
High Na+ Salt Diet and Remodeling of Vascular Smooth Muscle and Endothelial Cells


{kind=link}
{kind=link}
{kind=link}
Abstract
1. The Vascular System
2. Vascular Remodeling
3. Sodium and Sodium Transport in Vascular Smooth Muscle and Endothelial Cells
4. High Sodium Salt-Induced Salt-Sensitive Memory
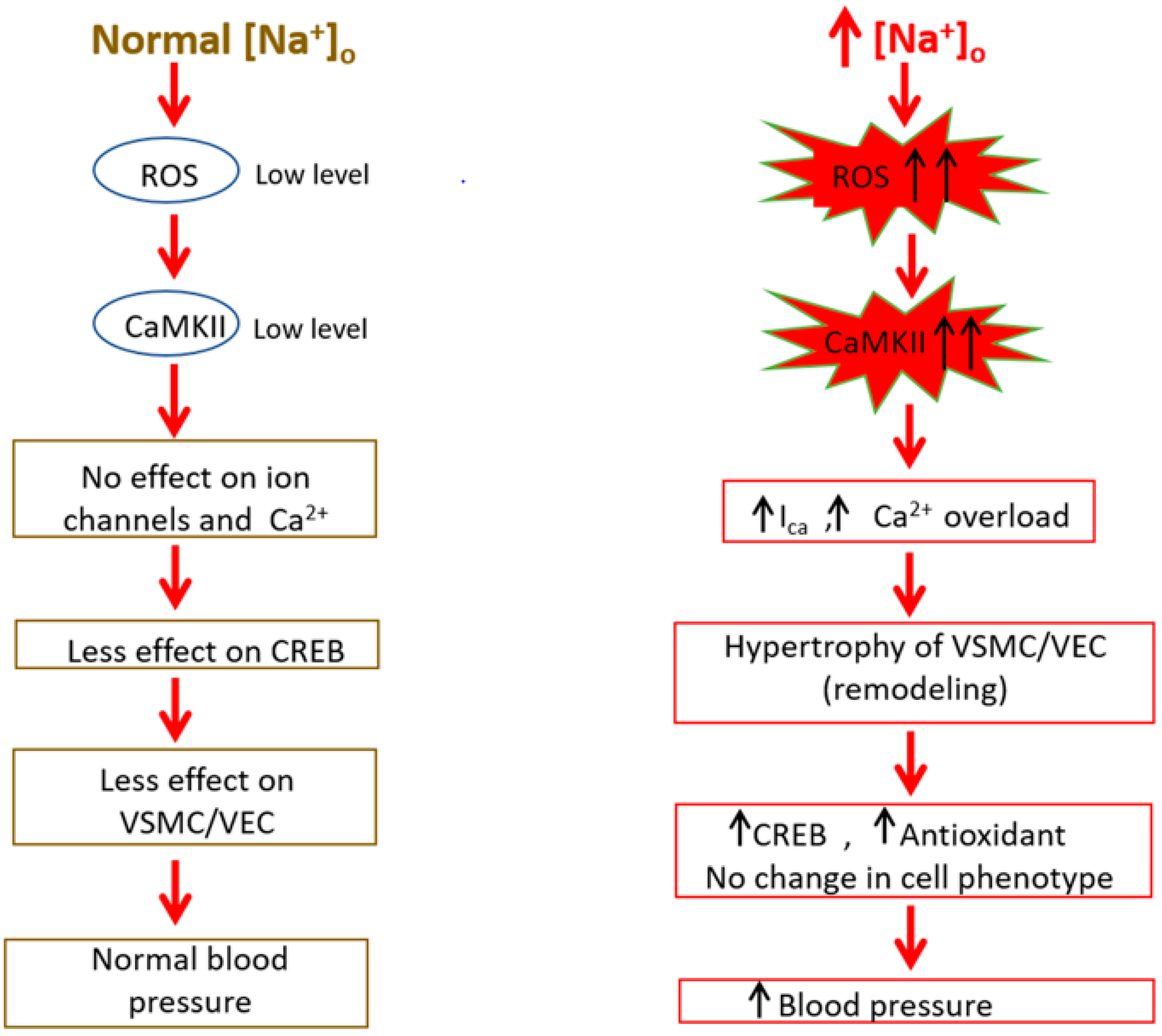
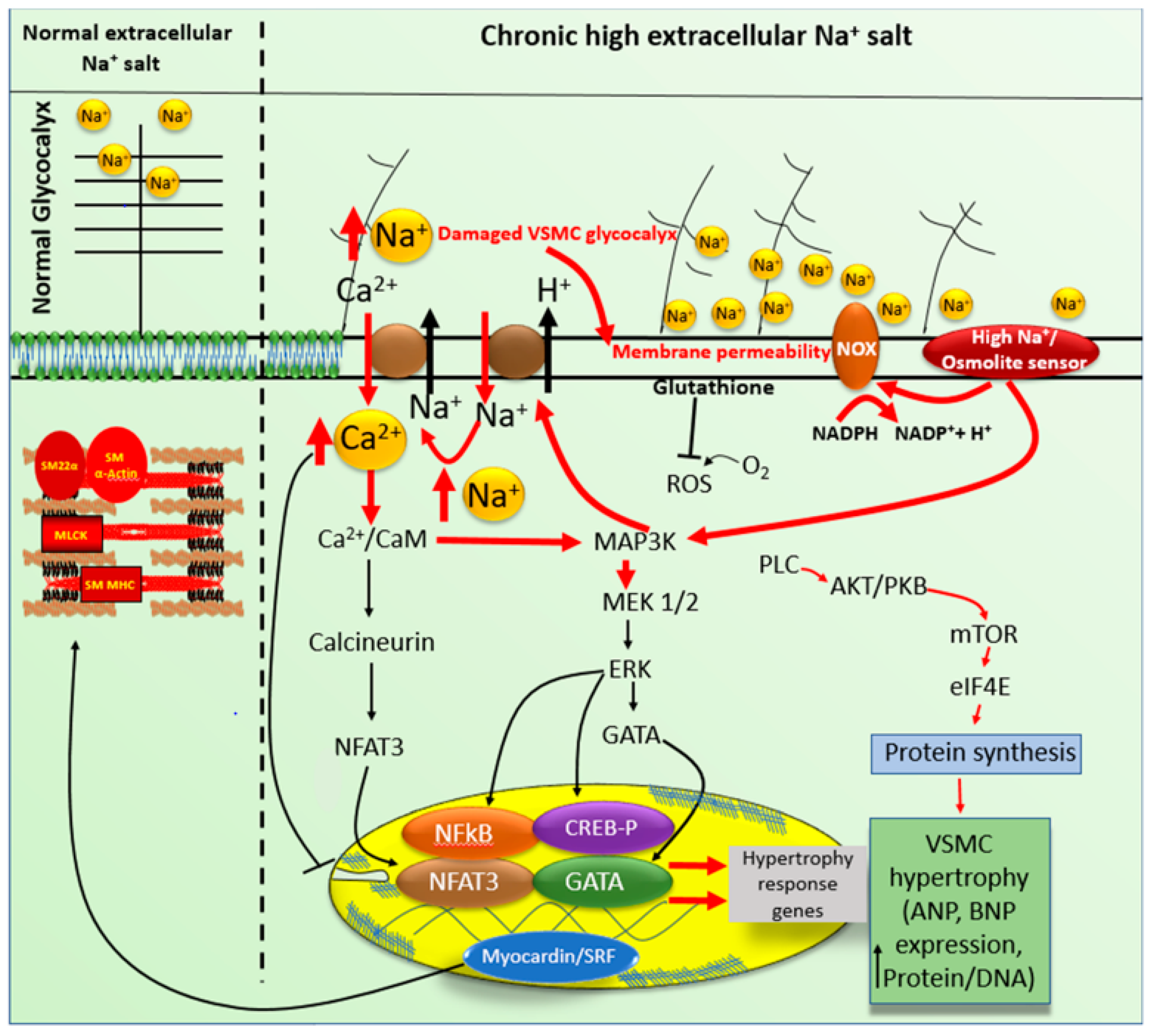
5. High Sodium Salt-Induced VSMCs and VECs Stress
6. High Na+ Salt-Induced Glycocalyx Remodeling
7. Adaptive Responses to High Sodium Salt Induced VSMCs Hypertrophy
8. Na+ Salt-Sensitive Hypertension
9. Implication of ROS/RNS in Na+ Sensitive Hypertension
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Pugsley, M.K.; Tabrizchi, R. The vascular system: An overview of structure and function. J. Pharmacol. Toxicol. 2000, 44, 333–340. [Google Scholar] [CrossRef]
- Bkaily, G.; Abou Abdallah, N.; Simon, Y.; Jazzar, A.; Jacques, D. Vascular smooth muscle remodeling in health and disease. Can. J. Physiol. Pharmacol. 2021, 99, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Bennett, H.S. Morphological aspects of extracellular polysaccharides. J. Histochem. Cytochem. 1963, 11, 14–23. [Google Scholar] [CrossRef]
- Wang, G.; Jacquet, L.; Karamariti, E.; Xu, Q. Origin and differentiation of vascular smooth muscle cells. J. Physiol. 2015, 593, 3013–3030. [Google Scholar] [CrossRef] [PubMed]
- Brozovich, F.V.; Nicholson, C.J.; Degen, C.V.; Gao, Y.Z.; Aggarwal, M.; Morgan, K.G. Mechanisms of Vascular Smooth Muscle Contraction and the Basis for Pharmacologic Treatment of Smooth Muscle Disorders. Pharmacol. Rev. 2016, 68, 476–532. [Google Scholar] [CrossRef] [PubMed]
- Chistiakov, D.A.; Orekhov, A.N.; Bobryshev, Y.V. Vascular smooth muscle cell in atherosclerosis. Acta Physiol. 2015, 214, 33–50. [Google Scholar] [CrossRef] [PubMed]
- Rzucidlo, E.M.; Martin, K.A.; Powell, R.J. Regulation of vascular smooth muscle cell differentiation. J. Vasc. Surg. 2007, 45, A25–A32. [Google Scholar] [CrossRef]
- Friedman, S.M. Sodium in blood vessels. J. Vasc. Res. 1979, 16, 2–16. [Google Scholar] [CrossRef]
- Owens, G.K. Control of hypertrophic versus hyperplastic growth of vascular smooth muscle cells. Am. J. Physiol. Heart Circ. Physiol. 1989, 257, H1755–H1765. [Google Scholar] [CrossRef] [PubMed]
- Bkaily, G.; Jaalouk, D.; Haddad, G.; Gros-Louis, N.; Simaan, M.; Naik, R.; Pothier, P. Modulation of cytosolic and nuclear Ca2+ and Na+ transport by taurine in heart cells. Mol. Cell. Biochem. 1997, 170, 1–8. [Google Scholar] [CrossRef]
- Baumbach, G.L.; Heistad, D.D. Remodeling of cerebral arterioles in chronic hypertension. Hypertension 1989, 13, 968–972. [Google Scholar] [CrossRef] [PubMed]
- Gibbons, G.H.; Dzau, V.J. The emerging concept of vascular remodeling. N. Engl. J. Med. 1994, 330, 1431–1438. [Google Scholar] [PubMed]
- Feihl, F.; Liaudet, L.; Waeber, B.; Levy, B.I. Hypertension: A disease of the microcirculation? Hypertension 2006, 48, 1012–1017. [Google Scholar] [CrossRef]
- Renna, N.F.; de las Heras, N.; Miatello, R.M. Pathophysiology of vascular remodeling in hypertension. Int. J. Hypertens. 2013, 2013, 808353. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; Dever, D.P.; Lee, C.M.; Azizi, A.; Pan, Y.; Camarena, J.; Köhnke, T.; Bao, G.; Porteus, M.H.; Majeti, R. The trace-seq method tracks recombination alleles and identifies clonal reconstitution dynamics of gene targeted human hematopoietic stem cells. Nat. Commun. 2021, 12, 472. [Google Scholar] [CrossRef]
- Lai, E.Y.; Onozato, M.L.; Solis, G.; Aslam, S.; Welch, W.J.; Wilcox, C.S. Myogenic responses of mouse isolated perfused renal afferent arterioles: Effects of salt intake and reduced renal mass. Hypertension 2010, 55, 983–989. [Google Scholar] [CrossRef] [PubMed]
- Raffetto, J.D.; Ligi, D.; Maniscalco, R.; Khalil, R.A.; Mannello, F. Why Venous Leg Ulcers Have Difficulty Healing: Overview on Pathophysiology, Clinical Consequences, and Treatment. J. Clin. Med. 2020, 24, 29. [Google Scholar] [CrossRef]
