
Zebrafish Blunt-Force TBI Induces Heterogenous Injury Pathologies That Mimic Human TBI and Responds with Sonic Hedgehog-Dependent Cell Proliferation across the Neuroaxis

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Fish Lines and Maintenance
2.2. TBI Induction via Modified Marmarou Weight Drop
2.3. Mortality and Early/Latent Seizure
2.4. Recovery Rate
2.5. Fluid Content Measurement
2.6. Tissue Processing
2.7. Vascular Injury
2.8. Sensorimotor Assay
2.9. Locomotion
2.10. Shuttle Box Assay Testing Apparatus
2.11. Learning
2.12. Immediate and Delayed Recall
2.13. Startle Response Habituation
2.14. Terminal Deoxynucleotidyl Transferase dUTP Nick-End Labeling (TUNEL) Assay
2.15. EdU Labeling
2.16. Immunohistochemistry
2.17. Image Acquisition
2.18. Tissue Clearing/EdU Labeling
2.19. Optical Density
2.20. Evans Blue Assay for Blood–Brain Barrier Disruption
2.21. Sonic Hedgehog Modulation
2.22. Quantitative Real-Time PCR (qRT-PCR)
2.23. Statistical Analysis
3. Results
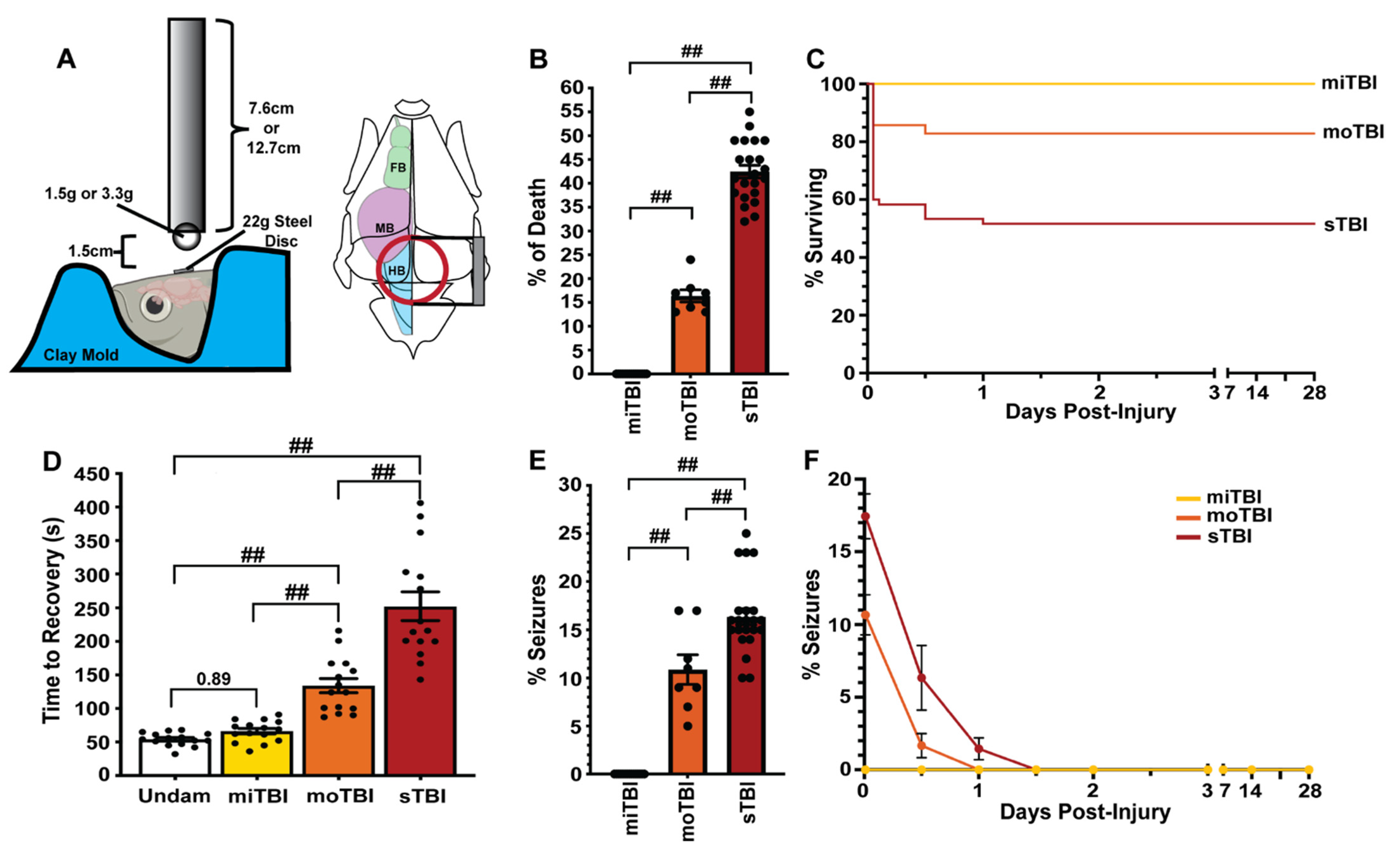
3.1. Modified Marmarou Weight Drop Results in a Reproducible and Scalable TBI
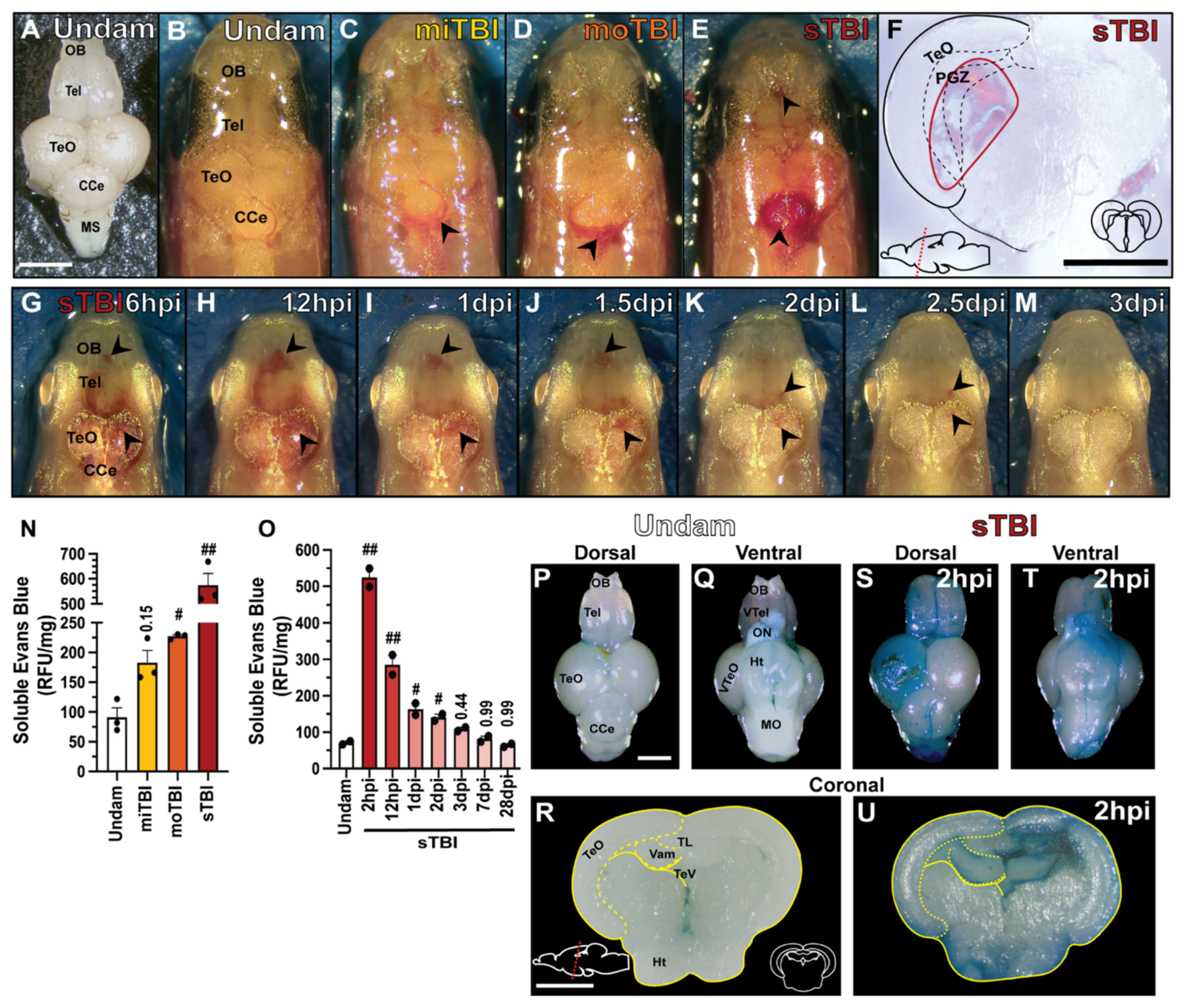
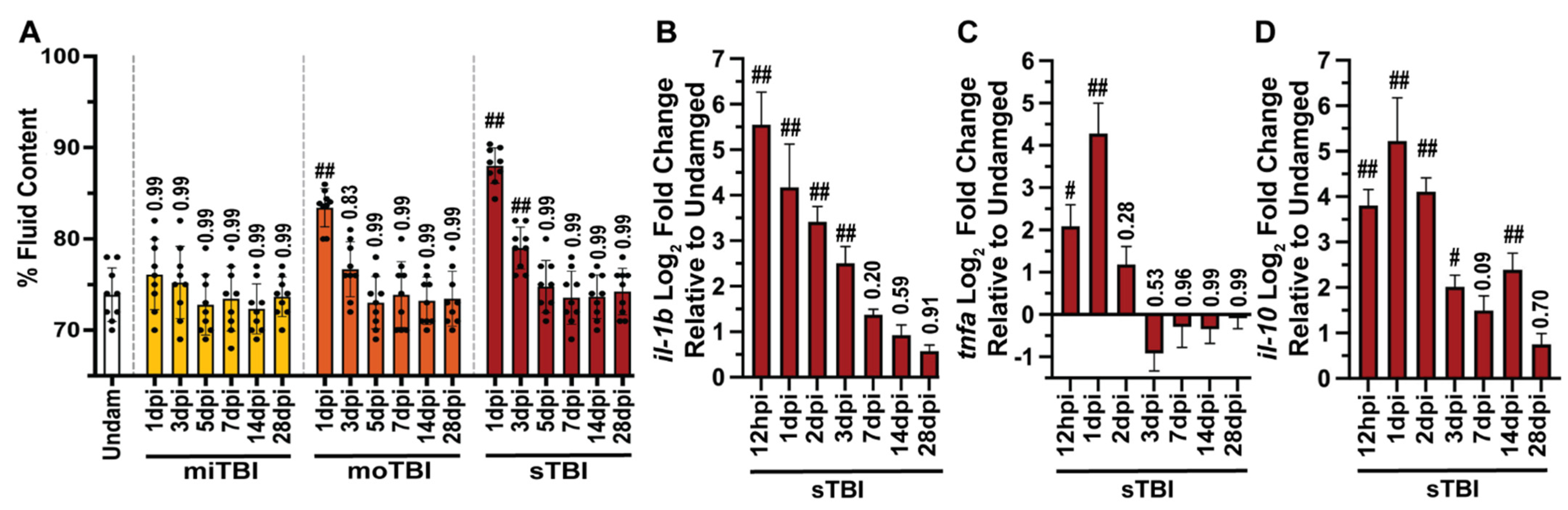
3.2. Blunt-Force TBI Induces Severity-Dependent Vascular Injury with Blood–Brain Barrier Disruption, Neuroinflammation, and Edema
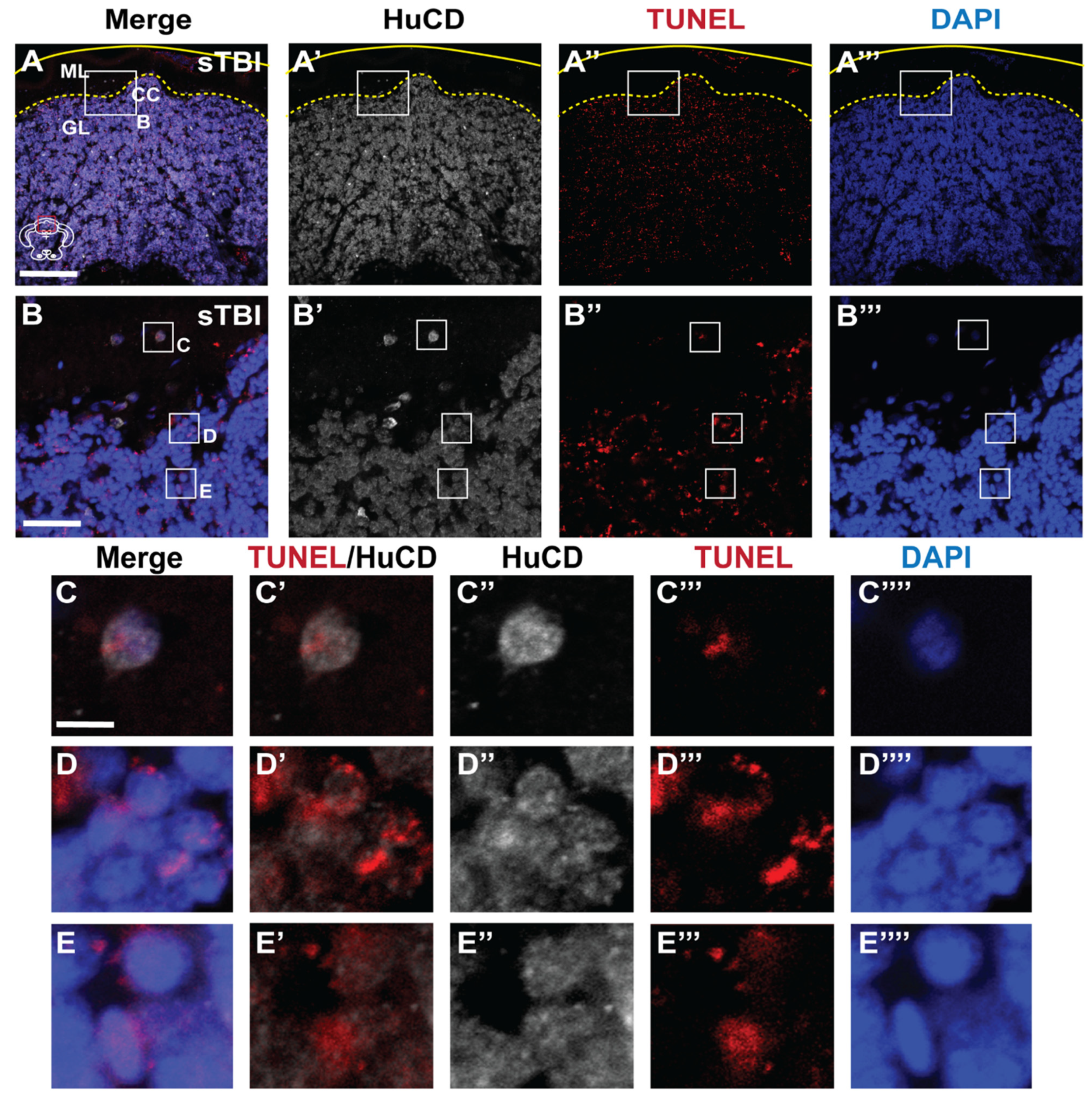
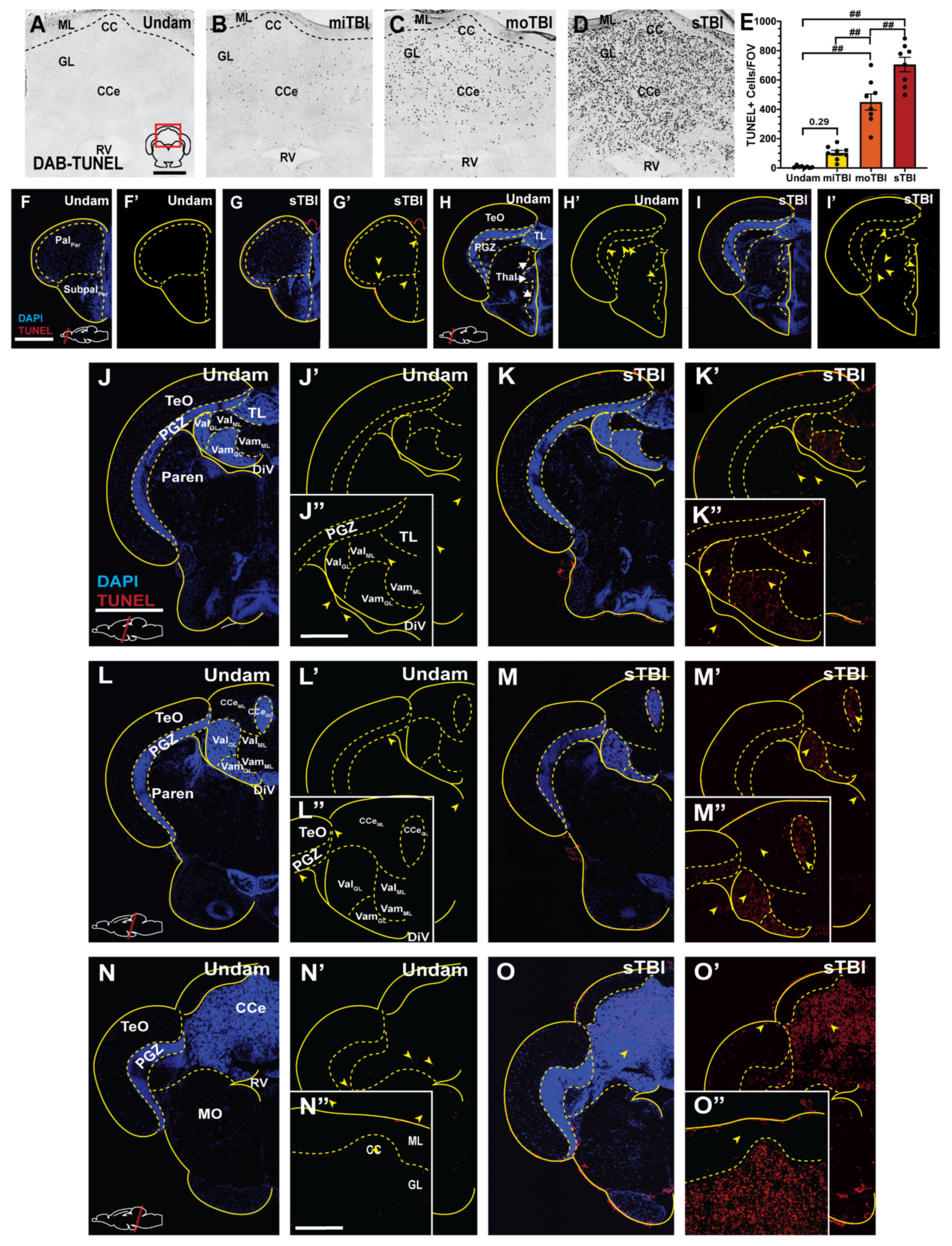
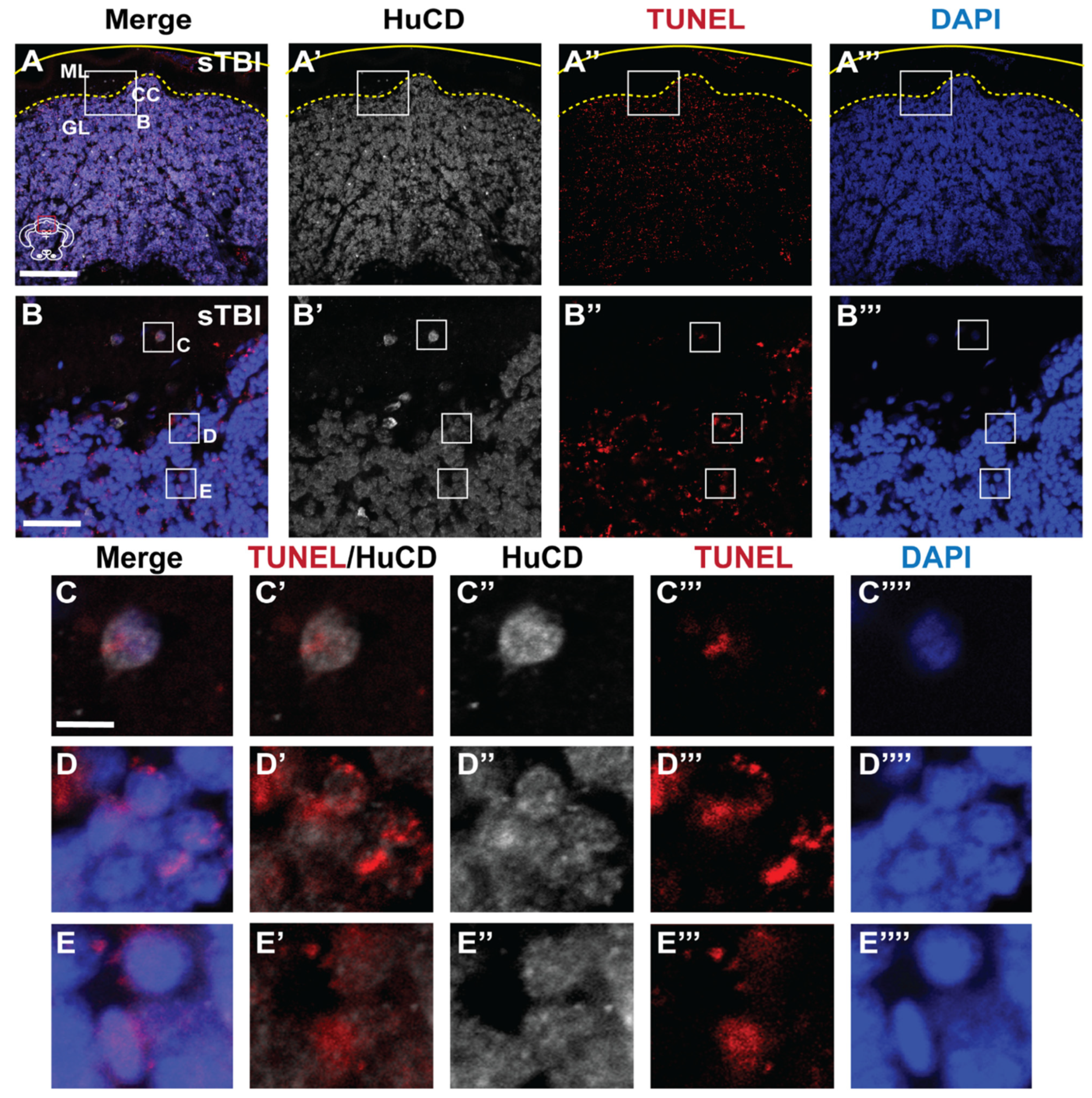
3.3. Blunt-Force TBI Results in Severity-Dependent Cell Death Spreading from the Impact Zone
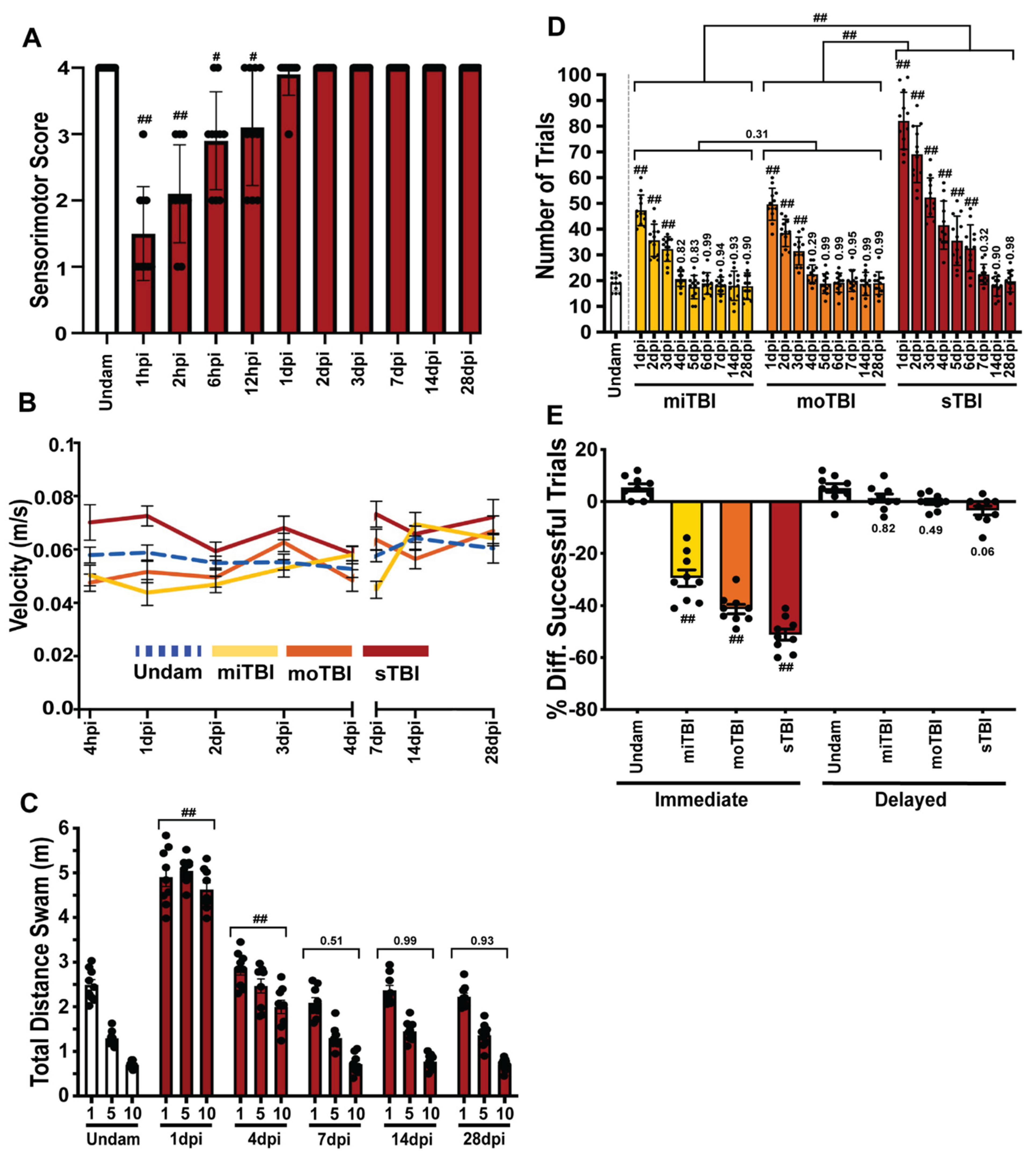
3.4. TBI Results in Sensorimotor Impairments and Associative Learning and Memory Deficits with Rapid Recovery
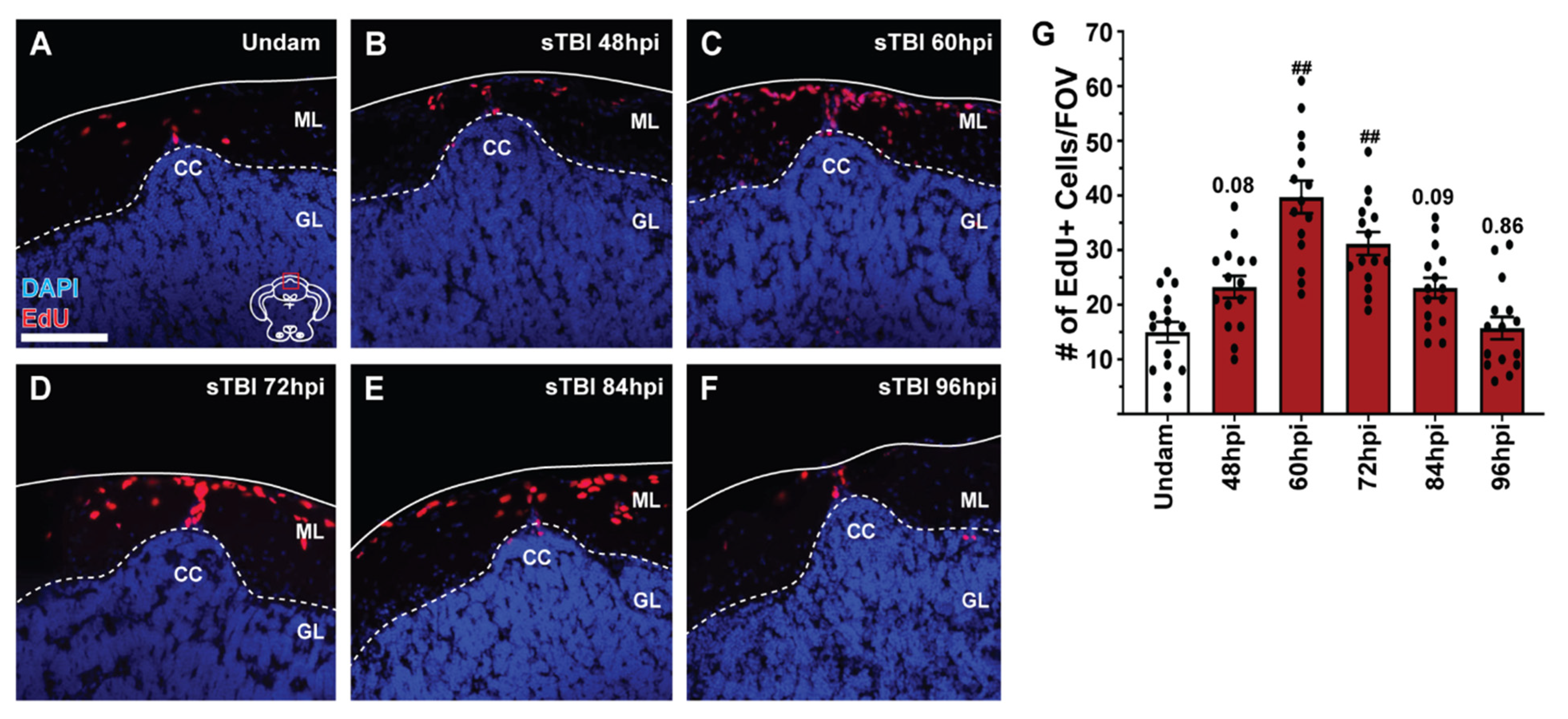
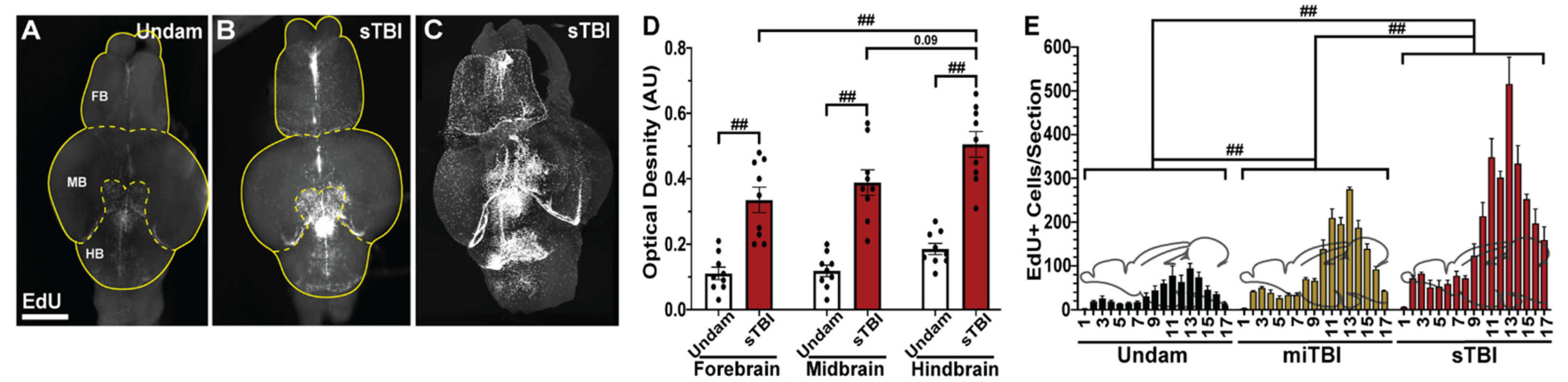
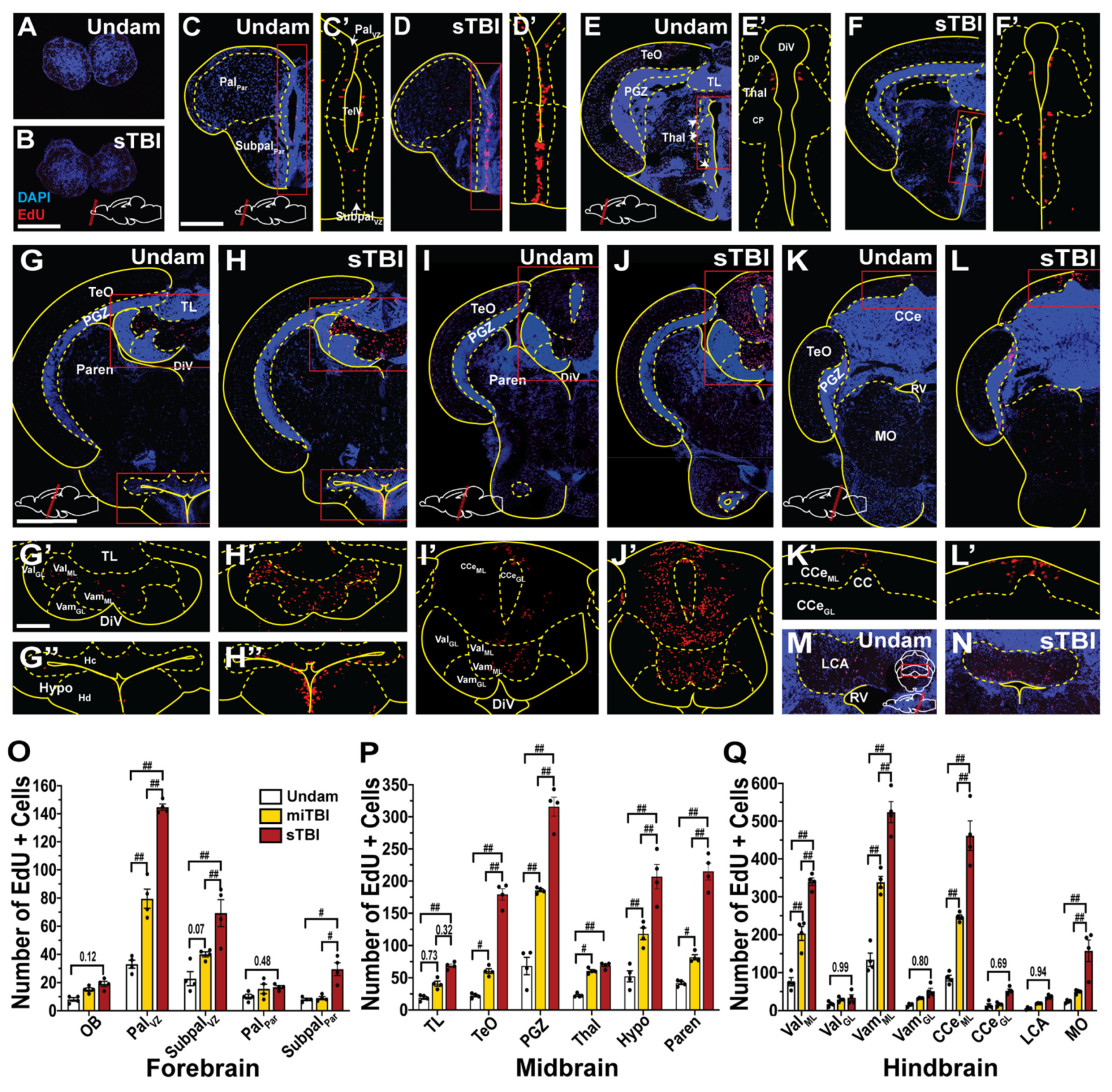
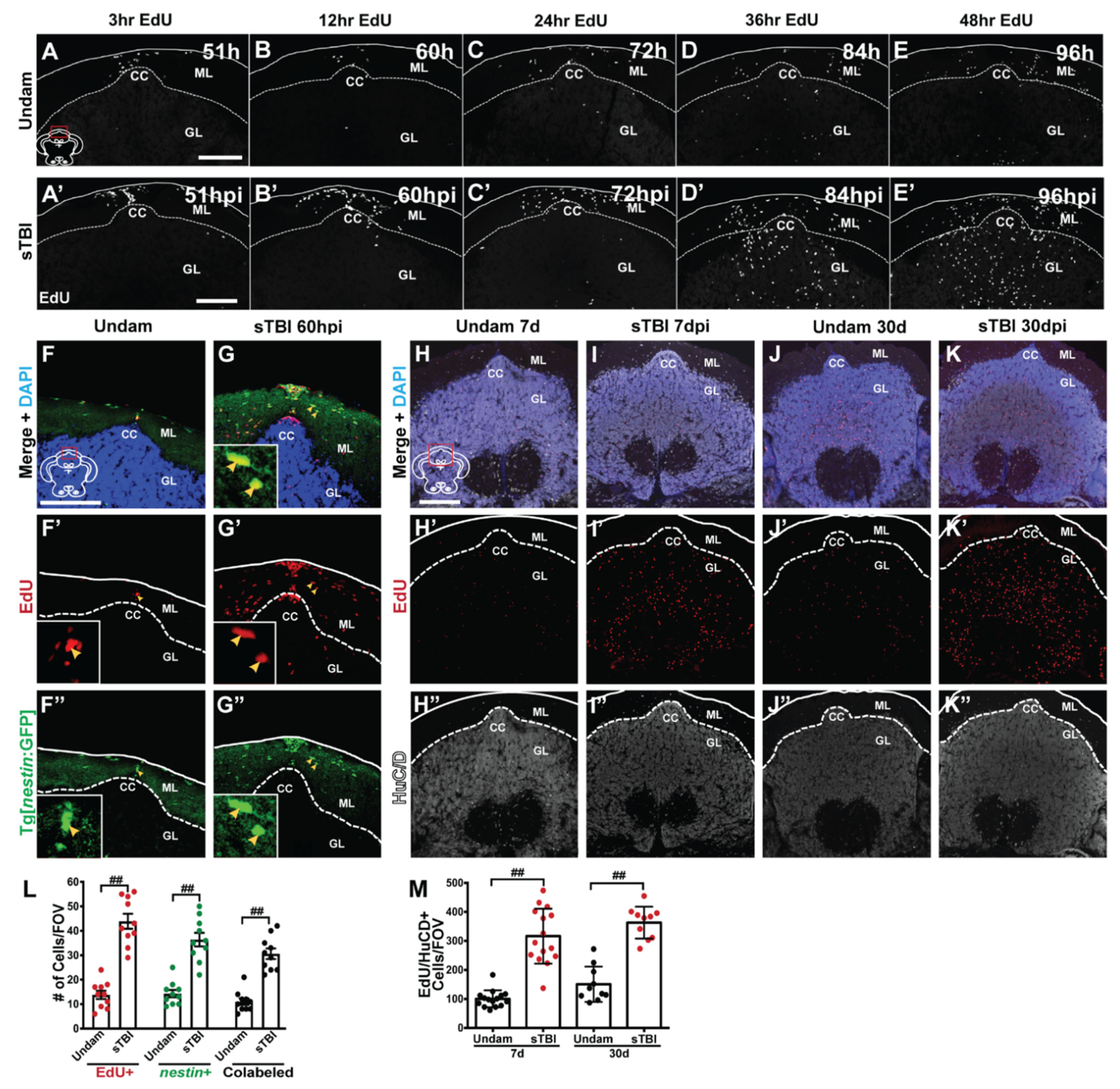
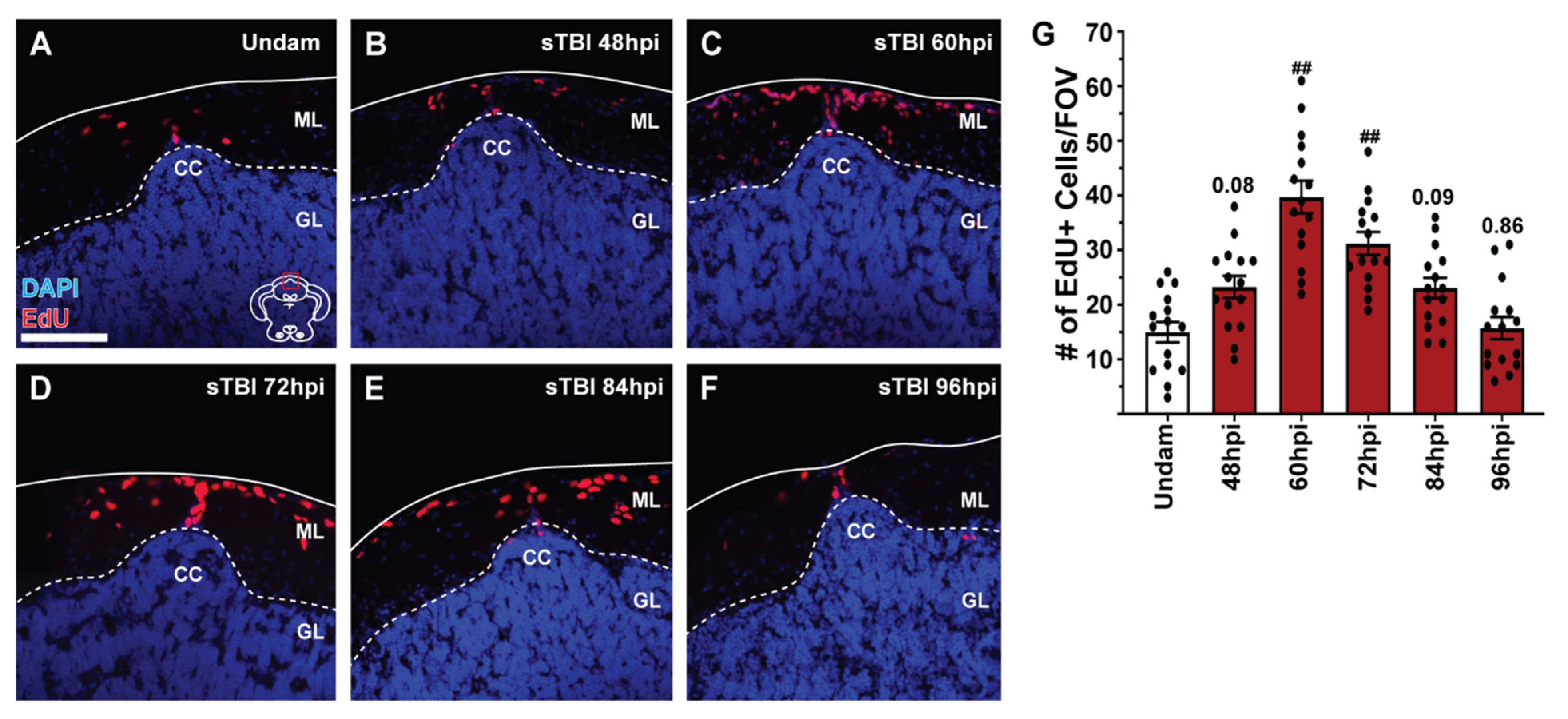
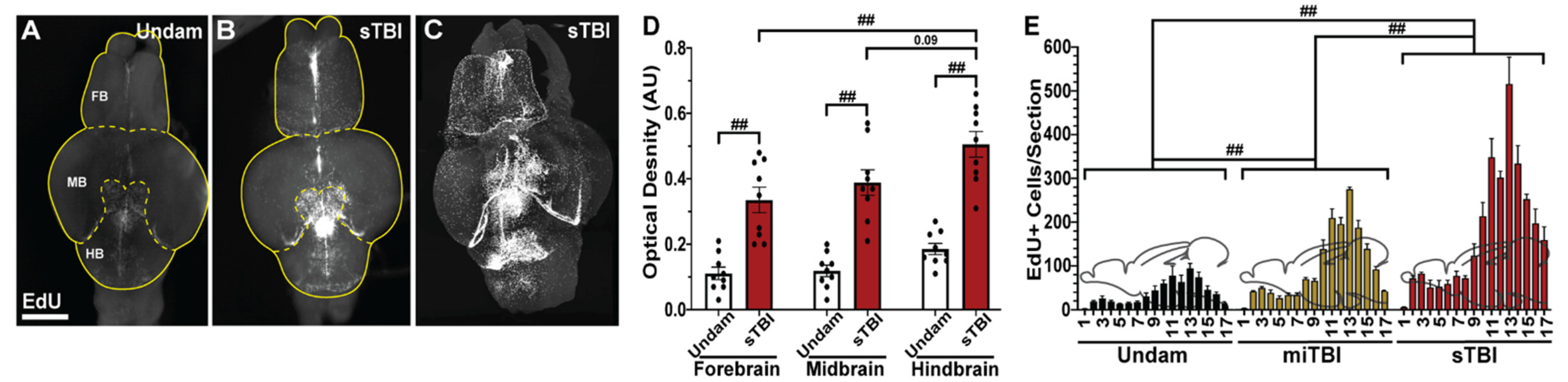
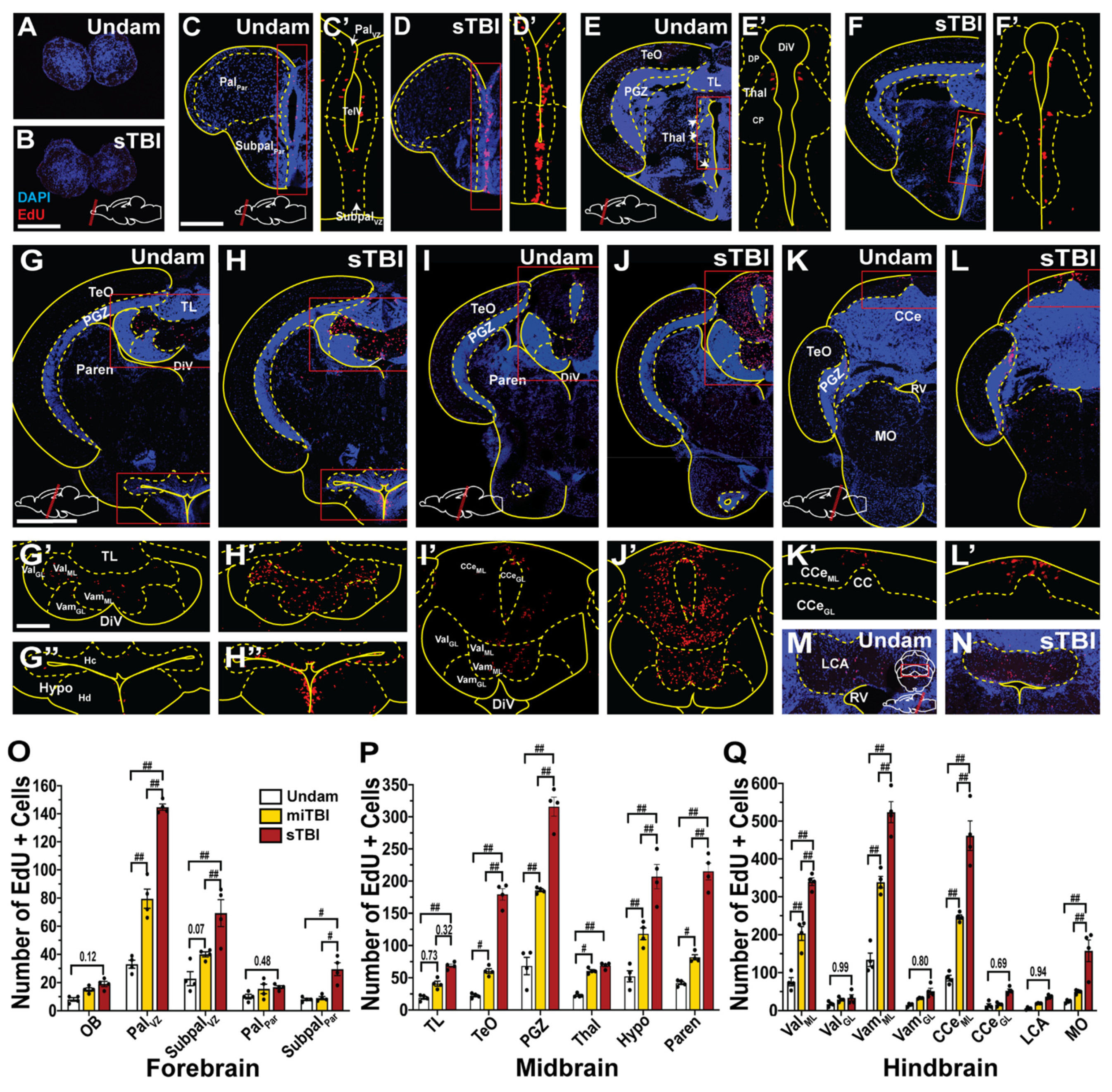
3.5. TBI Induces Cell Proliferation across the Neuroaxis in a Severity-Dependent Manner
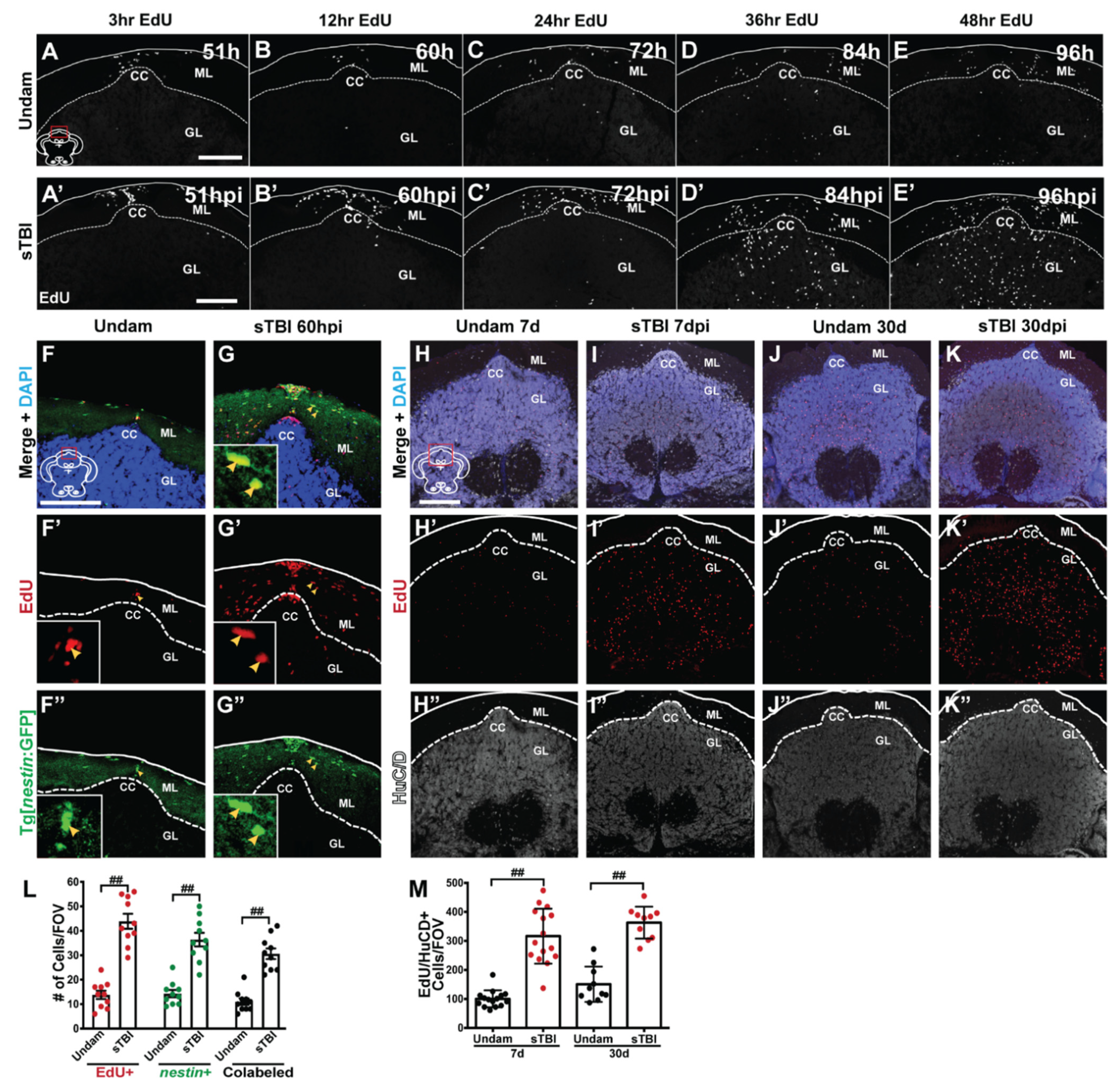
3.6. TBI Results in Injury-Induced Cerebellar Proliferation, Progenitor Migration, and Differentiation
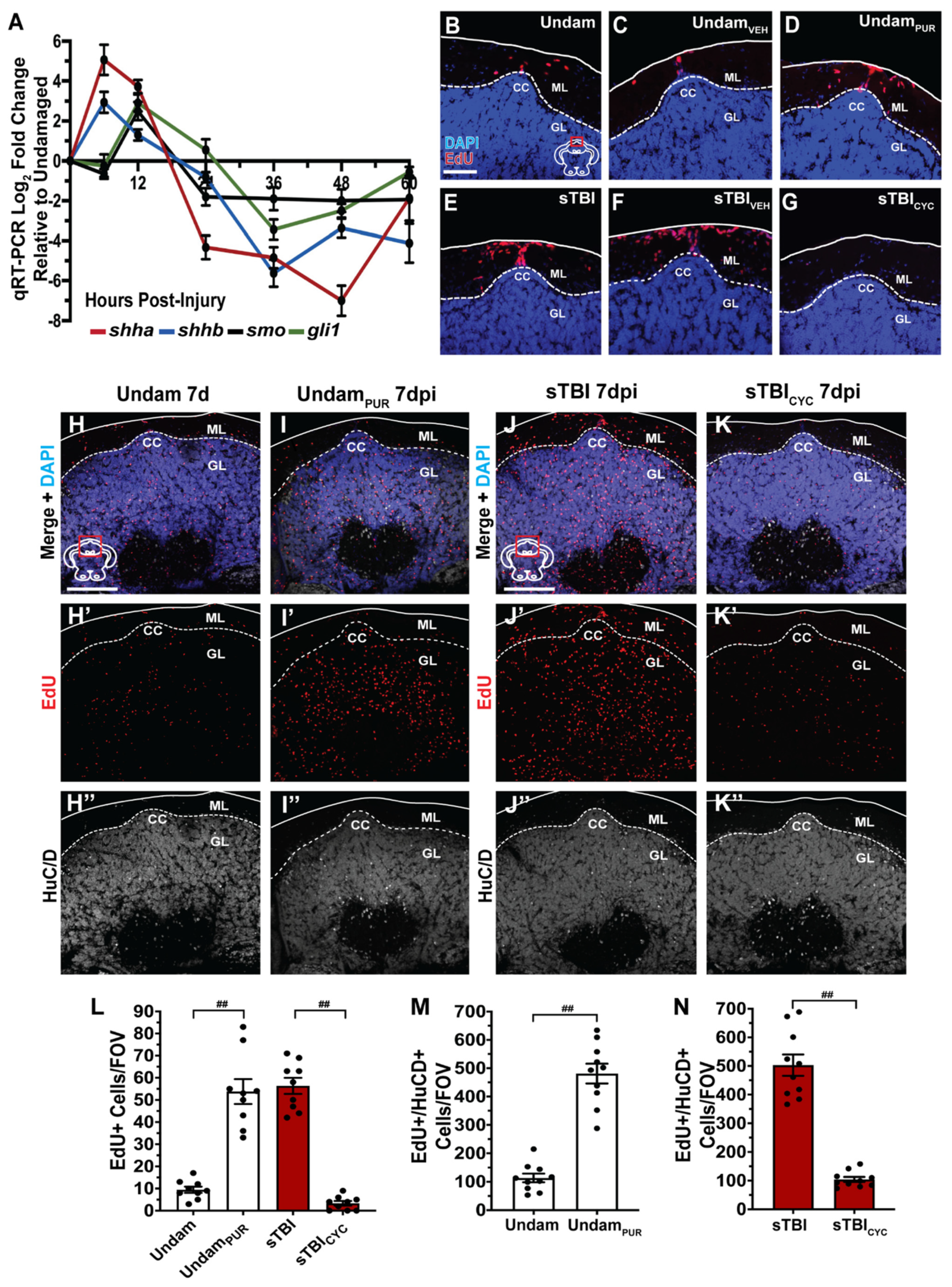
3.7. Sonic Hedgehog Regulates Injury-Induced Proliferation in the Cerebellum
4. Discussion
5. Conclusions
- 1.
- The modified TBI model for zebrafish is scalable for mild, moderate, and severe injury.
- 2.
- Zebrafish blunt-force TBI produces heterogeneous phenotypes replicating human injury.
- 3.
- Injury results in cognitive deficits that rapidly recover within 7 days.
- 4.
- Following injury, significant proliferation is observed across the entire brain.
- 5.
- Shh regulates injury-induced proliferation in the cerebellum.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Korley, F.K.; Kelen, G.D.; Jones, C.M.; Diaz-Arrastia, R. Emergency department evaluation of traumatic brain injury in the United States, 2009-2010. J. Head Trauma Rehabil. 2016, 31, 379–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corrigan, J.D.; Selassie, A.W.; Orman, J.A. The epidemiology of traumatic brain injury. J. Head Trauma Rehabil. 2010, 25, 72–80. [Google Scholar] [CrossRef]
