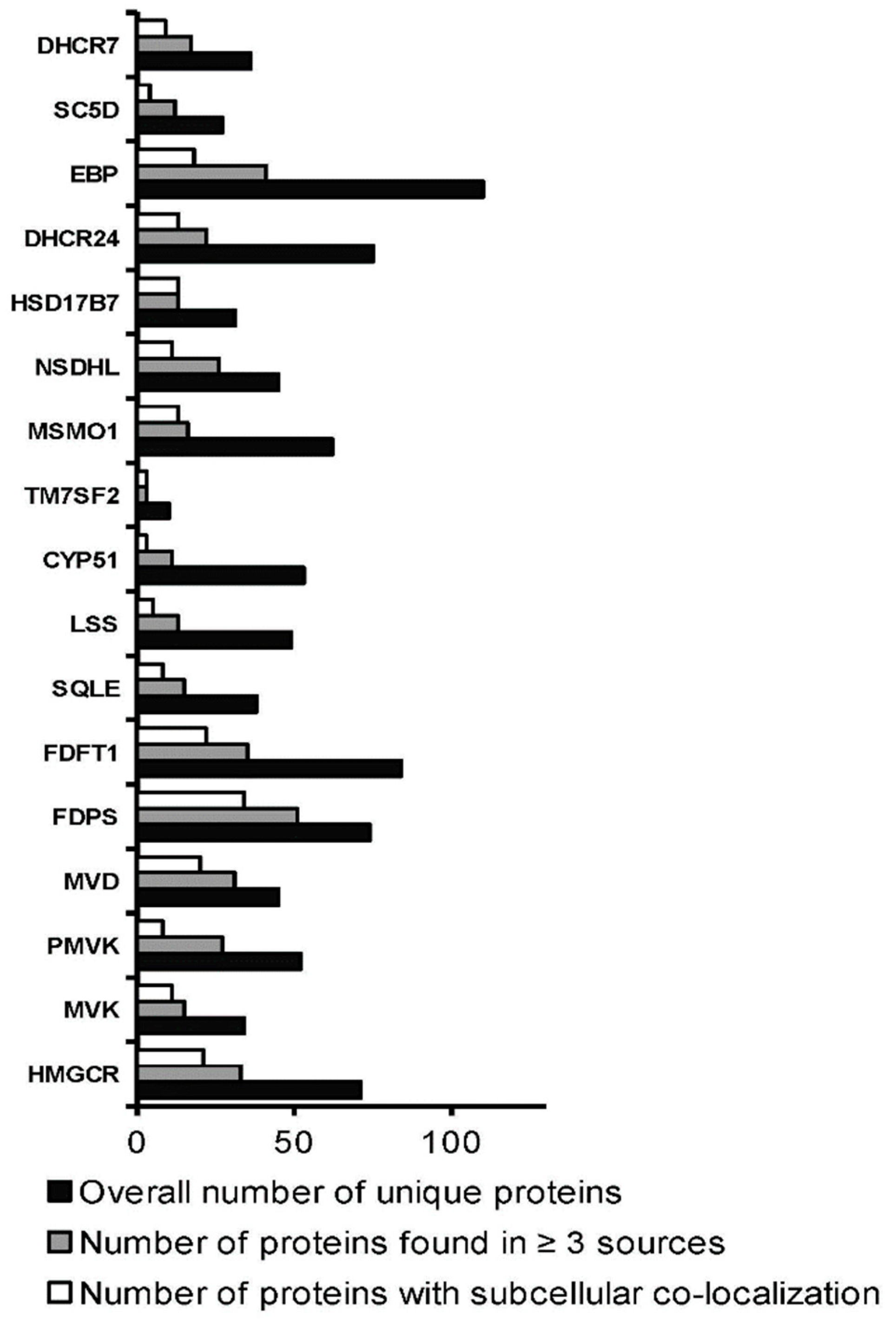
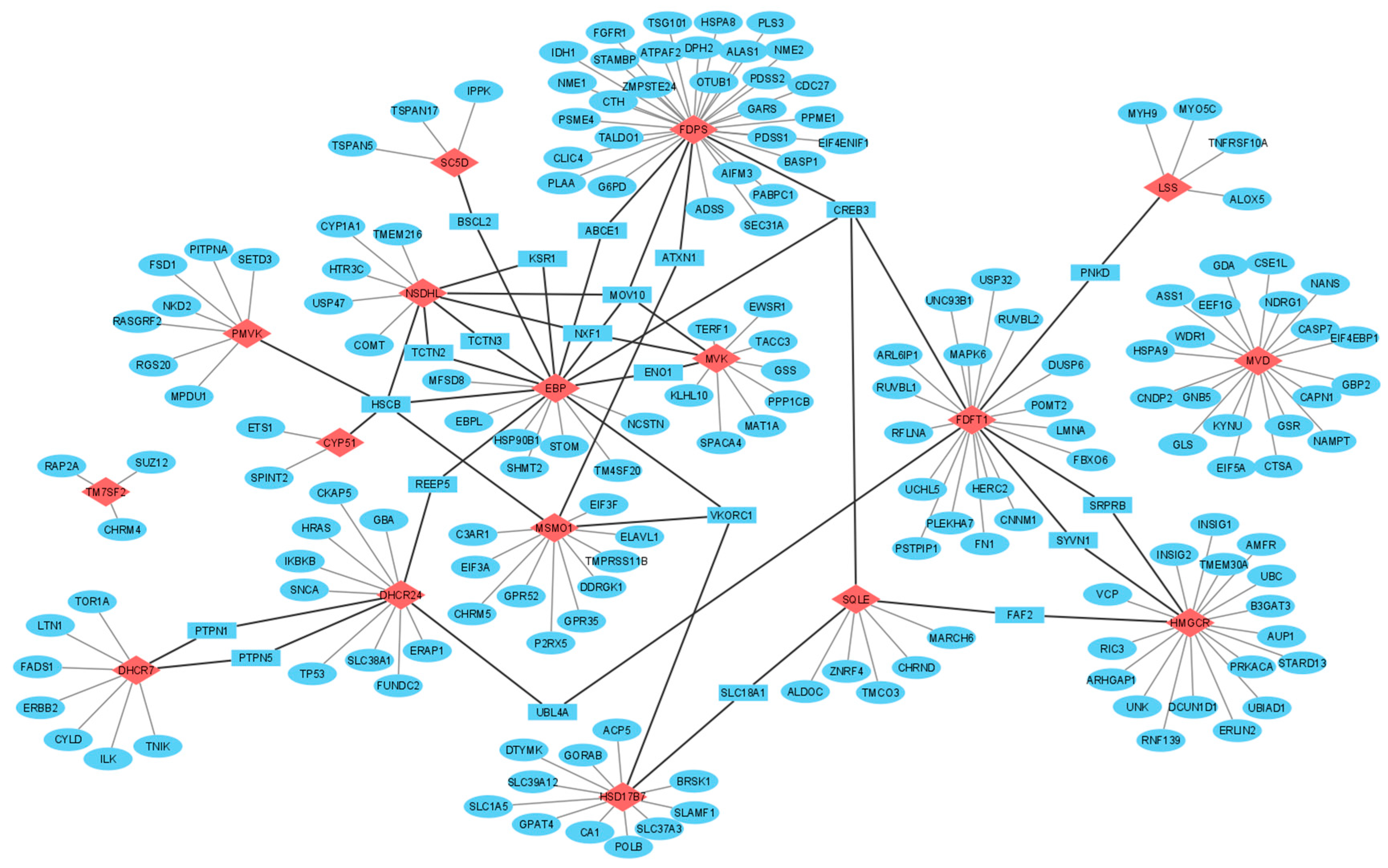
3.3. Interactomics Landscape of Cholesterol Synthesis Enzymes
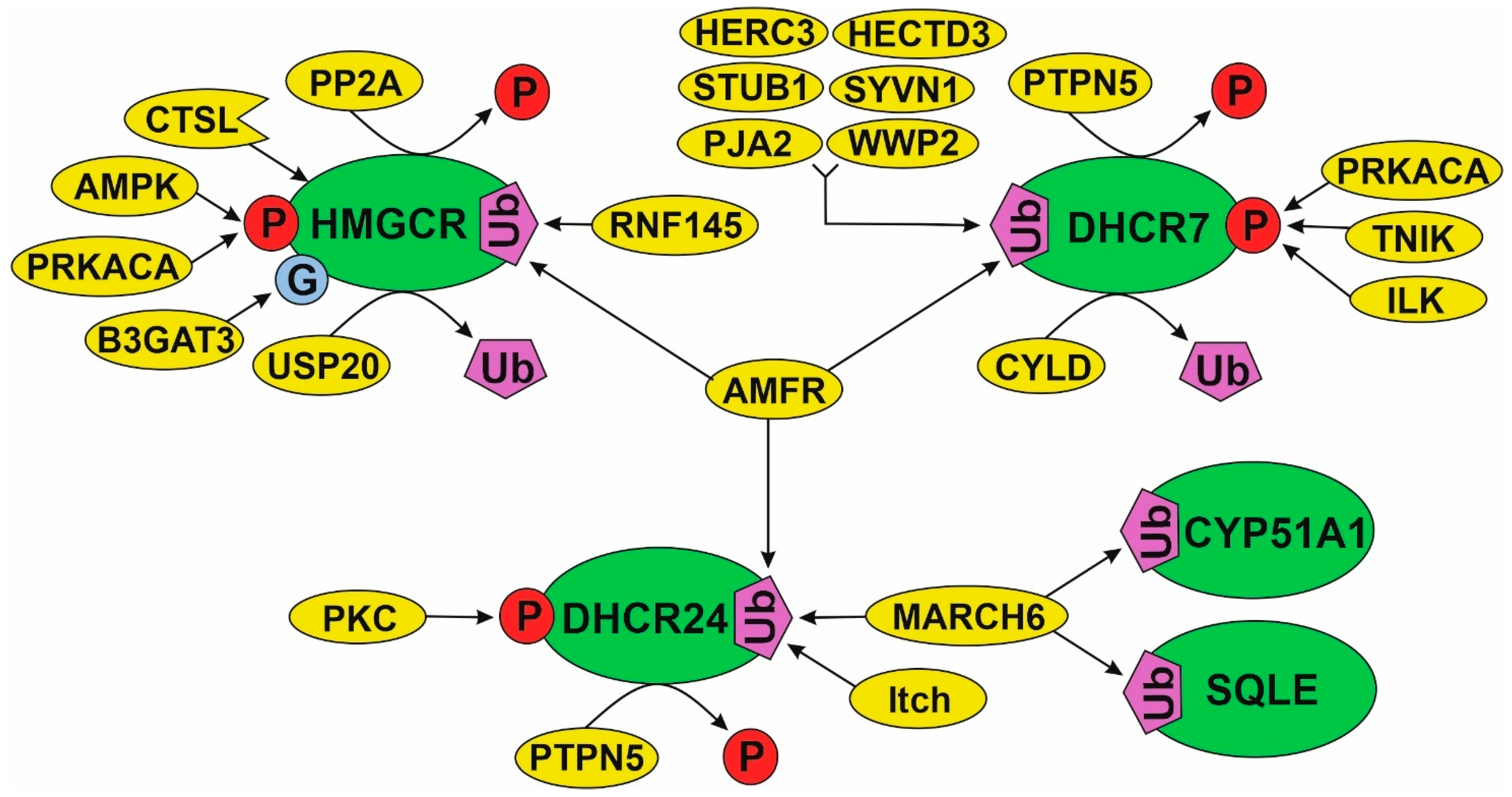
3.4. Post-Translational Modifications (PTM)
4. Discussion
4.1. Mapping of Protein–Protein Interaction
4.2. Protein–Protein Interactions of Cholesterol Synthesis Enzymes in the Cancer Context
4.2.1. SQLE
4.2.2. CYP51A1
4.2.3. TM7SF2
4.2.4. MVD
4.2.5. DHCR24
4.3. Common Protein Partners of Cholesterol Synthesis Enzymes
4.3.1. Molecular Chaperones
4.3.2. Ubiquitin-Protein Ligases
4.3.3. Metabolic Enzymes
4.3.4. Signaling Proteins
4.3.5. Transport Proteins
4.3.6. Modifying Proteins
4.4. Regulation of Cholesterol Synthesis Enzymes through Post-Translational Modifications (PTM)
4.4.1. PTMs of HMGCR Are the Most Studied
4.4.2. A Model of PTM Regulation of Different Parts of Cholesterol Synthesis Pathway
4.5. A Multiprotein Cholesterol “Metabolon”
4.6. A landscape of Cholesterol Precursors in the Cancer Context
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| PPI | protein–protein interaction |
| DEG | differentially expressed genes |
| ER | endoplasmic reticulum |
| ERAD | endoplasmic reticulum associated degradation |
| EMT | epithelial-mesenchymal transition |
| MAS | meiosis activating sterol |
| HMGCR | 3-hydroxy-3-methylglutaryl-CoA reductase |
| MVK | mevalonate kinase |
| PMVK | phosphomevalonate kinase |
| MVD | mevalonate diphosphate decarboxylase |
| FDPS | (2E,6E)-farnesyl diphosphate synthase |
| FDFT1 | farnesyl-diphosphate farnesyltransferase 1 |
| SQLE | squalene epoxidase |
| LSS | lanosterol synthase |
| DHCR24 | 24-dehydrocholesterol reductase |
| CYP51A1 | sterol 14-alpha-demethylase |
| TM7SF2 | delta(14)-sterol reductase |
| MSMO1 | methylsterol monooxygenase 1 |
| NSDHL | sterol-4alpha-carboxylate 3-dehydrogenase |
| HSD17B7 | 17-beta-estradiol 17-dehydrogenase |
| EBP | cholestenol delta-isomerase |
| SC5D | sterol-C5-desaturase |
| DHCR7 | 7-dehydrocholesterol reductase |
| ACC | adrenocortical carcinoma |
| BLCA | bladder urothelial carcinoma |
| BRCA | breast invasive carcinoma |
| CESC | cervical squamous cell carcinoma and endocervical adenocarcinoma |
| CHOL | cholangiocarcinoma |
| COAD | colon adenocarcinoma |
| DLBC | lymphoid neoplasm diffuse large B-cell lymphoma |
| ESCA | esophageal carcinoma |
| GBM | glioblastoma multiforme |
| HNSC | head and neck squamous cell carcinoma |
| KICH | kidney chromophobe renal cell carcinoma |
| KIRC | kidney renal clear cell carcinoma |
| KIRP | kidney renal papillary cell carcinoma |
| LAML | scute myeloid leukemia |
| LGG | brain lower grade glioma |
| LIHC | liver hepatocellular carcinoma |
| LUAD | lung adenocarcinoma |
| LUSC | lung squamous cell carcinoma |
| MESO | mesothelioma |
| OV | ovarian serous cystadenocarcinoma |
| PAAD | pancreatic adenocarcinoma |
| PCPG | pheochromocytoma and paraganglioma |
| PRAD | prostate adenocarcinoma, |
| READ | rectum adenocarcinoma |
| SARC | sarcoma |
| SKCM | skin cutaneous melanoma |
| STAD | stomach adenocarcinoma |
| TGCT | testicular germ cell tumors |
| THCA | thyroid carcinoma |
| THYM | thymoma |
| UCEC | uterine corpus endometrial carcinoma |
| UCS | uterine carcinosarcoma |
| UVM | uveal melanoma |
References
- Luo, J.; Yang, H.; Song, B.-L. Mechanisms and regulation of cholesterol homeostasis. Nat. Rev. Mol. Cell Biol. 2019, 21, 225–245. [Google Scholar] [CrossRef]
- Xu, H.; Zhou, S.; Tang, Q.; Xia, H.; Bi, F. Cholesterol metabolism: New functions and therapeutic approaches in cancer. Biochim. Biophys. Acta BBA Rev. Cancer 2020, 1874, 188394. [Google Scholar] [CrossRef] [PubMed]
- Kuzu, O.; Noory, M.A.; Robertson, G.P. The Role of Cholesterol in Cancer. Cancer Res. 2016, 76, 2063–2070. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Song, B.-L.; Xu, C. Cholesterol metabolism in cancer: Mechanisms and therapeutic opportunities. Nat. Metab. 2020, 2, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Riscal, R.; Skuli, N.; Simon, M.C. Even Cancer Cells Watch Their Cholesterol! Mol. Cell 2019, 76, 220–231. [Google Scholar] [CrossRef]
- Lyu, J.; Yang, E.J.; Shim, J.S. Cholesterol Trafficking: An Emerging Therapeutic Target for Angiogenesis and Cancer. Cells 2019, 8, 389. [Google Scholar] [CrossRef]
- Hu, J.; La Vecchia, C.; de Groh, M.; Negri, E.; Morrison, H.; Mery, L. Dietary cholesterol intake and cancer. Ann. Oncol. 2011, 23, 491–500. [Google Scholar] [CrossRef]
- Hryniewicz-Jankowska, A.; Augoff, K.; Sikorski, A.F. The role of cholesterol and cholesterol-driven membrane raft domains in prostate cancer. Exp. Biol. Med. 2019, 244, 1053–1061. [Google Scholar] [CrossRef]
- Ahmadi, M.; Amiri, S.; Pecic, S.; Machaj, F.; Rosik, J.; Łos, M.J.; Alizadeh, J.; Mahdian, R.; Rosa, S.C.D.S.; Schaafsma, D.; et al. Pleiotropic effects of statins: A focus on cancer. Biochim. et Biophys. Acta BBA Mol. Basis Dis. 2020, 1866, 165968. [Google Scholar] [CrossRef]
- Thomas, J.P.; Loke, Y.K.; Alexandre, L. Efficacy and safety profile of statins in patients with cancer: A systematic review of randomised controlled trials. Eur. J. Clin. Pharmacol. 2020, 76, 1639–1651. [Google Scholar] [CrossRef]
- Graaf, M.R.; Beiderbeck, A.B.; Egberts, T.; Richel, D.J.; Guchelaar, H.-J. The Risk of Cancer in Users of Statins. J. Clin. Oncol. 2004, 22, 2388–2394. [Google Scholar] [CrossRef] [PubMed]
- Farooqi, M.A.M.; Malhotra, N.; Mukherjee, S.D.; Sanger, S.; Dhesy-Thind, S.K.; Ellis, P.; Leong, D.P. Statin therapy in the treatment of active cancer: A systematic review and meta-analysis of randomized controlled trials. PLoS ONE 2018, 13, e0209486. [Google Scholar] [CrossRef]
- Emilsson, L.; García-Albéniz, X.; Logan, R.W.; Caniglia, E.C.; Kalager, M.; Hernán, M. Examining Bias in Studies of Statin Treatment and Survival in Patients With Cancer. JAMA Oncol. 2018, 4, 63–70. [Google Scholar] [CrossRef]
- Sharpe, L.J.; Coates, H.W.; Brown, A.J. Post-translational control of the long and winding road to cholesterol. J. Biol. Chem. 2020, 295, 17549–17559. [Google Scholar] [CrossRef]
- Wang, Z.; Jensen, M.; Zenklusen, J.C. A Practical Guide to The Cancer Genome Atlas (TCGA). In Statistical Genomics; Humana Press: New York, NY, USA, 2016; Volume 1418, pp. 111–141. [Google Scholar] [CrossRef]
- Weinstein, J.N.; The Cancer Genome Atlas Research Network; Lin, J.; Mills, G.B.; Shaw, K.R.M.; A Ozenberger, B.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Kang, B.; Li, C.; Chen, T.; Zhang, Z. GEPIA2: An enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res. 2019, 47, W556–W560. [Google Scholar] [CrossRef]
- Broom, B.M.; Ryan, M.C.; Brown, R.E.; Ikeda, F.; Stucky, M.; Kane, D.W.; Melott, J.; Wakefield, C.; Casasent, T.D.; Akbani, R.; et al. A Galaxy Implementation of Next-Generation Clustered Heatmaps for Interactive Exploration of Molecular Profiling Data. Cancer Res. 2017, 77, e23–e26. [Google Scholar] [CrossRef] [PubMed]
- Metsalu, T.; Vilo, J. ClustVis: A web tool for visualizing clustering of multivariate data using Principal Component Analysis and heatmap. Nucleic Acids Res. 2015, 43, W566–W570. [Google Scholar] [CrossRef]
- Alexeyenko, A.; Schmitt, T.; Tjärnberg, A.; Guala, D.; Frings, O.; Sonnhammer, E.L.L. Comparative interactomics with Funcoup 2.0. Nucleic Acids Res. 2011, 40, D821–D828. [Google Scholar] [CrossRef]
- Calderone, A.; Castagnoli, L.; Cesareni, G. Mentha: A resource for browsing integrated protein-interaction networks. Nat. Methods 2013, 10, 690–691. [Google Scholar] [CrossRef]
- Giurgiu, M.; Reinhard, J.; Brauner, B.; Dunger-Kaltenbach, I.; Fobo, G.; Frishman, G.; Montrone, C.; Ruepp, A. CORUM: The comprehensive resource of mammalian protein complexes—2019. Nucleic Acids Res. 2018, 47, D559–D563. [Google Scholar] [CrossRef] [PubMed]