- Boutouyrie, P.; Laurent, S. Remodelage des grosses et petites artères dans l’hypertension artérielle. Sang Thromb. Vaiss. 2004, 16, 81–89. [Google Scholar]
- Berk, B.C. Vascular smooth muscle growth: Autocrine growth mechanisms. Physiol. Rev. 2001, 81, 999–1030. [Google Scholar] [CrossRef]
- Bkaily, G.; Simon, Y.; Menkovic, I.; Bkaily, C.; Jacques, D. High salt-induced hypertrophy of human vascular smooth muscle cells associated with a decrease in glycocalyx. J. Mol. Cell. Cardiol. 2018, 125, 1–5. [Google Scholar] [CrossRef]
- Jacques, D.; Bkaily, G. Endocardial endothelial cell hypertrophy takes place during the development of hereditary cardiomyopathy. Mol. Cell. Biochem. 2019, 453, 157–161. [Google Scholar] [CrossRef]
- Shiraishi, Y.; Ishigami, N.; Kujiraoka, T.; Sato, A.; Fujita, M.; Ido, Y.; Adachi, T. Deletion of Superoxide Dismutase 1 Blunted Inflammatory Aortic Remodeling in Hypertensive Mice under Angiotensin II Infusion. Antioxidants 2021, 10, 471. [Google Scholar] [CrossRef] [PubMed]
- Walsh, K.; Shiojima, I.; Gualberto, A. DNA replication and smooth muscle cell hypertrophy. J. Clin. Investig. 1999, 104, 673–674. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Osol, G.; Moore, L.G. Maternal uterine vascular remodeling during pregnancy. Microcirculation 2014, 21, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Hellsten, Y.; Nyberg, M. Cardiovascular adaptations to exercise training. Compr. Physiol. 2015, 15, 1–32. [Google Scholar]
- Daou, G.B.; Srivastava, A.K. Reactive oxygen species mediate Endothelin-1-induced activation of ERK1/2, PKB, and Pyk2 signaling, as well as protein synthesis, in vascular smooth muscle cells. Free Radic. Biol. Med. 2004, 37, 208–215. [Google Scholar] [CrossRef]
- Bohr, D.F.; Webb, R.C. Vascular smooth muscle membrane in hypertension. Annu. Rev. Pharmacol. Toxicol. 1988, 28, 389–409. [Google Scholar] [CrossRef] [PubMed]
- Noon, J.P.; Rice, P.J.; Baldessarini, R.J. Calcium leakage as a cause of the high resting tension in vascular smooth muscle from the spontaneously hypertensive rat. Proc. Natl. Acad. Sci. USA 1978, 75, 1605–1607. [Google Scholar] [CrossRef] [PubMed]
- Orlov, S.; Resink, T.J.; Bernhardt, J.; Ferracin, F.; Buhler, F.R. Vascular smooth muscle cell calcium fluxes. Regulation by angiotensin II and lipoproteins. Hypertension 1993, 21, 195–203. [Google Scholar] [CrossRef]
- Wamhoff, B.R.; Bowles, D.K.; Owens, G.K. Excitation–transcription coupling in arterial smooth muscle. Circ. Res. 2006, 98, 868–878. [Google Scholar] [CrossRef]
- Wamhoff, B.R.; Bowles, D.K.; Dietz, N.J.; Hu, Q.; Sturek, M. Exercise training attenuates coronary smooth muscle phenotypic modulation and nuclear Ca2+ signaling. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H2397–H2410. [Google Scholar] [CrossRef]
- Geisterfer, A.A.; Peach, M.J.; Owens, G.K. Angiotensin II induces hypertrophy, not hyperplasia, of cultured rat aortic smooth muscle cells. Circ. Res. 1988, 62, 749–756. [Google Scholar] [CrossRef]
- Atef, M.E.; Anand-Srivastava, M.B. Enhanced expression of Gqα and PLC-β1 proteins contributes to vascular smooth muscle cell hypertrophy in SHR: Role of endogenous angiotensin II and endothelin-1. Am. J. Physiol. Cell Physiol. 2014, 307, C97–C106. [Google Scholar] [CrossRef] [PubMed]
- Ohtsu, H.; Higuchi, S.; Shirai, H.; Eguchi, K.; Suzuki, H.; Hinoki, A.; Brailoiu, E.; Eckhart, A.D.; Frank, G.D.; Eguchi, S. Central role of Gq in the hypertrophic signal transduction of angiotensin II in vascular smooth muscle cells. Endocrinology 2008, 149, 3569–3575. [Google Scholar] [CrossRef]
- Jacques, D.; Abdel-Karim Abdel-Malak, N.; Abou Abdallah, N.; Al-Khoury, J.; Bkaily, G. Difference in the response to angiotensin II between left and right ventricular endocardial endothelial cells. Can. J. Physiol. Pharmacol. 2017, 95, 1271–1282. [Google Scholar] [CrossRef] [PubMed]
- Bkaily, G. The possible role of Ca2+ and K+ channels in VSM pathophysiology. In Ion Channels in Vascular Smooth Muscle; Bkaily, G., Ed.; R.G. Landers Company: Austin, TX, USA, 1994; pp. 103–113. [Google Scholar]
