
A Novel Bispecific Antibody Targeting CD3 and Lewis Y with Potent Therapeutic Efficacy against Gastric Cancer

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Vectors Construction
2.2. Proteins Purification
2.3. Binding Activity to the Target Cells
2.4. Binding Affinity to the Target Cells
2.5. In Vitro Cytotoxicity Mediated by huPBMCs
2.6. T Cells Activation with CD69 and CD25 Expression
2.7. Detection of Cytokine Release
2.8. T Cell Proliferation Assay
2.9. Cell–Cell Association Mediated by m3s193 BsAb
2.10. In Vivo Activity of m3s193 BsAb with huPBMCs/Tumor Cells Co-Grafting Model
2.11. In Vivo Activity of m3s193 BsAb with huPBMCs Intravenous Injection Model
2.12. Histologic Analysis
3. Results
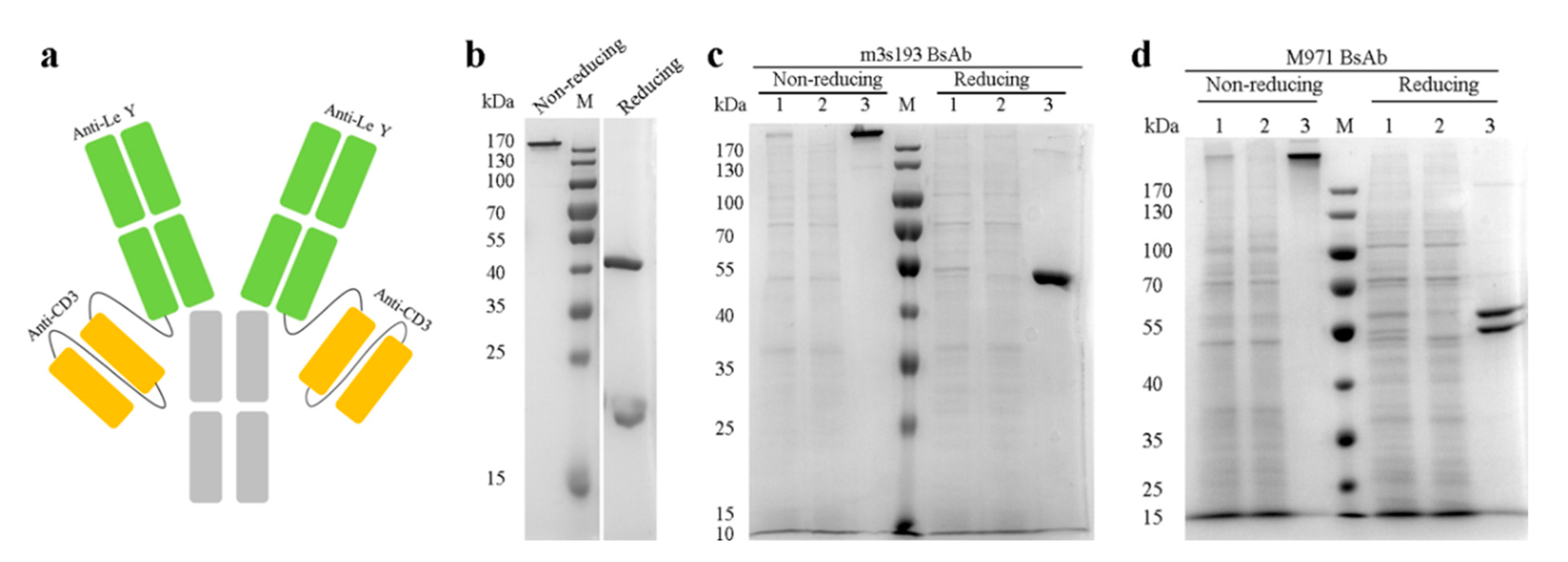
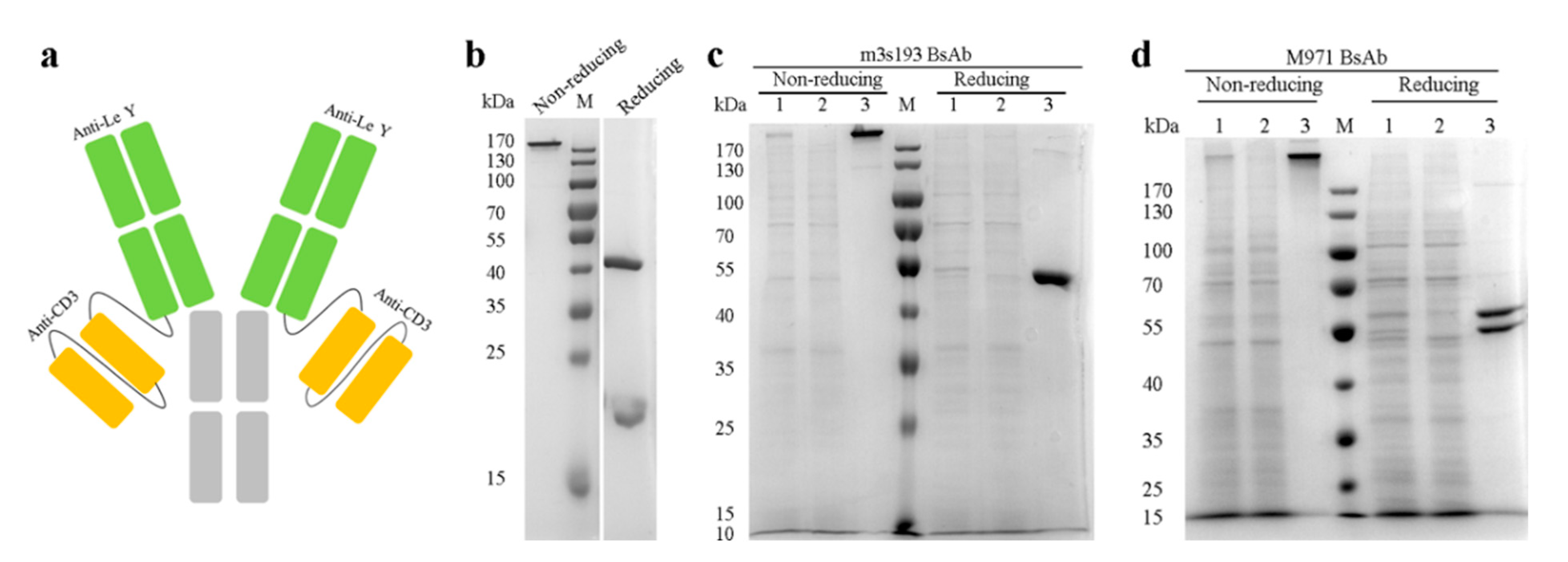
3.1. Expression and Purification of the Antibodies
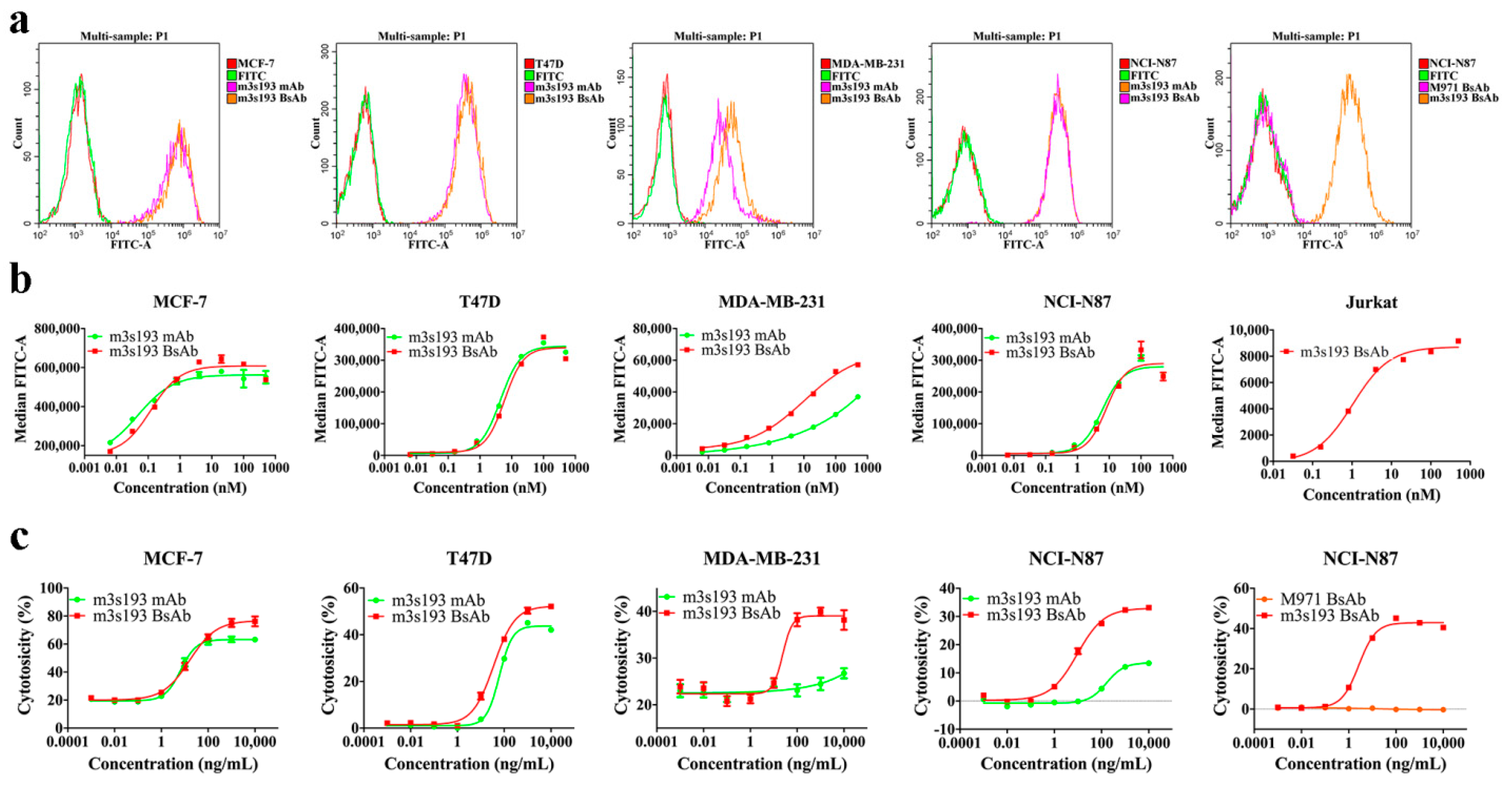