- Mathivanan, S.; Simpson, R.J. ExoCarta: A compendium of exosomal proteins and RNA. Proteomics 2009, 9, 4997–5000. [Google Scholar] [CrossRef]
- Alonso-López, D.; Laborie, F.J.C.; A Gutiérrez, M.; Lambourne, L.; A Calderwood, M.; Vidal, M.; Rivas, J.D.L. APID database: Redefining protein–protein interaction experimental evidences and binary interactomes. Database 2019, 2019. [Google Scholar] [CrossRef]
- Licata, L.; Briganti, L.; Peluso, D.; Perfetto, L.; Iannuccelli, M.; Galeota, E.; Sacco, F.; Palma, A.; Nardozza, A.P.; Santonico, E.; et al. MINT, the molecular interaction database: 2012 update. Nucleic Acids Res. 2011, 40, D857–D861. [Google Scholar] [CrossRef] [PubMed]
- Licata, L.; Surdo, P.L.; Iannuccelli, M.; Palma, A.; Micarelli, E.; Perfetto, L.; Peluso, D.; Calderone, A.; Castagnoli, L.; Cesareni, G. SIGNOR 2.0, the SIGnaling Network Open Resource 2.0: 2019 update. Nucleic Acids Res. 2019, 48, D504–D510. [Google Scholar] [CrossRef]
- Luck, K.; Kim, D.-K.; Lambourne, L.; Spirohn, K.; Begg, B.E.; Bian, W.; Brignall, R.; Cafarelli, T.; Laborie, F.J.C.; Charloteaux, B.; et al. A reference map of the human binary protein interactome. Nat. Cell Biol. 2020, 580, 402–408. [Google Scholar] [CrossRef]
- Kotlyar, M.; Pastrello, C.; Malik, Z.; Jurisica, I. IID 2018 update: Context-specific physical protein–protein interactions in human, model organisms and domesticated species. Nucleic Acids Res. 2018, 47, D581–D589. [Google Scholar] [CrossRef]
- Schweppe, D.K.; Huttlin, E.; Harper, J.; Gygi, S.P. BioPlex Display: An Interactive Suite for Large-Scale AP–MS Protein–Protein Interaction Data. J. Proteome Res. 2017, 17, 722–726. [Google Scholar] [CrossRef] [PubMed]
- Orii, N.; Ganapathiraju, M.K. Wiki-Pi: A Web-Server of Annotated Human Protein-Protein Interactions to Aid in Discovery of Protein Function. PLoS ONE 2012, 7, e49029. [Google Scholar] [CrossRef]
- Alanis-Lobato, G.; Andrade-Navarro, M.A.; Schaefer, M.H. HIPPIE v2.0: Enhancing meaningfulness and reliability of protein–protein interaction networks. Nucleic Acids Res. 2016, 45, D408–D414. [Google Scholar] [CrossRef]
- Das, J.; Yu, H. HINT: High-quality protein interactomes and their applications in understanding human disease. BMC Syst. Biol. 2012, 6, 92. [Google Scholar] [CrossRef]
- Wang, J.; Vasaikar, S.; Shi, Z.; Greer, M.; Zhang, B. WebGestalt 2017: A more comprehensive, powerful, flexible and interactive gene set enrichment analysis toolkit. Nucleic Acids Res. 2017, 45, W130–W137. [Google Scholar] [CrossRef] [PubMed]
- Tsherniak, A.; Vazquez, F.; Montgomery, P.G.; Weir, B.A.; Kryukov, G.; Cowley, G.S.; Gill, S.; Harrington, W.F.; Pantel, S.; Krill-Burger, J.; et al. Defining a Cancer Dependency Map. Cell 2017, 170, 564–576.e16. [Google Scholar] [CrossRef]
- Allot, A.; Chen, Q.; Kim, S.; Alvarez, R.V.; Comeau, D.C.; Wilbur, W.J.; Lu, Z. LitSense: Making sense of biomedical literature at sentence level. Nucleic Acids Res. 2019, 47, W594–W599. [Google Scholar] [CrossRef]
- Mani, M.; Chen, C.; Amblee, V.; Liu, H.; Mathur, T.; Zwicke, G.; Zabad, S.; Patel, B.; Thakkar, J.; Jeffery, C.J. MoonProt: A database for proteins that are known to moonlight. Nucleic Acids Res. 2014, 43, D277–D282. [Google Scholar] [CrossRef]
- Hu, L.-D.; Wang, J.; Chen, X.-J.; Yan, Y.-B. Lanosterol modulates proteostasis via dissolving cytosolic sequestosomes/aggresome-like induced structures. Biochim. Biophys. Acta BBA Mol. Cell Res. 2020, 1867, 118617. [Google Scholar] [CrossRef] [PubMed]
- Reilly, J.F.; Martinez, S.D.; Mickey, G.; Maher, P.A. A novel role for farnesyl pyrophosphate synthase in fibroblast growth factor-mediated signal transduction. Biochem. J. 2002, 366, 501–510. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Li, Y.; Wang, W.; Chen, S.; Liu, T.; Jia, D.; Quan, X.; Sun, D.; Chang, A.K.; Gao, B. 3 β-Hydroxysteroid-Δ 24 Reductase (DHCR24) Protects Neuronal Cells from Apoptotic Cell Death Induced by Endoplasmic Reticulum (ER) Stress. PLoS ONE 2014, 9, e86753. [Google Scholar] [CrossRef]
- Long, T.; Hassan, A.; Thompson, B.M.; McDonald, J.G.; Wang, J.; Li, X. Structural basis for human sterol isomerase in cholesterol biosynthesis and multidrug recognition. Nat. Commun. 2019, 10, 1–8. [Google Scholar] [CrossRef]
- Sharma, V.; Shing, B.; Hernandez-Alvarez, L.; Debnath, A.; Podust, L.M. Domain-Swap Dimerization of Acanthamoeba castellanii CYP51 and a Unique Mechanism of Inactivation by Isavuconazole. Mol. Pharmacol. 2020, 98, 770–780. [Google Scholar] [CrossRef]
- Potter, D.; Miziorko, H.M. Identification of Catalytic Residues in Human Mevalonate Kinase. J. Biol. Chem. 1997, 272, 25449–25454. [Google Scholar] [CrossRef]
- Padyana, A.K.; Gross, S.; Jin, L.; Cianchetta, G.; Narayanaswamy, R.; Wang, F.; Wang, R.; Fang, C.; Lv, X.; Biller, S.A.; et al. Structure and inhibition mechanism of the catalytic domain of human squalene epoxidase. Nat. Commun. 2019, 10, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Istvan, E.S.; Palnitkar, M.; Buchanan, S.K.; Deisenhofer, J. Crystal structure of the catalytic portion of human HMG-CoA reductase: Insights into regulation of activity and catalysis. EMBO J. 2000, 19, 819–830. [Google Scholar] [CrossRef]
- Hong, S.M.; Hwang, S.W.; Wang, T.; Park, C.W.; Ryu, Y.; Jung, J.; Shin, J.H.; Kim, S.; Lee, J.L.; Kim, C.W.; et al. Increased nicotinamide adenine dinucleotide pool promotes colon cancer progression by suppressing reactive oxygen species level. Cancer Sci. 2018, 110, 629–638. [Google Scholar] [CrossRef] [PubMed]
- Zou, L.; Li, L.; Porter, T.D. 7-Dehydrocholesterol reductase activity is independent of cytochrome P450 reductase. J. Steroid Biochem. Mol. Biol. 2011, 127, 435–438. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Porter, T.D. New insights into the role of cytochrome P450 reductase (POR) in microsomal redox biology. Acta Pharm. Sin. B 2012, 2, 102–106. [Google Scholar] [CrossRef][Green Version]
- Liu, D.; Wong, C.C.; Fu, L.; Chen, H.; Zhao, L.; Li, C.; Zhou, Y.; Zhang, Y.; Xu, W.; Yang, Y.; et al. Squalene epoxidase drives NAFLD-induced hepatocellular carcinoma and is a pharmaceutical target. Sci. Transl. Med. 2018, 10, eaap9840. [Google Scholar] [CrossRef]
- Yarmolinsky, J.; Bull, C.J.; Vincent, E.; Robinson, J.; Walther, A.; Smith, G.D.; Lewis, S.J.; Relton, C.; Martin, R.M. Association Between Genetically Proxied Inhibition of HMG-CoA Reductase and Epithelial Ovarian Cancer. JAMA 2020, 323, 646–655. [Google Scholar] [CrossRef]
- Zhang, T.; Bai, R.; Wang, Q.; Wang, K.; Li, X.; Liu, K.; Ryu, J.; Wang, T.; Chang, X.; Ma, W.; et al. Fluvastatin Inhibits HMG-CoA Reductase and Prevents Non–Small Cell Lung Carcinogenesis. Cancer Prev. Res. 2019, 12, 837–848. [Google Scholar] [CrossRef]
- Yun, U.-J.; Lee, J.-H.; Shim, J.; Yoon, K.; Goh, S.-H.; Yi, E.H.; Ye, S.-K.; Lee, J.-S.; Lee, H.; Park, J.; et al. Anti-cancer effect of doxorubicin is mediated by downregulation of HMG-Co A reductase via inhibition of EGFR/Src pathway. Lab. Investig. 2019, 99, 1157–1172. [Google Scholar] [CrossRef] [PubMed]
- Kozar, K.; Kaminski, R.; Legat, M.; Kopec, M.; Nowis, D.; Skierski, J.S.; Koronkiewicz, M.; Jakóbisiak, M.; Golab, J. Cerivastatin demonstrates enhanced antitumor activity against human breast cancer cell lines when used in combination with doxorubicin or cisplatin. Int. J. Oncol. 2004, 24, 1149–1157. [Google Scholar] [CrossRef] [PubMed]
- Fromigué, O.; Hamidouche, Z.; Marie, P.J. Statin-Induced Inhibition of 3-Hydroxy-3-Methyl Glutaryl Coenzyme A Reductase Sensitizes Human Osteosarcoma Cells to Anticancer Drugs. J. Pharmacol. Exp. Ther. 2008, 325, 595–600. [Google Scholar] [CrossRef] [PubMed]