- Bkaily, G.; Nader, M.; Avedanian, L.; Choufani, S.; Jacques, D.; D’Orléans-Juste, P.; Gobeil, F.; Chemtob, S.; Al-Khoury, J. G-protein-coupled receptors, channels, and Na+–H+ exchanger in nuclear membranes of heart, hepatic, vascular endothelial, and smooth muscle cells. Can. J. Physiol. Pharmacol. 2006, 84, 431–441. [Google Scholar] [CrossRef]
- Bkaily, G.; Avedanian, L.; Al-Khoury, J.; Ahmarani, L.; Perreault, C.; Jacques, D. Receptors and ionic transporters in nuclear membranes: New targets for therapeutical pharmacological interventions. Can. J. Physiol. Pharmacol. 2012, 90, 953–965. [Google Scholar] [CrossRef]
- Alexander, R.T.; Grinstein, S. Na+/H+ exchangers and the regulation of volume. Acta Physiol. 2006, 187, 159–167. [Google Scholar] [CrossRef]
- Stock, C.; Schwab, A. Role of the Na+/H+ exchanger NHE1 in cell migration. Acta Physiol. 2006, 187, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Pouysségur, J.; Sardet, C.; Franchi, A.; L’Allemain, G.; Paris, S. A specific mutation abolishing Na/H antiport activity in hamster fibroblasts precludes growth at neutral and acidic ph. Proc. Nat. Acad. Sci. USA 1984, 81, 4833–4837. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, S.F. The Na+/H+ exchanger NHE1 in stress-induced signal transduction: Implications for cell proliferation and cell death. Pflüg. Arch. Eur. J. Physiol. 2006, 45, 249–259. [Google Scholar] [CrossRef]
- Bkaily, G.; Avedanian, L.; Jacques, D. Nuclear membranes’ receptors and channels as targets for drug development in cardiovascular diseases. Can. J. Physiol. Pharmacol. 2009, 87, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Cingolani, H.E.; Irene, L. Sodium-Hydrogen exchanger, cardiac overload, and myocardial hypertrophy. Circulation 2007, 115, 1090–1100. [Google Scholar] [CrossRef] [PubMed]
- Oberleithner, H.; Schuricht, B.; Wünsch, S.; Schneider, S.; Püschel, B. Role of H+ ions in volume and voltage of epithelial cell nuclei. Pflug. Arch. Eur. J. Phsysiol. 1993, 423, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Oberleithner, H.; Schiller, H.; Wilhelmi, M.; Butzke, D.; Danker, T. Nuclear pores collapse in response to CO2 imaged with atomic force microscopy. Pflug. Arch. Eur. J. Physiol. 2000, 439, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Karmazyn, M.; Kilić, A.; Javadov, S. The role of NHE-1 in myocardial hypertrophy and remodeling. J. Mol. Cell. Cardiol. 2008, 44, 647–653. [Google Scholar] [CrossRef] [PubMed]
- Nicoll, D.A.; Longoni, S.; Philipson, K.D. Molecular cloning and functional expression of the cardiac sarcolemmal Na+-Ca2+ exchanger. Science 1990, 250, 562–565. [Google Scholar] [CrossRef] [PubMed]
- Blaustein, M.P.; Lederer, W.J. Sodium/calcium exchange: Its physiological implications. Physiol. Rev. 1999, 79, 763–854. [Google Scholar] [CrossRef] [PubMed]
- Brini, M.; Carafoli, E. The plasma membrane Ca2+ ATPase and the plasma membrane sodium calcium exchanger cooperate in the regulation of cell calcium. Cold Spring Harb. Perspect. Biol. 2011, 3, 1–15. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, M.E.; Owen, N.E. Regulation of ion pumps and carriers in vascular smooth muscle. Physiol. Rev. 1994, 74, 683–722. [Google Scholar] [CrossRef]
- Philipson, K.D.; Nicoll, D.A. Sodium-Calcium Exchange: À Molecular Perspective. Annu. Rev. Physiol. 2000, 62, 111–133. [Google Scholar] [CrossRef]
- Gill, D.L.; Grollman, E.F.; Kohn, L.D. Calcium transport mechanisms in membrane vesicles from guinea pig brain synaptosomes. J. Biol. Chem. 1981, 256, 184–192. [Google Scholar] [CrossRef]
- Bkaily, G.; Jaalouk, D.; Sader, S.; Shbaklo, H.; Pothier, P.; Jacques, D.; D’Orléans-Juste, P.; Cragoe, E.J., Jr.; Bose, R. Taurine indirectly increases [Ca]i by inducing Ca2+ influx through the Na+-Ca2+ Exchanger. In Molecular and Cellular Effects of Nutrition on Disease Processes; Pierce, G.N., Izumi, T., Rupp, H., Grynberg, A., Eds.; Springer: Boston, MA, USA, 1998; pp. 187–197. [Google Scholar]
- Bkaily, G.; Gros-Louis, N.; Naik, R.; Jaalouk, D.; Pothier, P. Implication of the nucleus in excitation contraction coupling of heart cells. Mol. Cell. Biochem. 1996, 154, 113–121. [Google Scholar] [CrossRef]