- Levin, H.S.; Diaz-Arrastia, R.R. Diagnosis, prognosis, and clinical management of mild traumatic brain injury. Lancet Neurol. 2015, 14, 506–517. [Google Scholar] [CrossRef]
- Deutsch, M.B.; Mendez, M.F.; Teng, E. Interactions between traumatic brain injury and frontotemporal degeneration. Dement. Geriatr. Cogn. Disord. 2015, 39, 143–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fleminger, S.; Oliver, D.L.; Lovestone, S.; Rabe-Hesketh, S.; Giora, A. Head injury as a risk factor for Alzheimer’s disease: The evidence 10 years on; a partial replication. J. Neurol. Neurosurg. Psychiatry 2003, 74, 857–862. [Google Scholar] [CrossRef] [PubMed]
- Gardner, R.C.; Burke, J.F.; Nettiksimmons, J.; Goldman, S.; Tanner, C.M.; Yaffe, K. Traumatic brain injury in later life increases risk for Parkinson disease. Ann. Neurol. 2015, 77, 987–995. [Google Scholar] [CrossRef]
- Marklund, N. Rodent models of traumatic brain injury: Methods and challenges. Methods Mol. Biol. 2016, 1462, 29–46. [Google Scholar]
- Xiong, Y.; Mahmood, A.; Chopp, M. Animal models of traumatic brain injuries. Nat. Rev. Neurosci. 2013, 14, 128–142. [Google Scholar] [CrossRef] [Green Version]
- Morganti-Kossmann, M.C.; Yan, E.; Bye, N. Animal models of traumatic brain injury: Is there an optimal model to reproduce human brain injury in the laboratory? Injury 2010, 41 (Suppl. 1), S10–S13. [Google Scholar] [CrossRef]
- Marmarou, A.; Foda, M.A.; van den Brink, W.; Campbell, J.; Kita, H.; Demetriadou, K. A new model of diffuse brain injury in rats. Part I: Pathophysiology and biomechanics. J. Neurosurg. 1994, 80, 291–300. [Google Scholar] [CrossRef] [Green Version]
- Flierl, M.A.; Stahel, P.F.; Beauchamp, K.M.; Morgan, S.J.; Smith, W.R.; Shohami, E. Mouse closed head injury model induced by a weight-drop device. Nat. Protoc. 2009, 4, 1328–1337. [Google Scholar] [CrossRef]
- Foda, M.A.; Marmarou, A. A new model of diffuse brain injury in rats. Part II: Morphological characterization. J. Neurosurg. 1994, 80, 301–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zohar, O.; Schreiber, S.; Getslev, V.; Schwartz, J.P.; Mullins, P.G.; Pick, C.G. Closed-head minimal traumatic brain injury produces long-term cognitive deficits in mice. Neuroscience 2003, 118, 949–955. [Google Scholar] [CrossRef]
- Babikian, T.; McArthur, D.; Asarnow, R.F. Predictors of 1-month and 1-year neurocognitive functioning from the UCLA longitudinal mild, uncomplicated, pediatric traumatic brain injury study. J. Int. Neuropsychol. Soc. 2013, 19, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Rabinowitz, A.R.; Levin, H.S. Cognitive sequelae of traumatic brain injury. Psychiatr. Clin. N. Am. 2014, 37, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McInnes, K.; Friesen, C.L.; MacKenzie, D.E.; Westwood, D.A.; Boe, S.G. Mild Traumatic Brain Injury (mTBI) and chronic cognitive impairment: A scoping review. PLoS ONE 2017, 12, e0174847. [Google Scholar] [CrossRef] [Green Version]
- Vihtelic, T.S.; Hyde, D.R. Light-induced rod and cone cell death and regeneration in the adult albino zebrafish (Danio rerio) retina. J. Neurobiol. 2000, 44, 289–307. [Google Scholar] [CrossRef]