- Roudier, E.; Mistafa, O.; Stenius, U. Statins induce mammalian target of rapamycin (mTOR)-mediated inhibition of Akt signaling and sensitize p53-deficient cells to cytostatic drugs. Mol. Cancer Ther. 2006, 5, 2706–2715. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hayashi, K.; Nakazato, Y.; Morito, N.; Sagi, M.; Fujita, T.; Anzai, N.; Chida, M. Fluvastatin is effective against thymic carcinoma. Life Sci. 2020, 240, 117110. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.I.; Jung, M.; Dan, K.; Lee, S.; Lee, C.; Kim, H.S.; Chung, H.H.; Kim, J.-W.; Park, N.H.; Song, Y.-S.; et al. Proteomic Discovery of Biomarkers to Predict Prognosis of High-Grade Serous Ovarian Carcinoma. Cancers 2020, 12, 790. [Google Scholar] [CrossRef]
- Shen, K.; Rice, S.D.; Gingrich, D.A.; Wang, D.; Mi, Z.; Tian, C.; Ding, Z.; Brower, S.L.; Ervin, P.R.; Gabrin, M.J.; et al. Distinct Genes Related to Drug Response Identified in ER Positive and ER Negative Breast Cancer Cell Lines. PLoS ONE 2012, 7, e40900. [Google Scholar] [CrossRef]
- Todenhöfer, T.; Hennenlotter, J.; Kühs, U.; Gerber, V.; Gakis, G.; Vogel, U.; Aufderklamm, S.; Merseburger, A.; Knapp, J.; Stenzl, A.; et al. Altered expression of farnesyl pyrophosphate synthase in prostate cancer: Evidence for a role of the mevalonate pathway in disease progression? World J. Urol. 2012, 31, 345–350. [Google Scholar] [CrossRef]
- Han, S.; Li, X.; Xia, Y.; Yu, Z.; Cai, N.; Malwal, S.R.; Han, X.; Oldfield, E.; Zhang, Y. Farnesyl Pyrophosphate Synthase as a Target for Drug Development: Discovery of Natural-Product-Derived Inhibitors and Their Activity in Pancreatic Cancer Cells. J. Med. Chem. 2019, 62, 10867–10896. [Google Scholar] [CrossRef] [PubMed]
- Qiu, L.; Yang, H.; Lv, G.; Li, K.; Liu, G.; Wang, W.; Wang, S.; Zhao, X.; Xie, M.; Lin, J. Insights into the mevalonate pathway in the anticancer effect of a platinum complex on human gastric cancer cells. Eur. J. Pharmacol. 2017, 810, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Li, M.; Lin, L.; Xu, X.; Jiang, G.; Wu, L. FPPS mediates TGF-β1-induced non-small cell lung cancer cell invasion and the EMT process via the RhoA/Rock1 pathway. Biochem. Biophys. Res. Commun. 2018, 496, 536–541. [Google Scholar] [CrossRef]
- Lin, Y.-S.; Park, J.; De Schutter, J.W.; Huang, X.F.; Berghuis, A.M.; Sebag, M.; Tsantrizos, Y.S. Design and Synthesis of Active Site Inhibitors of the Human Farnesyl Pyrophosphate Synthase: Apoptosis and Inhibition of ERK Phosphorylation in Multiple Myeloma Cells. J. Med. Chem. 2012, 55, 3201–3215. [Google Scholar] [CrossRef]
- Ha, N.T.; Lee, C.H. Roles of Farnesyl-Diphosphate Farnesyltransferase 1 in Tumour and Tumour Microenvironments. Cells 2020, 9, 2352. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.-F.; Jan, Y.-H.; Liu, Y.-P.; Yang, C.-J.; Su, C.-Y.; Chang, Y.-C.; Lai, T.-C.; Chiou, J.; Tsai, H.-Y.; Lu, J.; et al. Squalene Synthase Induces Tumor Necrosis Factor Receptor 1 Enrichment in Lipid Rafts to Promote Lung Cancer Metastasis. Am. J. Respir. Crit. Care Med. 2014, 190, 675–687. [Google Scholar] [CrossRef] [PubMed]
- Weng, M.-L.; Chen, W.-K.; Chen, X.-Y.; Lu, H.; Sun, Z.-R.; Yu, Q.; Sun, P.-F.; Xu, Y.-J.; Zhu, M.-M.; Jiang, N.; et al. Fasting inhibits aerobic glycolysis and proliferation in colorectal cancer via the Fdft1-mediated AKT/mTOR/HIF1α pathway suppression. Nat. Commun. 2020, 11, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Fukuma, Y.; Matsui, H.; Koike, H.; Sekine, Y.; Shechter, I.; Ohtake, N.; Nakata, S.; Ito, K.; Suzuki, K. Role of squalene synthase in prostate cancer risk and the biological aggressiveness of human prostate cancer. Prostate Cancer Prostatic Dis. 2012, 15, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Ge, H.; Zhao, Y.; Shi, X.; Tan, Z.; Chi, X.; He, M.; Jiang, G.; Ji, L.; Li, H. Squalene epoxidase promotes the proliferation and metastasis of lung squamous cell carcinoma cells though extracellular signal-regulated kinase signaling. Thorac. Cancer 2019, 10, 428–436. [Google Scholar] [CrossRef]
- Sui, Z.; Zhou, J.; Cheng, Z.; Lu, P. Squalene epoxidase (SQLE) promotes the growth and migration of the hepatocellular carcinoma cells. Tumor Biol. 2015, 36, 6173–6179. [Google Scholar] [CrossRef]
- Brown, D.N.; Caffa, I.; Cirmena, G.; Piras, D.; Garuti, A.; Gallo, M.; Alberti, S.; Nencioni, A.; Ballestrero, A.; Zoppoli, G. Squalene epoxidase is a bona fide oncogene by amplification with clinical relevance in breast cancer. Sci. Rep. 2016, 6, 19435. [Google Scholar] [CrossRef]
- Shen, T.; Lu, Y.; Zhang, Q. High Squalene Epoxidase in Tumors Predicts Worse Survival in Patients With Hepatocellular Carcinoma: Integrated Bioinformatic Analysis on NAFLD and HCC. Cancer Control. 2020, 27. [Google Scholar] [CrossRef]
- Jun, S.Y.; Brown, A.J.; Chua, N.K.; Yoon, J.-Y.; Lee, J.-J.; Yang, J.O.; Jang, I.; Jeon, S.-J.; Choi, T.-I.; Kim, C.-H.; et al. Reduction of Squalene Epoxidase by Cholesterol Accumulation Accelerates Colorectal Cancer Progression and Metastasis. Gastroenterology 2021, 160, 1194–1207.e28. [Google Scholar] [CrossRef]
- Li, L.; Zhang, Q.; Wang, X.; Li, Y.; Xie, H.; Chen, X. Squalene epoxidase-induced cholesteryl ester accumulation promotes nasopharyngeal carcinoma development by activating PI3K/AKT signaling. Cancer Sci. 2020, 111, 2275–2283. [Google Scholar] [CrossRef]
- Phillips, R.E.; Yang, Y.; Smith, R.C.; Thompson, B.; Yamasaki, T.; Soto-Feliciano, Y.M.; Funato, K.; Liang, Y.; Garcia-Bermudez, J.; Wang, X.; et al. Target identification reveals lanosterol synthase as a vulnerability in glioma. Proc. Natl. Acad. Sci. USA 2019, 116, 7957–7962. [Google Scholar] [CrossRef]
- Stäubert, C.; Krakowsky, R.; Bhuiyan, H.; Witek, B.; Lindahl, A.; Broom, O.; Nordström, A. Increased lanosterol turnover: A metabolic burden for daunorubicin-resistant leukemia cells. Med Oncol. 2015, 33, 1–10. [Google Scholar] [CrossRef]
- Howell, M.C.; Green, R.; Khalil, R.; Foran, E.; Quarni, W.; Nair, R.; Stevens, S.; Grinchuk, A.; Hanna, A.; Mohapatra, S.; et al. Lung cancer cells survive epidermal growth factor receptor tyrosine kinase inhibitor exposure through upregulation of cholesterol synthesis. FASEB BioAdv. 2019, 2, 90–105. [Google Scholar] [CrossRef]
- Kumarakulasingham, M.; Rooney, P.H.; Dundas, S.R.; Telfer, C.; Melvin, W.T.; Curran, S.; Murray, G.I. Cytochrome P450 Profile of Colorectal Cancer: Identification of Markers of Prognosis. Clin. Cancer Res. 2005, 11, 3758–3765. [Google Scholar] [CrossRef]
- Hargrove, T.Y.; Friggeri, L.; Wawrzak, Z.; Sivakumaran, S.; Yazlovitskaya, E.M.; Hiebert, S.W.; Guengerich, F.P.; Waterman, M.R.; Lepesheva, G.I. Human sterol 14α-demethylase as a target for anticancer chemotherapy: Towards structure-aided drug design. J. Lipid Res. 2016, 57, 1552–1563. [Google Scholar] [CrossRef]
- Bartoli, D.; Piobbico, D.; Bellet, M.M.; Bennati, A.M.; Roberti, R.; Della Fazia, M.A.; Servillo, G. Impaired cell proliferation in regenerating liver of 3 β-hydroxysterol Δ14-reductase (TM7SF2) knock-out mice. Cell Cycle 2016, 15, 2164–2173. [Google Scholar] [CrossRef]
- Bellezza, I.; Gatticchi, L.; DEL Sordo, R.; Peirce, M.J.; Sidoni, A.; Roberti, R.; Minelli, A. The loss of Tm7sf gene accelerates skin papilloma formation in mice. Sci. Rep. 2015, 5, 9471. [Google Scholar] [CrossRef]
- Yoon, S.-H.; Kim, H.S.; Kim, R.N.; Jung, S.-Y.; Hong, B.S.; Kang, E.J.; Lee, H.-B.; Moon, H.-G.; Noh, D.-Y.; Han, W. NAD(P)-dependent steroid dehydrogenase-like is involved in breast cancer cell growth and metastasis. BMC Cancer 2020, 20, 375. [Google Scholar] [CrossRef] [PubMed]
- Gabitova, L.; Restifo, D.; Gorin, A.; Manocha, K.; Handorf, E.; Yang, D.-H.; Cai, K.Q.; Klein-Szanto, A.J.; Cunningham, D.; Kratz, L.E.; et al. Endogenous Sterol Metabolites Regulate Growth of EGFR/KRAS-Dependent Tumors via LXR. Cell Rep. 2015, 12, 1927–1938. [Google Scholar] [CrossRef]
- Sukhanova, A.; Gorin, A.; Serebriiskii, I.G.; Gabitova, L.; Zheng, H.; Restifo, D.; Egleston, B.L.; Cunningham, D.; Bagnyukova, T.; Liu, H.; et al. Targeting C4-Demethylating Genes in the Cholesterol Pathway Sensitizes Cancer Cells to EGF Receptor Inhibitors via Increased EGF Receptor Degradation. Cancer Discov. 2012, 3, 96–111. [Google Scholar] [CrossRef]