- Jernigan, N.L.; Drummond, H.A. Vascular ENaC proteins are required for renal myogenic constriction. Am. J. Renal Physiol. 2005, 289, F891–F901. [Google Scholar] [CrossRef] [PubMed]
- Jernigan, N.L.; Lamarca, B.; Speed, J.; Galmiche, L.; Granger, J.P.; Drummond, H.A. Dietary salt enhances benzamil-sensitive component of myogenic constriction in mesenteric arteries. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H409–H420. [Google Scholar] [CrossRef]
- Kushe-Vihrog, K.; Jeggle, P.; Oberleithner, H. The role of ENaC in vascular endothelium. Pflug. Arch. Eur. J. Physiol. 2014, 466, 851–859. [Google Scholar] [CrossRef]
- Yang, X.; Niu, N.; Liang, C.; Wu, M.M.; Tang, L.L.; Wang, Q.S. Stimulation of epithelial sodium channels in endothelial cells by bone morphogenetic protein-4 contributes to salt-sensitive hypertension in rats. Oxid. Med. Cell. Longev. 2020, 2020, 3921897. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, R.M.; Padfield, P.L.; Seckl, J.R. Disorders of sodium balance. BMJ 2006, 332, 702–705. [Google Scholar] [CrossRef]
- Rose, B.D. New approach to disturbances in the plasma sodium concentration. Am. J. Med. 1986, 81, 1033–1040. [Google Scholar] [CrossRef]
- Shenouda, N.; Ramick, G.M.; Lennon, L.S.; Farquhar, B.W.; Edwards, G.D. High dietary sodium augments vascular tone and attenuates low-flow mediated constriction in salt-resistant adults. Eur. J. Appl. Physiol. 2020, 120, 1383–1389. [Google Scholar] [CrossRef] [PubMed]
- He, F.J.; MacGregor, G.A. Plasma sodium and hypertension. Kidney Int. 2004, 66, 2454–2466. [Google Scholar] [CrossRef] [PubMed]
- Uzan, A.; Delaveau, P. The salt content of food: A public health problem. Ann. Pharm. Fr. 2009, 67, 291–294. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, Z.; Yang, Z.; Yang, Y.; Yang, J.; Han, H.; Yang, H. The effect of different dietary levels of sodium and chloride on performance, blood parameters and excreta quality in goslings at 29 to 70 days of age. J. Anim. Physiol. Anim. Nutr. 2021. [Google Scholar] [CrossRef]
- Eaton, S.B.; Konner, M. Paleolithic nutrition. A consideration of its nature and current implications. N. Engl. J. Med. 1985, 312, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Meneton, P.; Jeunemaitre, X.; de Wardener, H.E.; Macgregor, G.A. Links between dietary salt intake, renal salt handling, blood pressure, and cardiovascular diseases. Physiol. Rev. 2005, 85, 679–715. [Google Scholar] [CrossRef] [PubMed]
- Oberleithner, H. Vascular endothelium: A vulnerable transit zone for merciless sodium. Nephrol. Dial. Transplant. 2013, 29, 240–246. [Google Scholar] [CrossRef]
- Chauveau, P.; Fouque, D.; Combe, C.; Aparicio, M. Évolution de l’alimentation du paléolithique à nos jours: Progression ou régression? Néphrologie Thérapeutique 2013, 9, 202–208. [Google Scholar] [CrossRef]
- Powles, J.; Fahimi, S.; Micha, R.; Khatibzadeh, S.; Shi, P.; Ezzati, M.; Engell, R.E.; Lim, S.S.; Danaei, G.; Mozaffarian, D. Global, regional and national sodium intakes in 1990 and 2010: A systematic analysis of 24 h urinary sodium excretion and dietary surveys worldwide. BMJ Open 2013, 3, e003733. [Google Scholar] [CrossRef] [PubMed]
- Luft, F.C.; Rankin, L.I.; Bloch, R.; Weyman, A.E.; Willis, L.R.; Murray, R.H.; Grim, C.E.; Weinberger, M.H. Cardiovascular and humoral responses to extremes of sodium intake in normal black and white men. Circulation 1979, 60, 697–706. [Google Scholar] [CrossRef]
- Sullivan, J.M.; Ratts, T.E.; Taylor, J.C.; Kraus, D.H.; Barton, B.R.; Patrick, D.R.; Reed, S.W. Hemodynamic effects of dietary sodium in man: A preliminary report. Hypertension 1980, 2, 506–514. [Google Scholar] [CrossRef]
- Sagnella, G.A.; Markandu, N.D.; Buckley, M.G.; Miller, M.A.; Singer, D.R.; MacGregor, G.A. Hormonal responses to gradual changes in dietary sodium intake in humans. Am. J. Physiol. 1989, 256, R1171–R1175. [Google Scholar] [CrossRef]