- Becker, C.G.; Becker, T. Adult zebrafish as a model for successful central nervous system regeneration. Restor. Neurol. Neurosci. 2008, 26, 71–80. [Google Scholar] [PubMed]
- Kaslin, J.; Ganz, J.; Geffarth, M.; Grandel, H.; Hans, S.; Brand, M. Stem cells in the adult zebrafish cerebellum: Initiation and maintenance of a novel stem cell niche. J. Neurosci. 2009, 29, 6142–6153. [Google Scholar] [CrossRef] [Green Version]
- Ito, Y.; Tanaka, H.; Okamoto, H.; Ohshima, T. Characterization of neural stem cells and their progeny in the adult zebrafish optic tectum. Dev. Biol. 2010, 342, 26–38. [Google Scholar] [CrossRef] [Green Version]
- Than-Trong, E.; Bally-Cuif, L. Radial glia and neural progenitors in the adult zebrafish central nervous system. Glia 2015, 63, 1406–1428. [Google Scholar] [CrossRef]
- Hentig, J.T.; Byrd-Jacobs, C.A. Exposure to Zinc Sulfate Results in Differential Effects on Olfactory Sensory Neuron Subtypes in Adult Zebrafish. Int. J. Mol. Sci. 2016, 17, 1445. [Google Scholar] [CrossRef] [Green Version]
- Lahne, M.; Brecker, M.; Jones, S.E.; Hyde, D.R. The Regenerating Adult Zebrafish Retina Recapitulates Developmental Fate Specification Programs. Front. Cell Dev. Biol. 2021, 8, 617923. [Google Scholar] [CrossRef] [PubMed]
- Kroehne, V.; Freudenreich, D.; Hans, S.; Kaslin, J.; Brand, M. Regeneration of the adult zebrafish brain from neurogenic radial glia-type progenitors. Development 2011, 138, 4831–4841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kishimoto, N.; Shimizu, K.; Sawamoto, K. Neuronal regeneration in a zebrafish model of adult brain injury. Dis. Model Mech. 2012, 5, 200–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skaggs, K.; Goldman, D.; Parent, J.M. Excitotoxic brain injury in adult zebrafish stimulates neurogenesis and long-distance neuronal integration. Glia 2014, 62, 2061–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaslin, J.; Kroehne, V.; Ganz, J.; Hans, S.; Brand, M. Distinct roles of neuroepithelial-like and radial glia-like progenitor cells in cerebellar regeneration. Development 2017, 144, 1462–1471. [Google Scholar] [CrossRef] [Green Version]
- McCutcheon, V.; Park, E.; Liu, E.; Sobhebidari, P.; Tavakkoli, J.; Wen, X.Y.; Baker, A.J. A novel model of traumatic brain injury in adult zebrafish demonstrates response to injury and treatment comparable with mammalian models. J. Neurotrauma. 2017, 34, 1382–1393. [Google Scholar] [CrossRef]
- Alyenbaawi, H.; Kanyo, R.; Locskai, L.F.; Kamali-Jamil, R.; DuVal, M.G.; Bai, Q.; Wille, H.; Burton, E.A.; Allison, W.T. Seizures are a druggable mechanistic link between TBI and subsequent tauopathy. Elife 2021, 10, e58744. [Google Scholar] [CrossRef]
- Doppenberg, E.M.; Choi, S.C.; Bullock, R. Clinical trials in traumatic brain injury: Lessons for the future. J. Neurosurg. Anesthesiol. 2004, 16, 87–94. [Google Scholar] [CrossRef]
- Marshall, L.F. Head injury: Recent past, present, and future. Neurosurgery 2000, 47, 546–561. [Google Scholar]
- Maheras, A.L.; Dix, B.; Carmo, O.M.S.; Young, A.E.; Gill, V.N.; Sun, J.L.; Booker, A.R.; Thomason, H.A.; Ibrahim, A.E.; Stanislaw, L.; et al. Genetic Pathways of Neuroregeneration in a Novel Mild Traumatic Brain Injury Model in Adult Zebrafish. eNeuro 2018, 5, ENEURO.0208-17.2017. [Google Scholar] [CrossRef]