- Xiao, Y.; Xie, J.; Liu, L.; Huang, W.; Han, Q.; Qin, J.; Liu, S.; Jiang, Z. NAD(P)-dependent steroid dehydrogenase-like protein and neutral cholesterol ester hydrolase 1 serve as novel markers for early detection of gastric cancer identified using quantitative proteomics. J. Clin. Lab. Anal. 2020, 35, e23652. [Google Scholar] [CrossRef] [PubMed]
- Xue, T.; Zhang, Y.; Zhang, L.; Yao, L.; Hu, X.; Xu, L.X. Proteomic Analysis of Two Metabolic Proteins with Potential to Translocate to Plasma Membrane Associated with Tumor Metastasis Development and Drug Targets. J. Proteome Res. 2013, 12, 1754–1763. [Google Scholar] [CrossRef] [PubMed]
- Qiu, T.; Cao, J.; Chen, W.; Wang, J.; Wang, Y.; Zhao, L.; Liu, M.; He, L.; Wu, G.; Li, H.; et al. 24-Dehydrocholesterol reductase promotes the growth of breast cancer stem-like cells through the Hedgehog pathway. Cancer Sci. 2020, 111, 3653–3664. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Guo, L.; Qiu, X.; Ren, Y.; Li, F.; Cui, W.; Song, S. Genkwadaphnin inhibits growth and invasion in hepatocellular carcinoma by blocking DHCR24-mediated cholesterol biosynthesis and lipid rafts formation. Br. J. Cancer 2020, 123, 1673–1685. [Google Scholar] [CrossRef]
- Liu, X.-P.; Yin, X.-H.; Meng, X.-Y.; Yan, X.-H.; Cao, Y.; Zeng, X.-T.; Wang, X.-H. DHCR24 predicts poor clinicopathological features of patients with bladder cancer. Medicine 2018, 97, e11830. [Google Scholar] [CrossRef] [PubMed]
- Berardi, F.; Abate, C.; Ferorelli, S.; De Robertis, A.F.; Leopoldo, M.; Colabufo, N.A.; Niso, M.; Perrone, R. Novel 4-(4-Aryl)cyclohexyl-1-(2-pyridyl)piperazines as Δ8−Δ7Sterol Isomerase (Emopamil Binding Protein) Selective Ligands with Antiproliferative Activity. J. Med. Chem. 2008, 51, 7523–7531. [Google Scholar] [CrossRef]
- Villalva, C.; Trempat, P.; Greenland, C.; Thomas, C.; Girard, J.P.; Moebius, F.; Delsol, G.; Brousset, P. Isolation of differentially expressed genes in NPM-ALK-positive anaplastic large cell lymphoma. Br. J. Haematol. 2002, 118, 791–798. [Google Scholar] [CrossRef] [PubMed]
- Theodoropoulos, P.C.; Wang, W.; Budhipramono, A.; Thompson, B.M.; Madhusudhan, N.; Mitsche, M.A.; McDonald, J.G.; De Brabander, J.K.; Nijhawan, D. A Medicinal Chemistry-Driven Approach Identified the Sterol Isomerase EBP as the Molecular Target of TASIN Colorectal Cancer Toxins. J. Am. Chem. Soc. 2020, 142, 6128–6138. [Google Scholar] [CrossRef]
- Berndt, N.; Hamilton, A.D.; Sebti, S.M. Targeting protein prenylation for cancer therapy. Nat. Rev. Cancer 2011, 11, 775–791. [Google Scholar] [CrossRef]
- Audet-Walsh, É; Vernier, M.; Yee, T.; Laflamme, C.; Li, S.; Chen, Y.; Giguère, V. SREBF1 Activity Is Regulated by an AR/mTOR Nuclear Axis in Prostate Cancer. Mol. Cancer Res. 2018, 16, 1396–1405. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.-A.; Xiong, X.; Zaytseva, Y.Y.; Napier, D.L.; Vallee, E.; Li, A.T.; Wang, C.; Weiss, H.L.; Evers, B.M.; Gao, T. Downregulation of SREBP inhibits tumor growth and initiation by altering cellular metabolism in colon cancer. Cell Death Dis. 2018, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Vellingiri, B.; Iyer, M.; Subramaniam, M.D.; Jayaramayya, K.; Siama, Z.; Giridharan, B.; Narayanasamy, A.; Dayem, A.A.; Cho, S.-G. Understanding the Role of the Transcription Factor Sp1 in Ovarian Cancer: From Theory to Practice. Int. J. Mol. Sci. 2020, 21, 1153. [Google Scholar] [CrossRef]
- Benatti, P.; Chiaramonte, M.L.; Lorenzo, M.; Hartley, J.A.; Hochhauser, D.; Gnesutta, N.; Mantovani, R.; Imbriano, C.; Dolfini, D. NF-Y activates genes of metabolic pathways altered in cancer cells. Oncotarget 2015, 7, 1633–1650. [Google Scholar] [CrossRef]
- Codini, M.; Garcia-Gil, M.; Albi, E. Cholesterol and Sphingolipid Enriched Lipid Rafts as Therapeutic Targets in Cancer. Int. J. Mol. Sci. 2021, 22, 726. [Google Scholar] [CrossRef]
- Vieira, A.; Schmitt, F. An Update on Breast Cancer Multigene Prognostic Tests—Emergent Clinical Biomarkers. Front. Med. 2018, 5, 248. [Google Scholar] [CrossRef] [PubMed]
- A Drabkin, H.; Gemmill, R.M. Cholesterol and the development of clear-cell renal carcinoma. Curr. Opin. Pharmacol. 2012, 12, 742–750. [Google Scholar] [CrossRef] [PubMed]
- Sundelin, J.P.; Ståhlman, M.; Lundqvist, A.; Levin, M.; Parini, P.; Johansson, M.E.; Borén, J. Increased Expression of the Very Low-Density Lipoprotein Receptor Mediates Lipid Accumulation in Clear-Cell Renal Cell Carcinoma. PLoS ONE 2012, 7, e48694. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Thompson, B.; Han, S.; Lotan, Y.; McDonald, J.G.; Ye, J. Uptake of HDL-cholesterol contributes to lipid accumulation in clear cell renal cell carcinoma. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2019, 1864, 158525. [Google Scholar] [CrossRef]
- Porta-Pardo, E.; Garcia-Alonso, L.; Hrabe, T.; Dopazo, J.; Godzik, A. A Pan-Cancer Catalogue of Cancer Driver Protein Interaction Interfaces. PLoS Comput. Biol. 2015, 11, e1004518. [Google Scholar] [CrossRef]
- Loh, K.; Tam, S.; Murray-Segal, L.; Huynh, K.; Meikle, P.; Scott, J.; Van Denderen, B.; Chen, Z.; Steel, R.; Leblond, N.D.; et al. Inhibition of Adenosine Monophosphate-Activated Protein Kinase-3-Hydroxy-3-Methylglutaryl Coenzyme A Reductase Signaling Leads to Hypercholesterolemia and Promotes Hepatic Steatosis and Insulin Resistance. Hepatol. Commun. 2018, 3, 84–98. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Song, Y.; Feng, M.; Zhou, X.; Lu, Y.; Gao, L.; Yu, C.; Jiang, X.; Zhao, J. Thyroid-stimulating hormone decreases HMG-CoA reductase phosphorylation via AMP-activated protein kinase in the liver. J. Lipid Res. 2015, 56, 963–971. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, N.; Liu, P.; Xie, X. AMPK and Cancer. Exp. Suppl. 2016, 107, 203–226. [Google Scholar] [CrossRef] [PubMed]
- Ching, Y.P.; Kobayashi, T.; Tamura, S.; Hardie, D.G. Specificity of different isoforms of protein phosphatase-2A and protein phosphatase-2C studied using site-directed mutagenesis of HMG-CoA reductase. FEBS Lett. 1997, 411, 265–268. [Google Scholar] [CrossRef]
- Wei, H.; Zhang, H.-L.; Xie, J.-Z.; Meng, D.-L.; Wang, X.-C.; Ke, D.; Zeng, J.; Liu, R. Protein Phosphatase 2A as a Drug Target in the Treatment of Cancer and Alzheimer’s Disease. Curr. Med Sci. 2020, 40, 1–8. [Google Scholar] [CrossRef]
- Moriyama, T.; Wada, M.; Urade, R.; Kito, M.; Katunuma, N.; Ogawa, T.; Simoni, R.D. 3-Hydroxy-3-methylglutaryl Coenzyme A Reductase Is Sterol-Dependently Cleaved by Cathepsin L-Type Cysteine Protease in the Isolated Endoplasmic Reticulum. Arch. Biochem. Biophys. 2001, 386, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.-Y.; Jiang, W.; Tian, N.; Xiong, Y.-N.; Liu, J.; Wei, J.; Wu, K.-Y.; Luo, J.; Shi, X.-J.; Song, B.-L. Ring finger protein 145 (RNF145) is a ubiquitin ligase for sterol-induced degradation of HMG-CoA reductase. J. Biol. Chem. 2018, 293, 4047–4055. [Google Scholar] [CrossRef] [PubMed]
- Song, B.-L.; Sever, N.; DeBose-Boyd, R.A. Gp78, a Membrane-Anchored Ubiquitin Ligase, Associates with Insig-1 and Couples Sterol-Regulated Ubiquitination to Degradation of HMG CoA Reductase. Mol. Cell 2005, 19, 829–840. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.-Y.; Shi, X.-J.; Hu, A.; Wang, J.-Q.; Ding, Y.; Jiang, W.; Sun, M.; Zhao, X.; Luo, J.; Qi, W.; et al. Feeding induces cholesterol biosynthesis via the mTORC1–USP20–HMGCR axis. Nat. Cell Biol. 2020, 588, 479–484. [Google Scholar] [CrossRef] [PubMed]