- Kliche, K.; Jeggle, P.; Pavenstädt, H.; Oberleithner, H. Role of cellular mechanics in the function and life span of vascular endothelium. Pflüg. Arch. Eur. J. Physiol. 2011, 462, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Elliott, P.; Walker, L.L.; Little, M.P.; Blair-West, J.R.; Shade, R.E.; Lee, D.R.; Rouquet, P.; Leroy, E.; Jeunemaitre, X.; Ardaillou, R.; et al. Change in salt intake affects blood pressure of chimpanzees: Implications for human populations. Circulation 2007, 116, 1563–1568. [Google Scholar] [CrossRef]
- He, F.J.; MacGregor, G.A. Reducing population salt intake worldwide: From evidence to implementation. Prog. Cardiovasc. Dis. 2010, 52, 363–382. [Google Scholar] [CrossRef] [PubMed]
- He, F.J.; Markandu, N.D.; Sagnella, G.A.; de Wardener, H.E.; MacGregor, G.A. Plasma Sodium: Ignored and Underestimated. Hypertension 2004, 4, 98–102. [Google Scholar] [CrossRef]
- Suckling, R.J.; He, F.J.; Markandu, N.D.; MacGregor, G.A. Dietary salt influences postprandial plasma sodium concentration and systolic blood pressure. Kidney Int. 2012, 81, 407–411. [Google Scholar] [CrossRef] [PubMed]
- Verbalis, J.G. Disorders of body water homeostasis. Best Pract. Res. Clin. Endocrinol. Metab. 2003, 17, 471–503. [Google Scholar] [CrossRef]
- Brocker, C.; Thompson, D.C.; Vasiliou, V. The role of hyperosmotic stress in inflammation and disease. Biomol. Concepts 2012, 3, 345–364. [Google Scholar] [CrossRef]
- Choi, H.Y.; Park, H.C.; Ha, S.K. Salt sensitivity and hypertension: A paradigm shift from kidney malfunction to vascular endothelial dysfunction. Electrolyte Blood Press. 2015, 13, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, T.; Delea, C.S.; Bartter, F.C.; Smith, H. The effect of high-sodium and low-sodium intakes on blood pressure and other related variables in human subjects with idiopathic hypertension. Am. J. Med. 1978, 64, 193–198. [Google Scholar] [CrossRef]
- Weinberger, M.H. Salt Sensitivity of Blood Pressure in Humans. Hypertension 1996, 27, 481–490. [Google Scholar] [CrossRef]
- De la Sierra, A.; Giner, V.; Bragulat, E.; Coca, A. Lack of correlation between two methods for the assessment of salt sensitivity in essential hypertension. J. Hum. Hypertens. 2002, 16, 255–260. [Google Scholar] [CrossRef]
- The GenSalt Collaborative Research Group. Genetic epidemiology network of salt sensitivity (GenSalt): Rationale, design, methods, and baseline characteristics of study participants. J. Hum. Hypertens. 2007, 21, 639–646. [Google Scholar] [CrossRef]
- Ando, K.; Fujita, T. Pathophysiology of salt sensitivity hypertension. Ann. Med. Hels. 2012, 44, S119–S126. [Google Scholar] [CrossRef]
- De Wardener, H.E. The primary role of the kidney and salt intake in the etiology of essential hypertension: Part I. Clin. Sci. 1990, 79, 193–200. [Google Scholar] [CrossRef]
- Campese, V.M.; Parise, M.; Karubian, F.; Bigazzi, R. Abnormal renal hemodynamics in black salt-sensitive patients with hypertension. Hypertension 1991, 18, 805–812. [Google Scholar] [CrossRef] [PubMed]
- Denton, D.; Weisinger, R.; Mundy, N.I.; Wickings, E.J.; Dixson, A.; Moisson, P.; Pingard, A.M.; Shade, R.; Carey, D.; Ardaillou, R. The effect of increased salt intake on blood pressure of chimpanzees. Nat. Med. 1995, 1, 1009–1016. [Google Scholar] [CrossRef] [PubMed]
- Paudel, P.; Mcdonald, J.F.; Fronius, M. The δ subunit of epithelial sodium channel in humans—A potential player in vascular physiology. Am. J. Physiol. Heart Circ. Physiol. 2021, 320, H487–H493. [Google Scholar] [CrossRef]
- Kusche-Vihrog, K.; Oberleithner, H. An emerging concept of vascular salt sensitivity. F1000 Biol. Rep. 2012, 4, 20. [Google Scholar] [CrossRef] [PubMed]
- Vestweber, D. Novel insights into leukocyte extravasation. Curr. Opin. Hematol. 2012, 19, 212–217. [Google Scholar] [CrossRef]
- Bevan, J.A. Flow regulation of vascular tone. Its sensitivity to changes in sodium and calcium. Hypertension 1993, 22, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Pries, A.R.; Secomb, T.W.; Gaehtgens, P. The endothelial surface layer. Pflüg. Arch. Eur. J. Physiol. 2000, 440, 653–666. [Google Scholar] [CrossRef]