- Kassen, S.C.; Ramanan, V.; Montgomery, J.E.; TBurket, C.; Liu, C.G.; Vihtelic, T.S.; Hyde, D.R. Time course analysis of gene expression during light-induced photoreceptor cell death and regeneration in albino zebrafish. Dev. Neurobiol. 2007, 67, 1009–1031. [Google Scholar] [CrossRef] [PubMed]
- Tsetskhladze, Z.R.; Canfield, V.A.; Ang, K.C.; Wentzel, S.M.; Reid, K.P.; Berg, A.S.; Johnson, S.L.; Kawakami, K.; Cheng, K.C. Functional assessment of human coding mutations affecting skin pigmentation using zebrafish. PLoS ONE 2012, 7, e47398. [Google Scholar] [CrossRef] [PubMed]
- White, R.M.; Sessa, A.; Burke, C.; Bowman, T.; LeBlanc, J.; Ceol, C.; Bourque, C.; Dovey, M.; Goessling, W.; Burns, C.E.; et al. Transparent adult zebrafish as a tool for in vivo transplantation analysis. Cell Stem Cell 2008, 2, 183–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hentig, J.; Cloghessy, K.; Dunseath, C.; Hyde, D.R. A scalable model to study the effects of blunt-force injury in adult zebrafish. J. Vis. Exp. 2021, 171. [Google Scholar] [CrossRef]
- Kalueff, A.V.; Gebhardt, M.; Stewart, A.M.; Cachat, J.M.; Brimmer, M.; Chawla, J.S.; Craddock, C.; Kyzar, E.J.; Roth, A.; Landsman, S.; et al. Zebrafish neuroscience research consortium. Towards a comprehensive catalog of zebrafish behavior 1.0 and beyond. Zebrafish 2013, 10, 70–86. [Google Scholar] [CrossRef] [PubMed]
- Hoshi, Y.; Okabe, K.; Shibasaki, K.; Funatsu, T.; Matsuki, N.; Ikegaya, Y.; Koyama, R. Ischemic brain injury leads to brain edema via hyperthermia-induced TRPV4 activation. J. Neurosci. 2018, 38, 5700–5709. [Google Scholar] [CrossRef] [PubMed]
- Truong, L.; Mandrell, D.; Mandrell, R.; Simonich, M.; Tanguay, R.L. A rapid throughput approach identifies cognitive deficits in adult zebrafish from developmental exposure to polybrominated flame retardants. Neurotoxicology 2014, 43, 134–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hentig, J.; Cloghessy, K.; Hyde, D.R. Shuttle box assay as an associative learning tool for cognitive assessment in learning and memory studies using adult zebrafish. J. Vis. Exp. 2021, 173. [Google Scholar] [CrossRef]
- Chanin, S.; Fryar, C.; Varga, D.; Raymond, J.; Kyzar, E.; Enriquez, J.; Bagawandoss, S.; Gaikwad, S.; Roth, A.; Pham, M. Assessing startle responses and their habituation in adult zebrafish. In Zebrafish Protocols for Neurobehavioral Research; Kalueff, A., Stewart, A., Eds.; Humana Press: Totowa, NJ, USA, 2012; Volume 66, pp. 287–300. [Google Scholar]
- Conner, C.; Ackerman, K.M.; Lahne, M.; Hobgood, J.S.; Hyde, D.R. Repressing notch signaling and expressing TNFα are sufficient to mimic retinal regeneration by inducing Müller glial proliferation to generate committed progenitor cells. J. Neurosci. 2014, 34, 14403–14419. [Google Scholar] [CrossRef] [Green Version]
- Lahne, M.; Li, J.; Marton, R.M.; Hyde, D.R. Actin-Cytoskeleton- and Rock-Mediated INM are required for photoreceptor regeneration in the adult zebrafish retina. J. Neurosci. 2015, 35, 15612–15634. [Google Scholar] [CrossRef] [Green Version]