3.2. Binding Activity of Purified Proteins
3.3. T Cell Cytotoxicity Mediated by m3s193 BsAbs
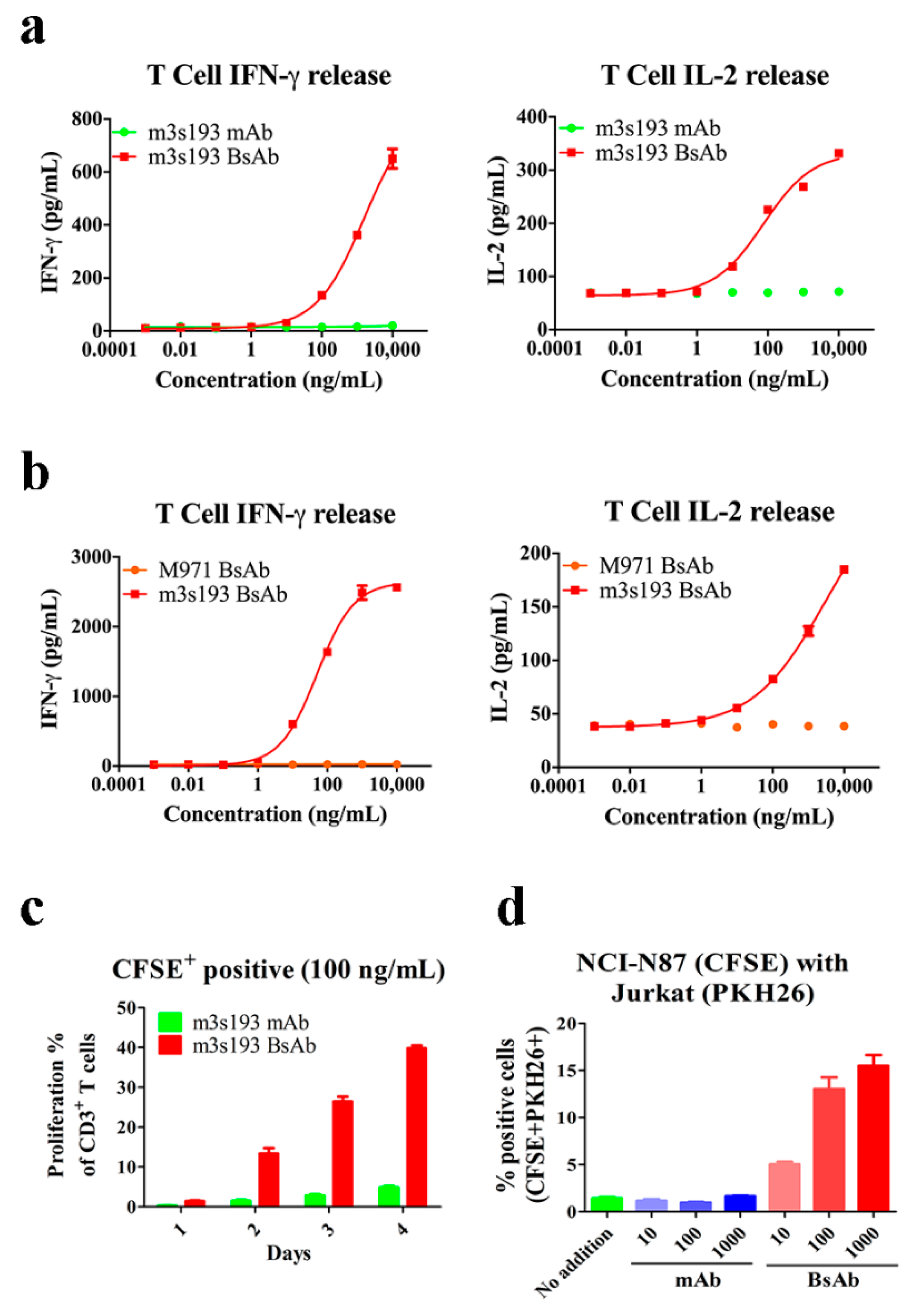
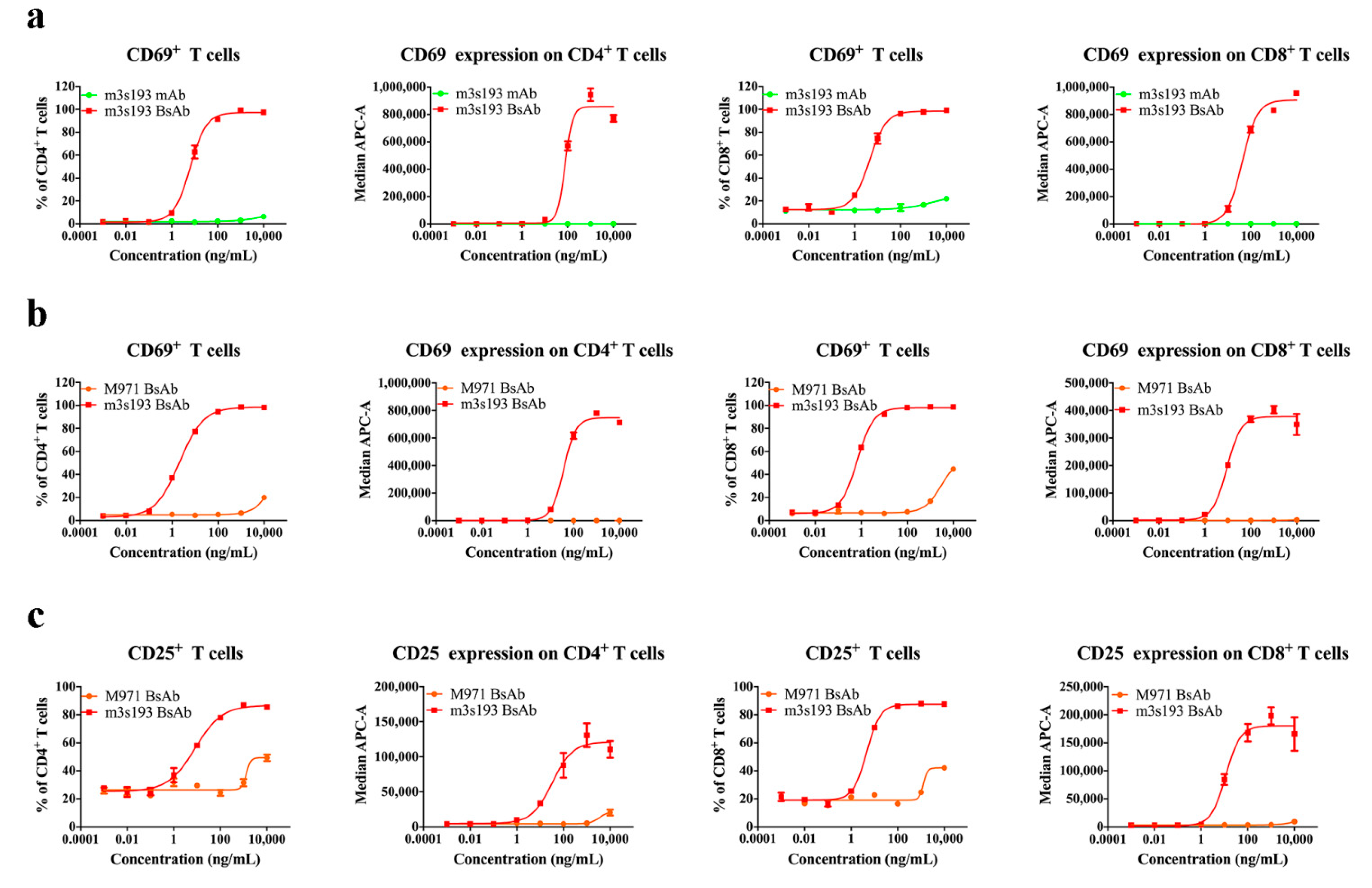