- Oberleithner, H. A physiological concept unmasking vascular salt sensitivity in man. Pflug. Arch. Eur. J. Physiol. 2012, 464, 287–293. [Google Scholar] [CrossRef][Green Version]
- Oberleithner, H.; Peters, W.; Kusche-Vihrog, K.S.; Schillers, H.; Kliche, K.; Oberleithner, K. Salt overload damages the glycocalyx sodium barrier of vascular endothelium. Pflüg. Arch. Eur. J. Physiol. 2011, 462, 519–528. [Google Scholar] [CrossRef]
- Lee, R.; Garfield, R.E.; Forrest, J.B.; Daniel, E.E. Morphometric study of structural changes in the mesenteric blood vessels of spontaneously hypertensive rats. J. Vasc. Res. 1983, 20, 57–71. [Google Scholar] [CrossRef] [PubMed]
- Mulvany, M.J.; Baandrup, U.; Gundersen, H.J.G. Evidence for hyperplasia in mesenteric resistance vessels of spontaneously hypertensive rats using a three-dimensional disector. Circ. Res. 1985, 57, 794–800. [Google Scholar] [CrossRef] [PubMed]
- Schiffrin, E.L. Vascular Remodeling in Hypertension. Hypertension 2012, 59, 367–374. [Google Scholar] [CrossRef]
- Kuper, C.; Beck, F.-X.; Neuhofer, W. Osmoadaptation of Mammalian cells-an orchestrated network of protective genes. Curr. Genom. 2007, 8, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Halterman, J.A.; Kwon, H.M.; Zargham, R.; Bortz, P.D.S.; Wamhoff, B.R. Nuclear factor of activated T cells 5 regulates vascular smooth muscle cell phenotypic modulation. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2287–2296. [Google Scholar] [CrossRef] [PubMed]
- Neuhofer, W. Role of NFAT5 in inflammatory disorders associated with osmotic stress. Curr. Genom. 2010, 11, 584–590. [Google Scholar] [CrossRef]
- Trama, J.; Go, W.Y.; Ho, S.N. The osmoprotective function of the NFAT5 transcription factor in T cell development and activation. J. Immunol. 2002, 169, 5477–5488. [Google Scholar] [CrossRef]
- Reinehr, R.; Häussinger, D. Hyperosmotic activation of the CD95 death receptor system. Acta Physiol. 2006, 187, 199–203. [Google Scholar] [CrossRef] [PubMed]
- Burg, M.B.; Ferraris, J.D.; Dmitrieva, N.I. Cellular response to hyperosmotic stresses. Physiol. Rev. 2007, 87, 1441–1474. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, T.; Kadowaki, H.; Okamoto, N.; Nagai, A.; Naguro, I.; Matsuzawa, A.; Shibuya, H.; Tanaka, K.; Murata, S.; Takeda, K. CHIP-dependent termination of MEKK2 regulates temporal ERK activation required for proper hyperosmotic response. EMBO J. 2010, 29, 2501–2514. [Google Scholar] [CrossRef]
- Umenishi, F.; Yoshihara, S.; Narikiyo, T.; Schrier, R.W. Modulation of hypertonicity-induced aquaporin-1 by sodium chloride, urea, betaine, and heat shock in murine renal medullary cells. J. Am. Soc. Nephrol. 2005, 16, 600–607. [Google Scholar] [CrossRef] [PubMed]
- Gaudette, S.; Hughes, D.; Boller, M. The endothelial glycocalyx: Structure and function in health and critical illness. J. Vet. Emerg. Crit. Care 2020, 30, 117–134. [Google Scholar] [CrossRef]
- Danielli, J.F. Capillary permeability and oedema in the perfused frog. J. Physiol. 1940, 98, 109–129. [Google Scholar] [CrossRef]
- Kusche-Vihrog, K.; Schmitz, B.; Brand, E. Salt controls endothelial and vascular phenotype. Pflug. Arch. Eur. J. Physiol. 2015, 467, 499–512. [Google Scholar] [CrossRef] [PubMed]
- Machin, D.R.; Bloom, S.I.; Campbell, R.A.; Phuong, T.T.; Gates, P.E.; Lesniewski, L.A.; Rondina, M.T.; Donato, A.J. Advanced age results in a diminished endothelial glycocalyx. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H531–H539. [Google Scholar] [CrossRef]
- Machin, D.R.; Phuong, T.T.; Donato, A.J. The role of the endothelial glycocalyx in advanced age and cardiovascular disease. Curr. Opin. Pharmacol. 2019, 45, 66–71. [Google Scholar] [CrossRef]
- Osuka, A.; Kusuki, H.; Yoneda, K.; Matsuura, H.; Matsumoto, H.; Ogura, H.; Ueyama, M. Glycocalyx shedding is enhanced by age and correlates with increased fluid requirement in patients with major burns. Shock 2018, 50, 60–65. [Google Scholar] [CrossRef]
- Dmitrieva, N.I.; Michea, L.F.; Rocha, G.M.; Burg, M.B. Cell cycle delay and apoptosis in response to osmotic stress. Comp. Biochem. Physiol. Part A Mol. Integr. Physiol. 2001, 130, 411–420. [Google Scholar] [CrossRef]