- Lindsey, B.W.; Douek, A.M.; Loosli, F.; Kaslin, J. A whole brain staining, embedding, and clearing pipeline for adult zebrafish to visualize cell proliferation and morphology in 3-Dimensions. Front. Neurosci. 2018, 11, 750. [Google Scholar] [CrossRef] [Green Version]
- Radu, M.; Chernoff, J. An in vivo assay to test blood vessel permeability. J. Vis. Exp. 2013, 73, e50062. [Google Scholar]
- Eliceiri, B.P.; Gonzalez, A.M.; Baird, A. Zebrafish model of the blood-brain barrier: Morphological and permeability studies. Methods Mol. Biol. 2011, 686, 371–378. [Google Scholar]
- Campbell, L.J.; Hobgood, J.S.; Jia, M.; Boyd, P.; Hipp, R.I.; Hyde, D.R. Notch3 and DeltaB maintain Müller glia quiescence and act as negative regulators of regeneration in the light-damaged zebrafish retina. Glia 2021, 69, 546–566. [Google Scholar] [CrossRef]
- Lund, S.B.; Gjeilo, K.H.; Moen, K.G.; Schirmer-Mikalsen, K.; Skandsen, T.; Vik, A. Moderate traumatic brain injury, acute phase course and deviations in physiological variables: An observational study. Scand. J. Trauma Resusc. Emerg. Med. 2016, 24, 77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, S.; Levin, H.S.; Prough, D.S. Mild, moderate and severe: Terminology implications for clinical and experimental traumatic brain injury. Curr. Opin. Neurol. 2018, 31, 672–680. [Google Scholar] [CrossRef] [PubMed]
- Wasserman, E.B.; Shah, M.N.; Jones, C.M.; Cushman, J.T.; Caterino, J.M.; Bazarian, J.J.; Gillespie, S.M.; Cheng, J.D.; Dozier, A. Identification of a neurologic scale that optimizes EMS detection of older adult traumatic brain injury patients who require transport to a trauma center. Prehosp. Emerg. Care. 2015, 19, 202–212. [Google Scholar] [CrossRef] [Green Version]
- Annegers, J.F.; Hauser, W.A.; Coan, S.P.; Rocca, W.A. A population-based study of seizures after traumatic brain injuries. N. Engl. J. Med. 1998, 338, 20–24. [Google Scholar] [CrossRef] [PubMed]
- Frey, L.C. Epidemiology of posttraumatic epilepsy: A critical review. Epilepsia 2003, 44, 11–17. [Google Scholar] [CrossRef]
- Servadei, F.; Nasi, M.T.; Cremonini, A.M.; Giuliani, G.; Cenni, P.; Nanni, A. Importance of a reliable admission Glasgow Coma Scale score for determining the need for evacuation of posttraumatic subdural hematomas: A prospective study of 65 patients. J. Trauma 1998, 44, 868–873. [Google Scholar] [CrossRef] [PubMed]
- Mutch, C.A.; Talbott, J.F.; Gean, A. Imaging Evaluation of Acute Traumatic Brain Injury. Neurosurg. Clin. 2016, 27, 409–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, K.N.; Parry-Jones, A.R.; Allan, S.M. Interleukin-1 and acute brain injury. Front. Cell Neurosci. 2015, 9, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gill, J.; Motamedi, V.; Osier, N.; Dell, K.; Arcurio, L.; Carr, W.; Walker, P.; Ahlers, S.; Lopresti, M.; Yarnell, A. Moderate blast exposure results in increased IL-6 and TNFα in peripheral blood. Brain Behav. Immun. 2017, 65, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Gill, J.; Mustapic, M.; Diaz-Arrastia, R.; Lange, R.; Gulyani, S.; Diehl, T.; Motamedi, V.; Osier, N.; Stern, R.A.; Kapogiannis, D. Higher exosomal tau, amyloid-beta 42 and IL-10 are associated with mild TBIs and chronic symptoms in military personnel. Brain Inj. 2018, 32, 1277–1284. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, T.; Hall, C.J.; Crosier, P.S.; Abe, G.; Kawakami, K.; Kudo, A.; Kawakami, A. Transient inflammatory response mediated by interleukin-1β is required for proper regeneration in zebrafish fin fold. Elife 2017, 6, e22716. [Google Scholar] [CrossRef]