- Adam, C.; Glück, L.; Ebert, R.; Goebeler, M.; Jakob, F.; Schmidt, M. The MEK5/ERK5 mitogen-activated protein kinase cascade is an effector pathway of bone-sustaining bisphosphonates that regulates osteogenic differentiation and mineralization. Bone 2018, 111, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Loregger, A.; Cook, E.C.L.; Nelson, J.K.; Moeton, M.; Sharpe, L.; Engberg, S.; Karimova, M.; Lambert, G.; Brown, A.J.; Zelcer, N. A MARCH6 and IDOL E3 Ubiquitin Ligase Circuit Uncouples Cholesterol Synthesis from Lipoprotein Uptake in Hepatocytes. Mol. Cell. Biol. 2016, 36, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.M.; Van Der Stoel, M.M.; Berg, M.V.D.; Van Loon, N.M.; Moeton, M.; Scholl, E.; Van Der Wel, N.N.; Kovačević, I.; Hordijk, P.L.; Loregger, A.; et al. The MARCH6-SQLE Axis Controls Endothelial Cholesterol Homeostasis and Angiogenic Sprouting. Cell Rep. 2020, 32, 107944. [Google Scholar] [CrossRef] [PubMed]
- Zelcer, N.; Sharpe, L.; Loregger, A.; Kristiana, I.; Cook, E.C.L.; Phan, L.; Stevenson, J.; Brown, A.J. The E3 Ubiquitin Ligase MARCH6 Degrades Squalene Monooxygenase and Affects 3-Hydroxy-3-Methyl-Glutaryl Coenzyme A Reductase and the Cholesterol Synthesis Pathway. Mol. Cell. Biol. 2014, 34, 1262–1270. [Google Scholar] [CrossRef] [PubMed]
- Foresti, O.; Ruggiano, A.; Hannibal-Bach, H.K.; Ejsing, C.S.; Carvalho, P. Sterol homeostasis requires regulated degradation of squalene monooxygenase by the ubiquitin ligase Doa10/Teb4. eLife 2013, 2, e00953. [Google Scholar] [CrossRef] [PubMed]
- Scott, N.A.; Sharpe, L.J.; Capell-Hattam, I.; Gullo, S.J.; Luu, W.; Brown, A.J. The cholesterol synthesis enzyme lanosterol 14α-demethylase is post-translationally regulated by the E3 ubiquitin ligase MARCH6. Biochem. J. 2020, 477, 541–555. [Google Scholar] [CrossRef]
- Luu, W.; Zerenturk, E.J.; Kristiana, I.; Bucknall, M.; Sharpe, L.; Brown, A.J. Signaling regulates activity of DHCR24, the final enzyme in cholesterol synthesis. J. Lipid Res. 2014, 55, 410–420. [Google Scholar] [CrossRef] [PubMed]
- Capell-Hattam, I.M.; Sharpe, L.J.; Qian, L.; Hart-Smith, G.; Prabhu, A.V.; Brown, A.J. Twin enzymes, divergent control: The cholesterogenic enzymes DHCR14 and LBR are differentially regulated transcriptionally and post-translationally. J. Biol. Chem. 2020, 295, 2850–2865. [Google Scholar] [CrossRef]
- Prabhu, A.; Luu, W.; Sharpe, L.; Brown, A.J. Phosphorylation regulates activity of 7-dehydrocholesterol reductase (DHCR7), a terminal enzyme of cholesterol synthesis. J. Steroid Biochem. Mol. Biol. 2017, 165, 363–368. [Google Scholar] [CrossRef] [PubMed]
- Marsh, J.A.; Teichmann, S. Structure, Dynamics, Assembly, and Evolution of Protein Complexes. Annu. Rev. Biochem. 2015, 84, 551–575. [Google Scholar] [CrossRef]
- Jones, S.; Thornton, J. Principles of protein-protein interactions. Proc. Natl. Acad. Sci. USA 1996, 93, 13–20. [Google Scholar] [CrossRef]
- Ershov, P.V.; Mezentsev, Y.V.; Kopylov, A.T.; Yablokov, E.O.; Svirid, A.; Lushchyk, A.Y.; Kaluzhskiy, L.A.; Gilep, A.A.; Usanov, S.A.; Medvedev, A.E.; et al. Affinity Isolation and Mass Spectrometry Identification of Prostacyclin Synthase (PTGIS) Subinteractome. Biology 2019, 8, 49. [Google Scholar] [CrossRef] [PubMed]
- Svirid, A.V.; Ershov, P.V.; Yablokov, E.O.; Kaluzhskiy, L.A.; Mezentsev, Y.V.; Florinskaya, A.V.; Sushko, T.A.; Strushkevich, N.V.; Gilep, A.A.; Usanov, S.A.; et al. Direct Molecular Fishing of New Protein Partners for Human Thromboxane Synthase. Acta Naturae 2017, 9, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Ershov, P.V.; Yablokov, E.; Zgoda, V.; Mezentsev, Y.; Gnedenko, O.; Kaluzhskiy, L.; Svirid, A.; Gilep, A.; Usanov, S.A.; Ivanov, A. A new insight into subinteractomes of functional antagonists: Thromboxane (CYP5A1) and prostacyclin (CYP8A1) synthases. Cell Biol. Int. 2021. [Google Scholar] [CrossRef] [PubMed]
- Ershov, P.; Mezentsev, Y.; Yablokov, E.O.; Kaluzhskiy, L.A.; Florinskaya, A.V.; Gnedenko, O.; Zgoda, V.G.; Vakhrushev, I.V.; Raeva, O.S.; Yarygin, K.N.; et al. Direct Molecular Fishing of Protein Partners for Proteins Encoded by Genes of Human Chromosome 18 in HepG2 Cell Lysate. Russ. J. Bioorg. Chem. 2018, 44, 759–768. [Google Scholar] [CrossRef]
- Ivanov, A.S.; Medvedev, A.; Ershov, P.; Molnar, A.; Mezentsev, Y.; Yablokov, E.; Kaluzhsky, L.; Gnedenko, O.; Buneeva, O.; Haidukevich, I.; et al. Protein interactomics based on direct molecular fishing on paramagnetic particles: Practical realization and further SPR validation. Proteomics 2014, 14, 2261–2274. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.S.; Ershov, P.V.; Molnar, A.A.; Mezentsev, Y.V.; Kaluzhskiy, L.A.; Yablokov, E.O.; Florinskaya, A.V.; Gnedenko, O.V.; Medvedev, A.E.; Kozin, S.A.; et al. Direct molecular fishing in molecular partners investigation in protein–protein and protein–peptide interactions. Russ. J. Bioorg. Chem. 2016, 42, 14–21. [Google Scholar] [CrossRef]
- Luck, K.; Sheynkman, G.M.; Zhang, I.; Vidal, M. Proteome-Scale Human Interactomics. Trends Biochem. Sci. 2017, 42, 342–354. [Google Scholar] [CrossRef]
- Vidal, M.; Cusick, M.E.; Barabási, A.-L. Interactome Networks and Human Disease. Cell 2011, 144, 986–998. [Google Scholar] [CrossRef] [PubMed]
- Hawe, J.; Theis, F.J.; Heinig, M. Inferring Interaction Networks From Multi-Omics Data. Front. Genet. 2019, 10, 535. [Google Scholar] [CrossRef]
- Gulfidan, G.; Turanli, B.; Beklen, H.; Sinha, R.; Arga, K.Y. Pan-cancer mapping of differential protein-protein interactions. Sci. Rep. 2020, 10, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Polycarpou-Schwarz, M.; Groß, M.; Mestdagh, P.; Schott, J.; Grund, S.E.; Hildenbrand, C.; Rom, J.; Aulmann, S.; Sinn, H.-P.; Vandesompele, J.; et al. The cancer-associated microprotein CASIMO1 controls cell proliferation and interacts with squalene epoxidase modulating lipid droplet formation. Oncogene 2018, 37, 4750–4768. [Google Scholar] [CrossRef] [PubMed]
- Hughes, A.; Powell, D.W.; Bard, M.; Eckstein, J.; Barbuch, R.; Link, A.J.; Espenshade, P.J. Dap1/PGRMC1 Binds and Regulates Cytochrome P450 Enzymes. Cell Metab. 2007, 5, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Asperger, H.; Stamm, N.; Gierke, B.; Pawlak, M.; Hofmann, U.; Zanger, U.M.; Marton, A.; Katona, R.L.; Buhala, A.; Vizler, C.; et al. Progesterone receptor membrane component 1 regulates lipid homeostasis and drives oncogenic signaling resulting in breast cancer progression. Breast Cancer Res. 2020, 22, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Yue, Z.; Jin, S.; Yang, C.; Levine, A.J.; Heintz, N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc. Natl. Acad. Sci. USA 2003, 100, 15077–15082. [Google Scholar] [CrossRef] [PubMed]
- Lei, N.; Wang, P.; Meng, Q. Tm7sf2 may participate in the healing of burn wounds. Mol. Med. Rep. 2016, 14, 1002–1006. [Google Scholar] [CrossRef] [PubMed]
- Vega-Rubín-De-Celis, S. The Role of Beclin 1-Dependent Autophagy in Cancer. Biology 2019, 9, 4. [Google Scholar] [CrossRef]
- Wadhwa, R.; Yaguchi, T.; Hasan, K.; Taira, K.; Kaul, S.C. Mortalin–MPD (mevalonate pyrophosphate decarboxylase) interactions and their role in control of cellular proliferation. Biochem. Biophys. Res. Commun. 2003, 302, 735–742. [Google Scholar] [CrossRef]
- Nishimura, T.; Kohara, M.; Izumi, K.; Kasama, Y.; Hirata, Y.; Huang, Y.; Shuda, M.; Mukaidani, C.; Takano, T.; Tokunaga, Y.; et al. Hepatitis C Virus Impairs p53 via Persistent Overexpression of 3β-Hydroxysterol Δ24-Reductase. J. Biol. Chem. 2009, 284, 36442–36452. [Google Scholar] [CrossRef] [PubMed]
- Puglisi, R.; Pastore, A. The role of chaperones in iron–sulfur cluster biogenesis. FEBS Lett. 2018, 592, 4011–4019. [Google Scholar] [CrossRef] [PubMed]
- Hoff, K.G.; Silberg, J.J.; Vickery, L.E. Interaction of the iron-sulfur cluster assembly protein IscU with the Hsc66/Hsc20 molecular chaperone system of Escherichiacoli. Proc. Natl. Acad. Sci. USA 2000, 97, 7790–7795. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Chen, X.-B.; Ding, B.-Q.; Liu, H.-L.; He, T. Down-regulation of ABCE1 inhibits temozolomide resistance in glioma through the PI3K/Akt/NF-κB signaling pathway. Biosci. Rep. 2018, 38. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Han, X.; Tian, D.-L. The biological regulation of ABCE1. IUBMB Life 2012, 64, 795–800. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, S.; Nishioka, K.; Nakajima, T.; Yagishita, N. The Roles of Synoviolin in Crosstalk Between Endoplasmic Reticulum Stress-Induced Apoptosis and p53 Pathway. Cell Cycle 2007, 6, 1319–1323. [Google Scholar] [CrossRef]