- Schaffer, S.; Takahashi, K.; Azuma, J. Role of osmoregulation in the actions of taurine. Amino Acids 2000, 19, 527–546. [Google Scholar] [CrossRef]
- Wehner, F.; Tinel, H. Osmolyte and Na+ transport balances of rat hepatocytes as a function of hypertonic stress. Pflüg. Arch. Eur. J. Physiol. 2000, 441, 12–24. [Google Scholar] [CrossRef] [PubMed]
- Wehner, F.; Olsen, H.; Tinel, H.; Kinne-Saffran, E.; Kinne, R.K. Cell volume regulation: Osmolytes, osmolyte transport, and signal transduction. Rev. Physiol. Biochem. Pharmacol. 2003, 148, 1–80. [Google Scholar]
- Alfieri, R.R.; Bonelli, M.A.; Cavazzoni, A.; Brigotti, M.; Fumarola, C.; Sestili, P.; Mozzoni, P.; De Palma, G.; Mutti, A.; Carnicelli, D. Creatine as a compatible osmolyte in muscle cells exposed to hypertonic stress. J. Physiol. 2006, 576, 391–401. [Google Scholar] [CrossRef] [PubMed]
- Yancey, P.H. Organic osmolytes as compatible, metabolic and counteracting cytoprotectants in high osmolarity and other stresses. J. Exp. Biol. 2005, 208, 2819–2830. [Google Scholar] [CrossRef] [PubMed]
- Natochin, Y.V. The physiological evolution of animals: Sodium is the clue to resolving contradictions. Her. Russ. Acad. Sci. 2007, 77, 581–591. [Google Scholar] [CrossRef]
- Ambard, L.; Beaujard, E. Causes of arterial hypertension. Arch. Gen. Med. 1904, 1, 520–533. [Google Scholar]
- Amiri, M.; Kelishadi, R. Can salt hypothesis explain the trends of mortality from stroke and stomach cancer in western Europe? Int. J. Prev. Med. 2012, 3, 377–378. [Google Scholar]
- Rapp, J.P. Dahl Salt-Susceptible and Salt-Resistant Rats. Hypertension 1982, 4, 753–763. [Google Scholar] [CrossRef]
- Freis, E.D. The role of salt in hypertension. Blood Press. 1992, 1, 196–200. [Google Scholar] [CrossRef]
- Rivasi, G.; Fedorowski, A. Hypertension, hypotension and syncope. Minerva Med. 2021. [Google Scholar] [CrossRef]
- Lim, S.S.; Vos, T.; Flaxman, A.D.; Danaei, G.; Shibuya, K.; Adair-Rohani, H.; Amann, M.; Anderson, H.R.; Andrews, K.G.; Aryee, M.; et al. A comparative risk assessment of burden of disease and injury attributable to 67 risk factors and risk factor clusters in 21 regions, 1990–2010: A systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012, 380, 2224–2260. [Google Scholar] [CrossRef]
- Chobanian, A.V.; Bakris, G.L.; Black, H.R.; Cushman, W.C.; Green, L.A.; Izzo, J.L.; Jones, D.W.; Materson, B.J.; Oparil, S.; Wright, J.T. Seventh report of the joint national committee on prevention, detection, evaluation, and treatment of high blood pressure. Hypertension 2003, 42, 1206–1252. [Google Scholar] [CrossRef]
- Marketou, M.E.; Maragkoudakis, S.; Anastasiou, I.; Nakou, H.; Plataki, M.; Vardas, P.E.; Parthenakis, F.I. Salt-induced effects on microvascular function: A critical factor in hypertension mediated organ damage. J. Clin. Hypertens. 2019, 21, 749–757. [Google Scholar] [CrossRef]
- Nijst, P.; Verbrugge, F.H.; Grieten, L.; Dupont, M.; Steels, P.; Tang, W.H.W.; Mullens, W. The pathophysiological role of interstitial sodium in heart failure. J. Am. Coll. Cardiol. 2015, 65, 378–388. [Google Scholar] [CrossRef] [PubMed]
- Dogné, S.L.; Flamion, B.; Caron, N. Endothelial Glycocalyx as a Shield Against Diabetic Vascular Complications: Involvement of Hyaluronan and Hyaluronidases. Arterioscler. Thromb. Vasc. Biol. 2018, 8, 1427–1439. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.W.; Anand, V.; Shek, E.W.; Moore, M.C.; Brady, A.L.; Kelly, W.C.; Adair, T.H. Sodium induces hypertrophy of cultured myocardial myoblasts and vascular smooth muscle cells. Hypertension 1998, 31, 1083–1087. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Naguro, I.; Ichijo, H.; Watanabe, K. Mitogen-activated protein kinases as key players in osmotic stress signaling. Biochim. Biophys. Acta 2016, 1860, 2037–2052. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; White, J.; Guo, L.; Zhao, X.; Wang, J.; Smart, E.J.; Li, X.A. Salt inactivates endothelial nitric oxide synthase in endothelial cells. J. Nutr. 2009, 139, 447–451. [Google Scholar] [CrossRef]