- Eierud, C.; Craddock, R.C.; Fletcher, S.; Aulakh, M.; King-Casas, B.; Kuehl, D.; LaConte, S.M. Neuroimaging after mild traumatic brain injury: Review and meta-analysis. Neuroimage Clin. 2014, 4, 283–294. [Google Scholar] [CrossRef] [Green Version]
- Gill, J.; Latour, L.; Diaz-Arrastia, R.; Motamedi, V.; Turtzo, C.; Shahim, P.; Mondello, S.; DeVoto, C.; Veras, E.; Hanlon, D.; et al. Glial fibrillary acidic protein elevations relate to neuroimaging abnormalities after mild TBI. Neurology 2018, 91, e1385–e1389. [Google Scholar] [CrossRef]
- Dikmen, S.S.; Corrigan, J.D.; Levin, H.S.; Machamer, J.; Stiers, W.; Weisskopf, M.G. Cognitive outcome following traumatic brain injury. J. Head Trauma Rehabil. 2009, 24, 430–438. [Google Scholar] [CrossRef]
- Vanderploeg, R.D.; Curtiss, G.; Belanger, H.G. Long-term neuropsychological outcomes following mild traumatic brain injury. J. Int. Neuropsychol. Soc. 2005, 11, 228–236. [Google Scholar] [CrossRef]
- Thompson, R.F.; Spencer, W.A. Habituation: A model phenomenon for the study of neuronal substrates of behavior. Psychol. Rev. 1966, 73, 16–43. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, K.; Yoshida, M.; Kawakami, K.; Hibi, M.; Shimizu, T. Granule cells control recovery from classical conditioned fear responses in the zebrafish cerebellum. Sci. Rep. 2017, 7, 11865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flood, N.C.; Overmier, J.B.; Savage, G.E. Teleost telencephalon and learning: An interpretive review of data and hypotheses. Physiol. Behav. 1976, 16, 783–788. [Google Scholar] [CrossRef]
- Xu, X.; Bazner, J.; Qi, M.; Johnson, E.; Freidhoff, R. The role of telencephalic NMDA receptors in avoidance learning in goldfish (Carassius auratus). Behav. Neurosci. 2003, 117, 548–554. [Google Scholar] [CrossRef]
- Greve, K.W.; Bianchini, K.J.; Mathias, C.W.; Houston, R.J.; Crouch, J.A. Detecting malingered performance on the Wechsler Adult Intelligence Scale. Validation of Mittenberg’s approach in traumatic brain injury. Arch. Clin. Neuropsychol. 2003, 18, 245–260. [Google Scholar] [CrossRef]
- Dunning, D.L.; Westgate, B.; Adlam, A.R. A meta-analysis of working memory impairments in survivors of moderate-to-severe traumatic brain injury. Neuropsychology 2016, 30, 811–819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wullimann, M.; Rupp, B.; Reichert, H. Neuroanatomy of the Zebrafish Brain. A Topological Atlas, 1st ed.; Brikhäuser: Basel, Switzerland, 1996; pp. 1–160. [Google Scholar]
- Kaslin, J.; Kroehne, V.; Benato, F.; Argenton, F.; Brand, M. Development and specification of cerebellar stem and progenitor cells in zebrafish: From embryo to adult. Neural Dev. 2013, 8, 9. [Google Scholar] [CrossRef] [Green Version]