- Kikkert, M.; Doolman, R.; Dai, M.; Avner, R.; Hassink, G.; van Voorden, S.; Thanedar, S.; Roitelman, J.; Chau, V.; Wiertz, E. Human HRD1 Is an E3 Ubiquitin Ligase Involved in Degradation of Proteins from the Endoplasmic Reticulum. J. Biol. Chem. 2004, 279, 3525–3534. [Google Scholar] [CrossRef] [PubMed]
- Phan, V.T.; Ding, V.W.; Li, F.; Chalkley, R.J.; Burlingame, A.; McCormick, F. The RasGAP Proteins Ira2 and Neurofibromin Are Negatively Regulated by Gpb1 in Yeast and ETEA in Humans. Mol. Cell. Biol. 2010, 30, 2264–2279. [Google Scholar] [CrossRef]
- Loregger, A.; Raaben, M.; Tan, J.; Scheij, S.; Moeton, M.; Berg, M.V.D.; Gelberg-Etel, H.; Stickel, E.; Roitelman, J.; Brummelkamp, T.; et al. Haploid Mammalian Genetic Screen Identifies UBXD8 as a Key Determinant of HMGCR Degradation and Cholesterol Biosynthesis. Arter. Thromb. Vasc. Biol. 2017, 37, 2064–2074. [Google Scholar] [CrossRef] [PubMed]
- Almaguel, F.A.; Sanchez, T.W.; Ortiz-Hernandez, G.L.; Casiano, C.A. Alpha-Enolase: Emerging Tumor-Associated Antigen, Cancer Biomarker, and Oncotherapeutic Target. Front. Genet. 2021, 11. [Google Scholar] [CrossRef] [PubMed]
- Novelli, F. Alpha-Enolase i ENO1 i a potential target in novel immunotherapies. Front. Biosci. 2017, 22, 944–959. [Google Scholar] [CrossRef] [PubMed]
- Wajih, N.; Hutson, S.M.; Wallin, R. Disulfide-dependent Protein Folding Is Linked to Operation of the Vitamin K Cycle in the Endoplasmic Reticulum. J. Biol. Chem. 2007, 282, 2626–2635. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, H.; Dong, J.; Ma, L.; Liao, A.; Rong, Z.; Zhou, Z.; Cao, L.; Wang, F.; Wang, J.; et al. mTOR and ERK regulate VKORC1 expression in both hepatoma cells and hepatocytes which influence blood coagulation. Clin. Exp. Med. 2018, 19, 121–132. [Google Scholar] [CrossRef]
- Schaafhausen, A.; Rost, S.; Oldenburg, J.; Muller, C.R. Identification of VKORC1 interaction partners by split-ubiquitin system and coimmunoprecipitation. Thromb. Haemost. 2011, 105, 285–294. [Google Scholar] [CrossRef]
- Ma, Q.; Wu, X.; Wu, J.; Liang, Z.; Liu, T. SERP1 is a novel marker of poor prognosis in pancreatic ductal adenocarcinoma patients via anti-apoptosis and regulating SRPRB/NF-κB axis. Int. J. Oncol. 2017, 51, 1104–1114. [Google Scholar] [CrossRef][Green Version]
- Legate, K.; Andrews, D.; Farkas, L.; Málnási-Csizmadia, A.; Nakamura, A.; Kohama, K.; Nyitray, L. The β-Subunit of the Signal Recognition Particle Receptor Is a Novel GTP-binding Protein without Intrinsic GTPase Activity. J. Biol. Chem. 2003, 278, 27712–27720. [Google Scholar] [CrossRef]
- Wang, J.; Zhao, W.; Liu, H.; He, H.; Shao, R. Myofibrillogenesis regulator 1 (MR-1): A potential therapeutic target for cancer and PNKD. J. Drug Target. 2017, 26, 643–648. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; He, H.; Liu, H.; Zhang, C.; Zhao, W.; Shao, R.-G. Phosphorylation of myofibrillogenesis regulator-1 activates the MAPK signaling pathway and induces proliferation and migration in human breast cancer MCF7 cells. FEBS Lett. 2014, 588, 2903–2910. [Google Scholar] [CrossRef] [PubMed]
- Park, C.R.; You, N.-J.; Park, S.; Mander, S.; Jang, D.-E.; Yeom, S.-C.; Oh, S.-H.; Ahn, C.; Lee, S.H.; Seong, J.Y.; et al. The accessory proteins REEP5 and REEP6 refine CXCR1-mediated cellular responses and lung cancer progression. Sci. Rep. 2016, 6, 39041. [Google Scholar] [CrossRef]
- Salo, V.; Li, S.; Vihinen, H.; Hölttä-Vuori, M.; Szkalisity, A.; Horvath, P.; Belevich, I.; Peränen, J.; Thiele, C.; Somerharju, P.; et al. Seipin Facilitates Triglyceride Flow to Lipid Droplet and Counteracts Droplet Ripening via Endoplasmic Reticulum Contact. Dev. Cell 2019, 50, 478–493.e9. [Google Scholar] [CrossRef] [PubMed]
- Salo, V.; Belevich, I.; Li, S.; Karhinen, L.; Vihinen, H.; Vigouroux, C.; Magré, J.; Thiele, C.; Hölttä-Vuori, M.; Jokitalo, E.; et al. Seipin regulates ER –lipid droplet contacts and cargo delivery. EMBO J. 2016, 35, 2699–2716. [Google Scholar] [CrossRef] [PubMed]
- He, R.-J.; Yu, Z.-H.; Zhang, R.-Y.; Zhang, Z.-Y. Protein tyrosine phosphatases as potential therapeutic targets. Acta Pharmacol. Sin. 2014, 35, 1227–1246. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.; Ha, J.; Park, S.-H. Identification of PTPN1 as a novel negative regulator of the JNK MAPK pathway using a synthetic screening for pathway-specific phosphatases. Sci. Rep. 2017, 7, 1–9. [Google Scholar] [CrossRef]
- A Menzies, S.; Volkmar, N.; Boomen, D.J.V.D.; Timms, R.T.; Dickson, A.S.; A Nathan, J.; Lehner, P.J. The sterol-responsive RNF145 E3 ubiquitin ligase mediates the degradation of HMG-CoA reductase together with gp78 and Hrd1. eLife 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, J.; Huang, E.Y.; Olzmann, J.A. Endoplasmic Reticulum–Associated Degradation and Lipid Homeostasis. Annu. Rev. Nutr. 2016, 36, 511–542. [Google Scholar] [CrossRef]
- Hartman, I.Z.; Liu, P.; Zehmer, J.K.; Luby-Phelps, K.; Jo, Y.; Anderson, R.G.W.; DeBose-Boyd, R.A. Sterol-induced Dislocation of 3-Hydroxy-3-methylglutaryl Coenzyme A Reductase from Endoplasmic Reticulum Membranes into the Cytosol through a Subcellular Compartment Resembling Lipid Droplets. J. Biol. Chem. 2010, 285, 19288–19298. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Xue, L.; Zhang, C.; Li, H.; Cai, Z.; Guo, R. HSP90 interacts with HMGCR and promotes the progression of hepatocellular carcinoma. Mol. Med. Rep. 2018, 19, 524–532. [Google Scholar] [CrossRef]
- Jiang, S.-Y.; Tang, J.-J.; Xiao, X.; Qi, W.; Wu, S.; Jiang, C.; Hong, J.; Xu, J.; Song, B.-L.; Luo, J. Schnyder corneal dystrophy-associated UBIAD1 mutations cause corneal cholesterol accumulation by stabilizing HMG-CoA reductase. PLoS Genet. 2019, 15, e1008289. [Google Scholar] [CrossRef]
- Zou, Y.; Ma, D.; Wang, Y. The PROTAC technology in drug development. Cell Biochem. Funct. 2019, 37, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Mo, C.; Valachovic, M.; Randall, S.; Nickels, J.T.; Bard, M. Protein-protein interactions among C-4 demethylation enzymes involved in yeast sterol biosynthesis. Proc. Natl. Acad. Sci. USA 2002, 99, 9739–9744. [Google Scholar] [CrossRef] [PubMed]
- Luu, W.; Hart-Smith, G.; Sharpe, L.; Brown, A.J. The terminal enzymes of cholesterol synthesis, DHCR24 and DHCR7, interact physically and functionally. J. Lipid Res. 2015, 56, 888–897. [Google Scholar] [CrossRef]
- Koczok, K.; Gurumurthy, C.B.; Balogh, I.; Korade, Z.; Mirnics, K. Subcellular localization of sterol biosynthesis enzymes. J. Mol. Histol. 2018, 50, 63–73. [Google Scholar] [CrossRef]
- Nalewajska, M.; Marchelek-Myśliwiec, M.; Opara-Bajerowicz, M.; Dziedziejko, V.; Pawlik, A. Connexins—Therapeutic Targets in Cancers. Int. J. Mol. Sci. 2020, 21, 9119. [Google Scholar] [CrossRef]
- Göbel, A.; Zinna, V.M.; Dell’Endice, S.; Jaschke, N.; Kuhlmann, J.D.; Wimberger, P.; Rachner, T.D. Anti-tumor effects of mevalonate pathway inhibition in ovarian cancer. BMC Cancer 2020, 20, 1–17. [Google Scholar] [CrossRef]
- Cho, Y.; Kim, M.S.; Nam, C.M.; Kang, E.S. Statin Use is Associated with Decreased Hepatocellular Carcinoma Recurrence in Liver Transplant Patients. Sci. Rep. 2019, 9, 1–8. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, J.; Xu, D.; Zhang, T.; Hu, W.; Feng, Z. Gain-of-function mutant p53 in cancer progression and therapy. J. Mol. Cell Biol. 2020, 12, 674–687. [Google Scholar] [CrossRef]