- Touyz, R.M.; Schiffrin, E.L. Signal transduction mechanisms mediating the physiological and pathophysiological actions of angiotensin II in vascular smooth muscle cells. Pharmacol. Rev. 2000, 52, 639–672. [Google Scholar] [PubMed]
- Oguchi, H.; Sasamura, H.; Shinoda, K.; Morita, S.; Kono, H.; Nakagawa, K.; Ishiguro, K.; Hayashi, K.; Nakamura, M.; Azegami, T.; et al. Renal arteriolar injury by salt intake contributes to salt memory for the development of hypertension. Hypertension 2014, 64, 784–791. [Google Scholar] [CrossRef]
- Di Meo, S.; Reed, T.T.; Venditti, P.; Victor, V.M. Role of ROS and RNS sources in physiological and pathological conditions. Oxid. Med. Cell. Longev. 2016, 2016, 1245049. [Google Scholar] [CrossRef] [PubMed]
- Ahmarani, L.; Avedanian, L.; Al-Khoury, J.; Perreault, C.; Jacques, D.; Bkaily, G. Whole-cell and nuclear NADPH oxidases levels and distribution in human endocardial endothelial, vascular smooth muscle, and vascular endothelial cells. Can. J. Physiol. Pharmacol. 2013, 91, 71–79. [Google Scholar] [CrossRef]
- Bayorh, M.A.; Ganafa, A.A.; Socci, R.R.; Silvestrov, N.; Abukhalaf, I.K. The role of oxidative stress in salt-induced hypertension. Am. J. Hypertens. 2004, 17, 31–36. [Google Scholar]
- Cai, H. Hydrogen peroxide regulation of endothelial function: Origins, mechanisms, and consequences. Cardiovasc. Res. 2005, 68, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Lai, E.Y.; Luo, Z.; Solis, G.; Mendonca, M.; Griendling, K.K.; Wellstein, A.; Welch, W.J.; Wilcox, C.S. High Salt Enhances Reactive Oxygen Species and Angiotensin II Contractions of Glomerular Afferent Arterioles from Mice with Reduced Renal Mass. Hypertension 2018, 72, 1208–1216. [Google Scholar] [CrossRef] [PubMed]
- Koga, Y.; Hirooka, Y.; Araki, S.; Nozoe, M.; Kishi, T.; Sunagawa, K. High salt intake enhances blood pressure increase during development of hypertension via oxidative stress in rostral ventrolateral medulla of spontaneously hypertensive rats. Hypertens. Res. 2008, 31, 2075–2083. [Google Scholar] [CrossRef]
- Dornas, W.C.; Cardoso, L.M.; Silva, M.; Machado, N.L.S.; Chianca, D.A., Jr.; Alzamora, A.C.; Lima, W.G.; Lagente, V.; Silva, M.E. Oxidative stress causes hypertension and activation of nuclear factor-κB after high-fructose and salt treatments. Sci. Rep. 2017, 11, 46051. [Google Scholar] [CrossRef]
- Abais-Battad, J.M.; Lund, H.; Dasinger, J.H.; Fehrenbach, D.J.; Cowley, A.W., Jr.; Mattson, D.L. NOX2-derived reactive oxygen species in immune cells exacerbates salt-sensitive hypertension. Free Radic. Biol. Med. 2020, 146, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Montiel, V.; Bella, R.; Michel, L.Y.M.; Esfahani, H.; Mulder, D.D.; Robinson, E.L.; Deglasse, J.P.; Tiburcy, M.; Chow, P.H.; Jonas, J.C.; et al. Inhibition of aquaporin-1 prevents myocardial remodeling by blocking the transmembrane transport of hydrogen peroxide. Sci. Transl. Med. 2020, 12, eaay2176. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bkaily, G.; Simon, Y.; Jazzar, A.; Najibeddine, H.; Normand, A.; Jacques, D. High Na+ Salt Diet and Remodeling of Vascular Smooth Muscle and Endothelial Cells. Biomedicines 2021, 9, 883. https://doi.org/10.3390/biomedicines9080883
Bkaily G, Simon Y, Jazzar A, Najibeddine H, Normand A, Jacques D. High Na+ Salt Diet and Remodeling of Vascular Smooth Muscle and Endothelial Cells. Biomedicines. 2021; 9(8):883. https://doi.org/10.3390/biomedicines9080883
Chicago/Turabian StyleBkaily, Ghassan, Yanick Simon, Ashley Jazzar, Houssein Najibeddine, Alexandre Normand, and Danielle Jacques. 2021. "High Na+ Salt Diet and Remodeling of Vascular Smooth Muscle and Endothelial Cells" Biomedicines 9, no. 8: 883. https://doi.org/10.3390/biomedicines9080883
APA StyleBkaily, G., Simon, Y., Jazzar, A., Najibeddine, H., Normand, A., & Jacques, D. (2021). High Na+ Salt Diet and Remodeling of Vascular Smooth Muscle and Endothelial Cells. Biomedicines, 9(8), 883. https://doi.org/10.3390/biomedicines9080883