- Grandel, H.; Kaslin, J.; Ganz, J.; Wenzel, I.; Brand, M. Neural stem cells and neurogenesis in the adult zebrafish brain: Origin, proliferation dynamics, migration and cell fate. Dev. Biol. 2006, 295, 263–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kani, S.; Bae, Y.K.; Shimizu, T.; Tanabe, K.; Satou, C.; Parsons, M.J.; Scott, E.; Higashijima, S.; Hibi, M. Proneural gene-linked neurogenesis in zebrafish cerebellum. Dev. Biol. 2010, 343, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Ueda, Y.; Shimizu, Y.; Shimizu, N.; Ishitani, T.; Ohshima, T. Involvement of sonic hedgehog and notch signaling in regenerative neurogenesis in adult zebrafish optic tectum after stab injury. J. Comp. Neurol. 2018, 526, 2360–2372. [Google Scholar] [CrossRef]
- Thomas, J.L.; Morgan, G.W.; Dolinski, K.M.; Thummel, R. Characterization of the pleiotropic roles of Sonic Hedgehog during retinal regeneration in adult zebrafish. Exp. Eye Res. 2018, 166, 106–115. [Google Scholar] [CrossRef]
- Binder, S.; Corrigan, J.D.; Langlois, J.A. The public health approach to traumatic brain injury: An overview of CDC’s research and programs. J. Head Trauma Rehabil. 2005, 20, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Cassidy, J.D.; Carroll, L.; Peloso, P.; Borg, J.; Von Holst, H.; Holm, L.; Kraus, J.; Coronado, V. Incidence, risk factors and prevention of mild traumatic brain injury: Results of the WHO Collaborating Centre Task Force on Mild Traumatic Brain Injury. J. Rehabil. Med. 2004, 43, 28–60. [Google Scholar] [CrossRef] [Green Version]
- Marklund, N.; Hillered, L. Animal modelling of traumatic brain injury in preclinical drug development: Where do we go from here? Br. J. Pharmacol. 2011, 164, 1207–1229. [Google Scholar] [CrossRef] [Green Version]
- Ruff, R.M.; Iverson, G.L.; Barth, J.T.; Bush, S.S.; Broshek, D.K.; NAN Policy and Planning Committee. Recommendations for diagnosing a mild traumatic brain injury: A National Academy of Neuropsychology education paper. Arch. Clin. Neuropsychol. 2009, 24, 3–10. [Google Scholar] [CrossRef] [Green Version]
- Nasution, R.A.; Islam, A.A.; Hatta, M. Decreased neutrophil levels in mice with traumatic brain injury after cape administration. Ann. Med. Surg. 2020, 54, 89–92. [Google Scholar] [CrossRef]
- Gemberling, M.; Bailey, T.J.; Hyde, D.R.; Poss, K.D. The zebrafish as a model for complex tissue regeneration. Trends Genet. 2013, 29, 611–620. [Google Scholar] [CrossRef] [Green Version]
- Lahne, M.; Nagashima, M.; Hyde, D.R.; Hitchcock, P.F. Reprogramming Müller Glia to regenerate retinal neurons. Annu. Rev. Vis. Sci. 2020, 6, 171–193. [Google Scholar] [CrossRef] [PubMed]
- Amamoto, R.; Huerta, V.G.; Takahashi, E.; Dai, G.; Grant, A.K.; Fu, Z.; Arlotta, P. Adult axolotls can regenerate original neuronal diversity in response to brain injury. Elife 2016, 5, e13998. [Google Scholar] [CrossRef] [PubMed]
- McFarland, K.A.; Topczewska, J.M.; Weidinger, G.; Dorsky, R.I.; Appel, B. Hh and Wnt signaling regulate formation of olig2+ neurons in the zebrafish cerebellum. Dev. Biol. 2008, 318, 162–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaplin, N.; Tendeng, C.; Wingate, R.J. Absence of an external germinal layer in zebrafish and shark reveals a distinct, anamniote ground plan of cerebellum development. J. Neurosci. 2010, 30, 3048–3057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganz, J.; Brand, M. Adult Neurogenesis in Fish. Cold Spring Harb. Perspect. Biol. 2016, 8, a019018. [Google Scholar] [CrossRef] [Green Version]