- Safwat, S.; Ishak, R.; Hathout, R.M.; Mortada, N. Statins anticancer targeted delivery systems: Re-purposing an old molecule. J. Pharm. Pharmacol. 2017, 69, 613–624. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.; Seo, W.; Silwal, P.; Jo, E.-K. Crosstalks between inflammasome and autophagy in cancer. J. Hematol. Oncol. 2020, 13, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.; Battafarano, G.; D’Agostini, M.; Del Fattore, A. The Role of Extracellular Vesicles in Bone Metastasis. Int. J. Mol. Sci. 2018, 19, 1136. [Google Scholar] [CrossRef]
- El-Sahli, S.; Wang, L. Cancer Stem Cell-Associated Pathways in the Metabolic Reprogramming of Breast Cancer. Int. J. Mol. Sci. 2020, 21, 9125. [Google Scholar] [CrossRef]
- Al Dujaily, E.; Baena, J.; Das, M.; Sereno, M.; Smith, C.; Kamata, T.; Officer, L.; Pritchard, C.; Le Quesne, J. Reduced Protumorigenic Tumor-Associated Macrophages With Statin Use in Premalignant Human Lung Adenocarcinoma. JNCI Cancer Spectr. 2019, 4, pkz101. [Google Scholar] [CrossRef]
- Parrales, A.; Thoenen, E.; Iwakuma, T. The interplay between mutant p53 and the mevalonate pathway. Cell Death Differ. 2017, 25, 460–470. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Bermudez, J.; Baudrier, L.; Bayraktar, E.; Shen, Y.; La, K.; Guarecuco, R.; Yucel, B.; Fiore, D.; Tavora, B.; Freinkman, E.; et al. Squalene accumulation in cholesterol auxotrophic lymphomas prevents oxidative cell death. Nat. Cell Biol. 2019, 567, 118–122. [Google Scholar] [CrossRef]
- Paolicelli, R.C.; Widmann, C. Squalene: Friend or foe for cancers. Curr. Opin. Lipidol. 2019, 30, 353–354. [Google Scholar] [CrossRef] [PubMed]
- Rao, C.V.; Newmark, H.L.; Reddy, B.S. Chemopreventive effect of farnesol and lanosterol on colon carcinogenesis. Cancer Detect. Prev. 2002, 26, 419–425. [Google Scholar] [CrossRef]
- Gabitova, L.; Gorin, A.; Astsaturov, I. Molecular Pathways: Sterols and Receptor Signaling in Cancer. Clin. Cancer Res. 2013, 20, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Gelzo, M.; Granato, G.; Albano, F.; Arcucci, A.; Russo, A.D.; De Vendittis, E.; Ruocco, M.R.; Corso, G. Evaluation of cytotoxic effects of 7-dehydrocholesterol on melanoma cells. Free. Radic. Biol. Med. 2014, 70, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhong, F.; Cao, L.; Zhu, R.; Qu, J.; Yang, L.; Chen, T.; Hu, Y.; Wang, Y.; Yao, M.; et al. 7-dehydrocholesterol suppresses melanoma cell proliferation and invasion via Akt1/NF-κB signaling. Oncol. Lett. 2020, 20, 398. [Google Scholar] [CrossRef]
- Chandler, P.D.; Chen, W.Y.; Ajala, O.N.; Hazra, A.; Cook, N.; Bubes, V.; Lee, I.-M.; Giovannucci, E.L.; Willett, W.; Buring, J.E.; et al. Effect of Vitamin D3 Supplements on Development of Advanced Cancer. JAMA Netw. Open 2020, 3, e2025850. [Google Scholar] [CrossRef] [PubMed]
- Danesi, R.; Nardini, D.; Basolo, F.; Del Tacca, M.; Samid, D.; E Myers, C. Phenylacetate inhibits protein isoprenylation and growth of the androgen-independent LNCaP prostate cancer cells transfected with the T24 Ha-ras oncogene. Mol. Pharmacol. 1996, 49, 972–979. [Google Scholar] [PubMed]
- Shepelin, D.; Korzinkin, M.; Vanyushina, A.; Aliper, A.; Borisov, N.; Vasilov, R.; Zhukov, N.; Sokov, D.; Prassolov, V.; Gaifullin, N.; et al. Molecular pathway activation features linked with transition from normal skin to primary and metastatic melanomas in human. Oncotarget 2015, 7, 656–670. [Google Scholar] [CrossRef] [PubMed]
- Popják, G.; Meenan, A.; Parish, E.J.; Nes, W.D. Inhibition of Cholesterol Synthesis and Cell Growth by 24(R, S),25-Iminolanosterol and Triparanol in Cultured Rat Hepatoma Cells. J. Biol. Chem. 1989, 264, 6230–6238. [Google Scholar] [CrossRef]
- Fukao, K.; Tanimoto, Y.; Kayata, Y.; Yoshiga, K.; Takada, K.; Ohyama, Y.; Okuda, K. Alteration of cholesterol biosynthetic pathways in the skin of mice administered polycyclic aromatic hydrocarbons. Cancer Res. 1988, 48, 2555–2560. [Google Scholar] [PubMed]
- Tanimoto, Y.; Fukao, K.-I.; Yoshiga, K.; Takada, K.; Ohyama, Y.; Okuda, K. Effect of chemical carcinogens on cholesterol biosynthetic pathways in the skin of mice. Carcinogenesis 1990, 11, 1647–1651. [Google Scholar] [CrossRef] [PubMed]
- Likus, W.; Siemianowicz, K.; Bieńk, K.; Pakuła, M.; Pathak, H.; Dutta, C.; Wang, Q.; Shojaei, S.; Assaraf, Y.G.; Ghavami, S.; et al. Could drugs inhibiting the mevalonate pathway also target cancer stem cells? Drug Resist. Updat. 2016, 25, 13–25. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Name and Ref. | Site | Notes |
|---|---|---|---|
| 1 | Funcoup 5.0 [20] | http://FunCoup.sbc.su.se (accessed on 15 March 2021) | Evidence type—protein interactions, LLR Score > 2, confidence > 0.9, network: “PPI”, “Complex” |
| 2 | Mentha [21] | https://mentha.uniroma2.it/ (accessed on 15 March 2021) | Evidence type: physical interactions |
| 3 | CORUM [22] | http://mips.helmholtz-muenchen.de/corum/ (accessed on 15 March 2021) | - |
| 4 | ExoCarta [23] | http://www.exocarta.org/ (accessed on 15 March 2021) | - |
| 5 | APID [24] | http://apid.dep.usal.es (accessed on 15 March 2021) | Methods (binary, indirect), |
| ≥1 publication | |||
| 6 | MINT [25] | https://mint.bio.uniroma2.it/ (accessed on 16 March 2021) | Interaction type: association, physical association |
| 7 | SIGNOR 2.0 [26] | https://signor.uniroma2.it/ (accessed on 16 March 2021) | - |
| 8 | HuRI [27] | http://www.interactome-atlas.org/ (accessed on 16 March 2021) | - |
| 9 | IID [28] | http://iid.ophid.utoronto.ca (accessed on 16 March 2021) | Interaction type: experimental |
| 10 | Bioplex Explorer 3.0 [29] | https://bioplex.hms.harvard.edu/explorer/ (accessed on 16 March 2021) | Interaction probability > 0.9 |
| 11 | Wiki-Pi [30] | https://hagrid.dbmi.pitt.edu/wiki-pi/ (accessed on 16 March 2021) | - |
| 12 | HIPPIE 2.0 [31] | http://cbdm-01.zdv.uni-mainz.de/~mschaefer/hippie/ (accessed on 16 March 2021) | Score > 0.6 |
| 13 | HINT [32] | http://hint.yulab.org/ (accessed on 16 March 2021) | Binary interaction |
| Enzyme | Thesis | Ref. |
|---|---|---|
| HMGCR | Genetically proxied inhibition of HMGCR is significantly associated with lower odds of epithelial ovarian cancer ([odds ratio 0.60 [95% CI, 0.43–0.83]). | [49] |
| Inhibition of HMGCR by fluvastatin disrupts the non-small cell lung cancer (NSCLC) tumorigenesis. Knockdown of HMGCR in NSCLC cells induces apoptosis in vitro and in vivo models. | [50] | |
| Statin drugs, inhibiting HMGCR, increase the efficacy of some genotoxic anti-cancer drugs. | [51,52,53,54] | |
| HMGCR is expressed on carcinoma cells but not on normal epithelial cells in thymic tissue. Inhibition of HMGCR by fluvastatin suppresses cell proliferation and induces the carcinoma cell death. | [55] | |
| PMVK | PMVK can be considered as a novel prognostic biomarker for high-grade serous ovarian carcinoma (HGSOC). High expression of PMVK is significantly improves the survival of patients with HGSOC (adjusted hazard ratio, 0.430; [95% CI, 0.228–0.809]). | [56] |
| PMVK expression is positively correlated with drug response in estrogen receptor (ER) positive cells and negatively correlated in ER negative cells | [57] | |
| FDPS | Increased FDPS expression is an independent risk factor of prostate cancer (PC) for early biochemical recurrence. | [58] |
| FDPS inhibitors, the carnosic acid derivatives, induces apoptosis in pancreatic cancer cell lines. | [59] | |
| Inhibition of the FDPS in the mevalonate pathway mediates the cytotoxic effects of a platinum (II) complex with zoledronic acid against human gastric cancer cell line SGC7901. | [60] | |
| FDPS expression significantly correlates with TNM stage and metastasis in non-small cell lung cancer (NSCLC). Inhibition or knockdown of FDPS disrupts the TGF-β1-induced cell invasion and epithelial-mesenchymal transition (EMT). | [61] | |
| FDPS inhibitors improve survival of multiple myeloma (MM) patients and results in down-regulation of ERK phosphorylation in human MM cell lines. | [62] | |