- Vaillant, C.; Monard, D. SHH pathway and cerebellar development. Cerebellum 2009, 8, 291–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time (n = 10) | Tilt Avg ZBC 1.83, 1.164, 1.175 | Pain Stimuli Avg ZBC 1.104 | Escape Avg ZBC 1.5, 1.52 | Avoidance Avg ZBC 1.12 | Total Score |
|---|---|---|---|---|---|
| Undam | 1 | 1 | 1 | 1 | 4 |
| sTBI 1 hpi | 0.4 | 1 | 0.1 | 0 | 1.5 |
| sTBI 2 hpi | 0.7 | 1 | 0.4 | 0 | 2.1 |
| sTBI 6 hpi | 1 | 1 | 0.7 | 0.2 | 2.9 |
| sTBI 12 hpi | 1 | 1 | 0.7 | 0.4 | 3.1 |
| sTBI 1 dpi | 1 | 1 | 1 | 0.9 | 3.9 |
| sTBI 2 dpi | 1 | 1 | 1 | 1 | 4 |
| sTBI 3 dpi | 1 | 1 | 1 | 1 | 4 |
| sTBI 7 dpi | 1 | 1 | 1 | 1 | 4 |
| sTBI 14 dpi | 1 | 1 | 1 | 1 | 4 |
| sTBI 28 dpi | 1 | 1 | 1 | 1 | 4 |
| Group n = 9 | Iteration | ||
|---|---|---|---|
| 1 | 5 | 10 | |
| Undam Startle Velocity | 0.256 ± 0.02 m/s | 0.190 ± 0.01 m/s | 0.138 ± 0.01 m/s |
| (Recovery Time) | (22 ± 1.59 s) | (15 ± 1.24 s) | (10 ± 0.60 s) |
| sTBI 1 dpi Startle Velocity | 0.253 ±0.01 m/s | 0.233 ± 0.02 m/s | 0.246 ± 0.01 m/s |
| (Recovery Time) | (30 s+) | (30 s+) | (30 s+) |
| sTBI 4 dpi Startle Velocity | 0.246 ± 0.01 m/s | 0.231 ± 0.01 m/s | 0.216 ± 0.01 m/s |
| (Recovery Time) | (23 ± 1.02 s) | (24 ± 1.88 s) | (21 ± 0.95 s) |
| sTBI 7 dpi Startle Velocity | 0.236 ± 0.02 m/s | 0.153 ± 0.01 m/s | 0.100 ± 0.01 m/s |
| (Recovery Time) | (21 ± 1.25 s) | (16 ± 1.14 s) | (13 ± 1.03 s) |
| sTBI 14 dpi Startle Velocity | 0.248 ± 0.02 m/s | 0.171 ± 0.01 m/s | 0.144 ± 0.02 m/s |
| (Recovery Time) | (23 ± 1.12 s) | (17 ± 1.09 s) | (12 ± 1.31 s) |
| sTBI 28 dpi Startle Velocity | 0.251 ± 0.01 m/s | 0.182 ± 0.01 m/s | 0.124 ± 0.01 m/s |
| (Recovery Time) | (21 ± 1.71 s) | (15 ± 1.22 s) | (11 ± 0.89 s) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hentig, J.; Cloghessy, K.; Lahne, M.; Jung, Y.J.; Petersen, R.A.; Morris, A.C.; Hyde, D.R. Zebrafish Blunt-Force TBI Induces Heterogenous Injury Pathologies That Mimic Human TBI and Responds with Sonic Hedgehog-Dependent Cell Proliferation across the Neuroaxis. Biomedicines 2021, 9, 861. https://doi.org/10.3390/biomedicines9080861
Hentig J, Cloghessy K, Lahne M, Jung YJ, Petersen RA, Morris AC, Hyde DR. Zebrafish Blunt-Force TBI Induces Heterogenous Injury Pathologies That Mimic Human TBI and Responds with Sonic Hedgehog-Dependent Cell Proliferation across the Neuroaxis. Biomedicines. 2021; 9(8):861. https://doi.org/10.3390/biomedicines9080861
Chicago/Turabian StyleHentig, James, Kaylee Cloghessy, Manuela Lahne, Yoo Jin Jung, Rebecca A. Petersen, Ann C. Morris, and David R. Hyde. 2021. "Zebrafish Blunt-Force TBI Induces Heterogenous Injury Pathologies That Mimic Human TBI and Responds with Sonic Hedgehog-Dependent Cell Proliferation across the Neuroaxis" Biomedicines 9, no. 8: 861. https://doi.org/10.3390/biomedicines9080861
APA StyleHentig, J., Cloghessy, K., Lahne, M., Jung, Y. J., Petersen, R. A., Morris, A. C., & Hyde, D. R. (2021). Zebrafish Blunt-Force TBI Induces Heterogenous Injury Pathologies That Mimic Human TBI and Responds with Sonic Hedgehog-Dependent Cell Proliferation across the Neuroaxis. Biomedicines, 9(8), 861. https://doi.org/10.3390/biomedicines9080861