| FDFT1 | FDFT1 is highly expressed in liver, lung, prostate, breast, ovary, small intestine, bladder, cervix, thyroid, and esophageal cancers. FDFT1 regulates cell cycle progression and is directly or indirectly associated with apoptotic signals. | [63] |
| High expression of FDFT1 is associated with poor prognosis and promotes metastasis of lung cancer. Loss of function of FDFT1 or its knockdown significantly inhibits invasion/migration and metastasis in cell and animal models. | [64] | |
| FDFT1 acts as a critical tumor suppressor in colorectal cancer (CRC). Down-regulation of FDFT1 is correlated with CRC malignant progression and poor prognosis. | [65] | |
| The knockdown of expression or activity inhibition of FDFT1 leads to a significant decrease in prostate cancer cell proliferation. | [66] | |
| SQLE | Overexpression of SQLE promotes lung squamous-cell carcinoma (SCC) proliferation, migration and invasion, whereas knockdown of SQLE expression shows the opposite effect. High expression of SQLE corresponds with poor prognosis in lung SCC. | [67] |
| The expression of SQLE is upregulated in the hepatocellular carcinoma (HCC) tissues and its overexpression promotes cell proliferation and migration. Downregulation of SQLE inhibits the tumorigenicity of HCC cells in vitro and in vivo. | [68] | |
| SQLE overexpression is more prevalent in aggressive breast cancer (BC) and is an independent prognostic factor of unfavorable outcome. | [69] | |
| SQLE epoxidase serves as a novel prognostic biomarker for patients with HCC. Overexpression of SQLE in non-alcoholic fatty liver disease HCC tumors is significantly associated with worse overall survival and disease-free survival. | [48,70] | |
| SQLE reduction helps colorectal cancer cells to overcome constraints by inducing the EMT required for generation cancer stem cells. SQLE depletion disrupts the GSK3B/p53 complex, resulting in a metastatic phenotype. | [71] | |
| SQLE promotes nasopharyngeal carcinoma (NPC) proliferation by cholesteryl ester accumulation instead of cholesterol. | [72] | |
| LSS | Lanosterol synthase is a molecular target for menin inhibitor leading to the loss of cholesterol homeostasis and cell death in glioma. | [73] |
| LSS activity increases in the daunorubicin-resistant leukemia cell line (CEM/R2). | [74] | |
| CYP51A1 | CYP51A1 is significantly upregulated in the drug-tolerant (DT) human lung cancer cell lines. The CYP51A1 inhibitor, ketoconazole, shows the synergy in apoptosis induction with tyrosine kinase inhibitors of epidermal growth factor receptor. | [75] |
| CYP51 is present at a significantly higher level in primary colorectal cancer, compared with normal colon. The strong CYP51 immunoreactivity is associated with poor prognosis. | [76] | |
| The VFV, a potent non-azole inhibitor of human CYP51A1, decreases the proliferation rates of lung cancer, hormone-responsive and -nonresponsive breast and skin cancer cells in a concentration-dependent manner. | [77] | |
| TM7SF2 | TM7SF2 knockout (KO) mice show no alteration in cholesterol content. However, delayed cell cycle progression to the G1/S phase was shown in TM7SF2 KO mice, resulting in reduced cell division. | [78] |
| Loss of TM7SF2 increases incidence and multiplicity of skin papillomas. The null genotype shows reduced expression of nur77, a gene associated with resistance to neoplastic transformation. | [79] | |
| NSDHL | NSDHL knockdown affects the cell cycle, survival, proliferation, and migration of breast cancer cells, resulting in suppression of breast tumor progression and metastasis. High NSDHL expression is a potential predictor of poor prognosis in breast cancer patients. | [80] |
| Inhibition NSDHL can be an effective strategy against carcinomas with activated EGFR-KRAS signaling. | [81] | |
| The inactivation of NSDHL or its partner SC4MOL sensitizes tumor cells to EGFR inhibitors. | [82] | |
| NSDHL is significantly overexpressed in gastric cancer tissues that correlates with local tumor invasion, histological grade and TNM II-IV staging. | [83] | |
| The NSDHL up-regulated in the metastasizing mammalian mouse cell line 4T1 compared to the non-metastasizing 67NR. | [84] | |
| DHCR24 | DHCR24 knockdown reduces whereas DHCR24 overexpression enhances breast cancer stem-like cell populations, mammosphere and aldehyde dehydrogenase positive cell. | [85] |
| High expression of DHCR24 in human HCC specimens correlates with poor clinical outcome. Interfering DHCR24 alters growth and migration of HCC cells. | [86] | |
| DHCR24 is up-regulated in bladder cancer (BC) cells compared with that in normal tissues. DHCR24 might promote the proliferation of BC cells through several cancer-associated processes. | [87] | |
| EBP | The EBP inhibitors show the good potency and efficacy in inhibiting proliferation of human prostate cancer PC-3 cell line. | [88] |
| mRNA and protein accumulation are observed in anaplastic lymphoma kinase (ALK+) tumors. | [89] | |
| Inhibition of the EBP leads to cancer cell death via depletion of downstream sterols. | [90] | |
| SC5D | Decreased SC5D activity in cancer might increase prenylation of RAS, RAC or RHOC thereby promoting cancer progression. | [91] |
| Tumors | Protein–Protein Interactions |
|---|---|
| DLBC | MVK **—GSS (L—L) ***, MVK—ENO1 (L—M), MVD—EIF4EBP1 (L—M), SQLE—FAF2 (M—L), SQLE—TMCO3 (M—n/d), LSS—MYO5C (M—n/d), CYP51A1—ETS1 (n/d—H), MSMO1—C3AR1 (n/d—n/d), MSMO1—ELAVL1 (n/d—M, MSMO1—P2RX5 (n/d—M), EBP—VKORC1 (n/d—n/d), EBP—ENO1 (n/d—M) |
| PAAD | MSMO1—GPR35(n/d—n/d), EBP—MOV10 (L—n/d), DHCR7—FADS1 (H—M) |
| PRAD | DHCR24—ERAP1(n/d—M), DHCR24—CKAP5 (n/d—M), DHCR24—PTPN1 (n/d—n/d), DHCR24—REEP5 (n/d—M), DHCR24—UBL4A (n/d—M) |
| READ | HMGCR—VCP (M—M), DHCR24—CKAP5 (M—M), HMGCR—INSIG1 (M—n/d), DHCR7—FADS1 (H—M) |
| THYM | MVK—TACC3, FDPS—ATXN1, FDPS—PSME4, FDPS—TALDO1, FDPS—SEC31A, SQLE—MARCH6, SQLE—CHRND, LSS—ALOX5, LSS—MYH9, CYP51A1—SPINT2, MSMO1—ATXN1, MSMO1—ELAVL1, HSD17B7—GORAB, SC5D—IPPK, EBP—HSP90B1 (neg *), EBP—ABCE1 (neg), EBP—KSR1 (neg), EBP—TCTN2 (neg), EBP—BSCL2 (neg), EBP—MFSD8 (neg), FDPS—NME1, SQLE—FAF2, SQLE—TMCO3, MSMO1—EIF3A, EBP—NCSTN (neg) |
| Metabolite | References |
|---|---|
| Group I (metabolites promoting tumors) | |
| Mevalonate | [50,171,172,173,174,175,176,177,178,179] |
| Mevalonate-5-phosphate | [180] |
| Squalene | [181,182] |
| Group II (metabolites with protective effects) | |
| Lanosterol | [183] |
| 4,4 -dimethyl-cholesta-8,14,24-trienol | [184] |
| 14-demethyl-lanosterol | [184] |
| 7-dehydrocholesterol | [185,186,187] |
| Group III (metabolites with no significant effects) | |
| (E,E)-farnesyl-pyrophophate | [188] |
| Zymosterol | [189,190] |
| Lathosterol | [191,192] |
| 7-dehydrodesmosterol | [191,192] |
| Desmosterol | [190,191,192] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ershov, P.; Kaluzhskiy, L.; Mezentsev, Y.; Yablokov, E.; Gnedenko, O.; Ivanov, A. Enzymes in the Cholesterol Synthesis Pathway: Interactomics in the Cancer Context. Biomedicines 2021, 9, 895. https://doi.org/10.3390/biomedicines9080895
Ershov P, Kaluzhskiy L, Mezentsev Y, Yablokov E, Gnedenko O, Ivanov A. Enzymes in the Cholesterol Synthesis Pathway: Interactomics in the Cancer Context. Biomedicines. 2021; 9(8):895. https://doi.org/10.3390/biomedicines9080895
Chicago/Turabian StyleErshov, Pavel, Leonid Kaluzhskiy, Yuri Mezentsev, Evgeniy Yablokov, Oksana Gnedenko, and Alexis Ivanov. 2021. "Enzymes in the Cholesterol Synthesis Pathway: Interactomics in the Cancer Context" Biomedicines 9, no. 8: 895. https://doi.org/10.3390/biomedicines9080895
APA StyleErshov, P., Kaluzhskiy, L., Mezentsev, Y., Yablokov, E., Gnedenko, O., & Ivanov, A. (2021). Enzymes in the Cholesterol Synthesis Pathway: Interactomics in the Cancer Context. Biomedicines, 9(8), 895. https://doi.org/10.3390/biomedicines9080895