3.4. T Cell Activation Mediated by m3s193 BsAb
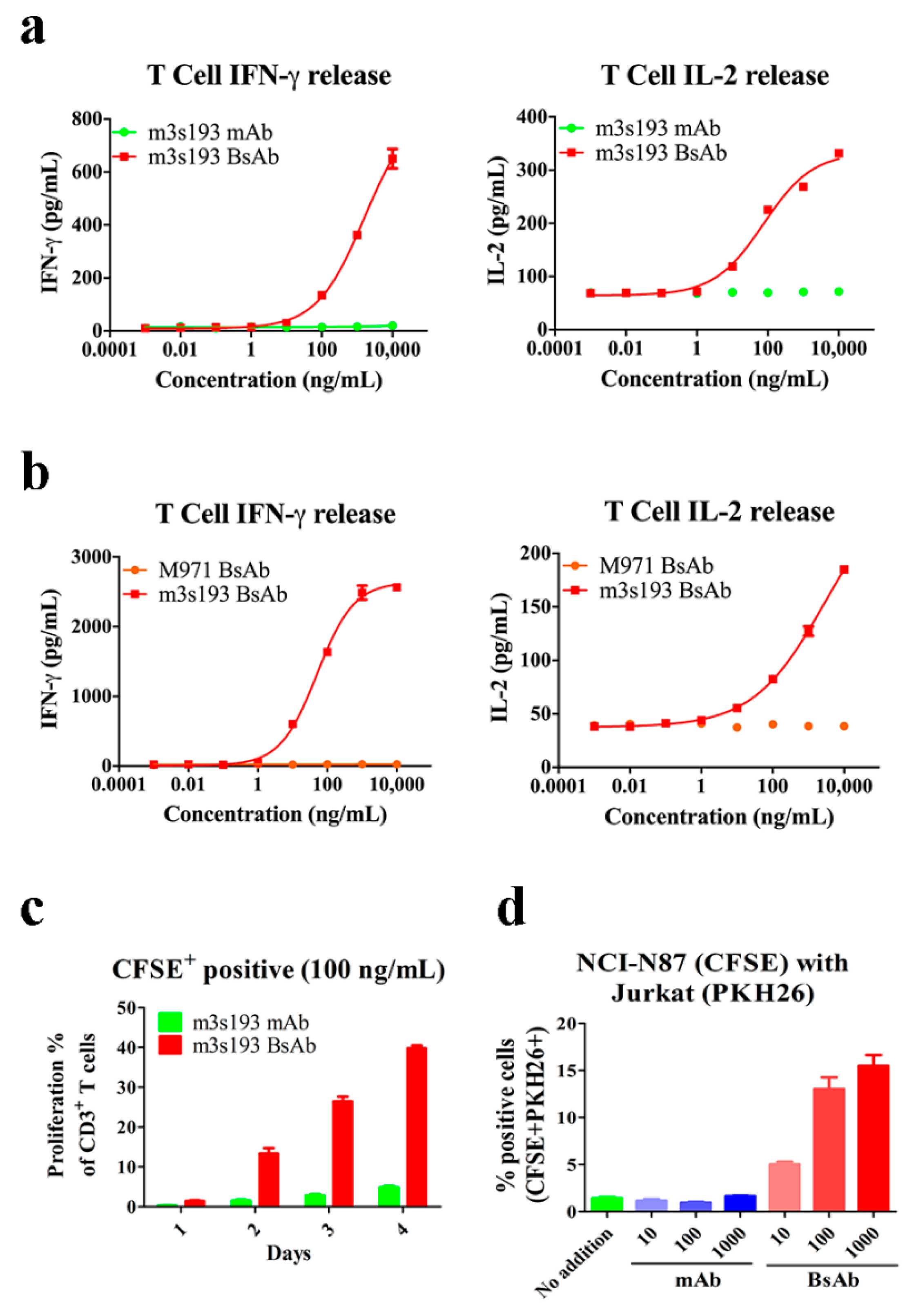
3.5. Cytokine Release, Proliferation, and Recruiting Mediated by m3s193 BsAb
3.6. In Vivo Anti-Tumor Activity
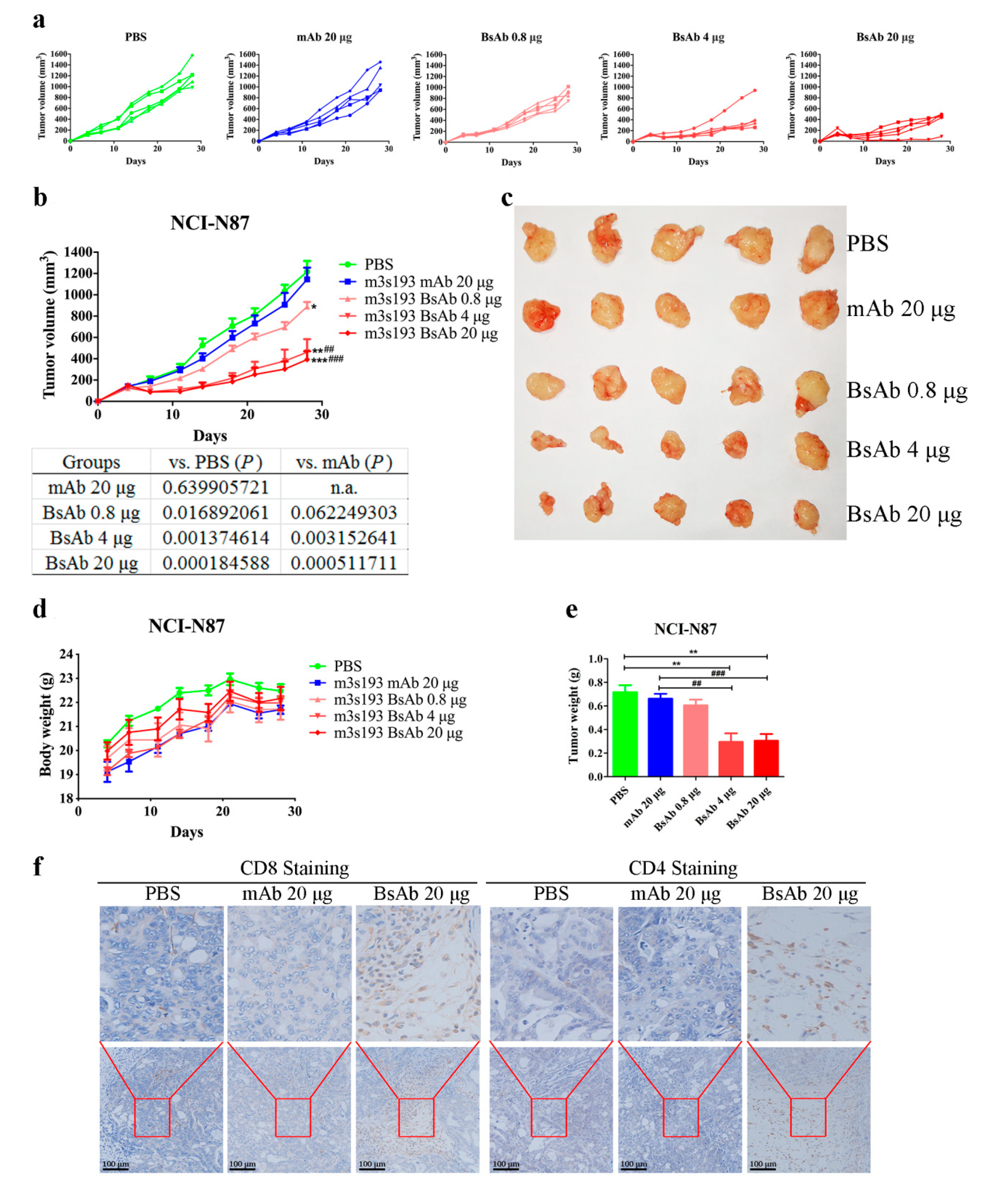
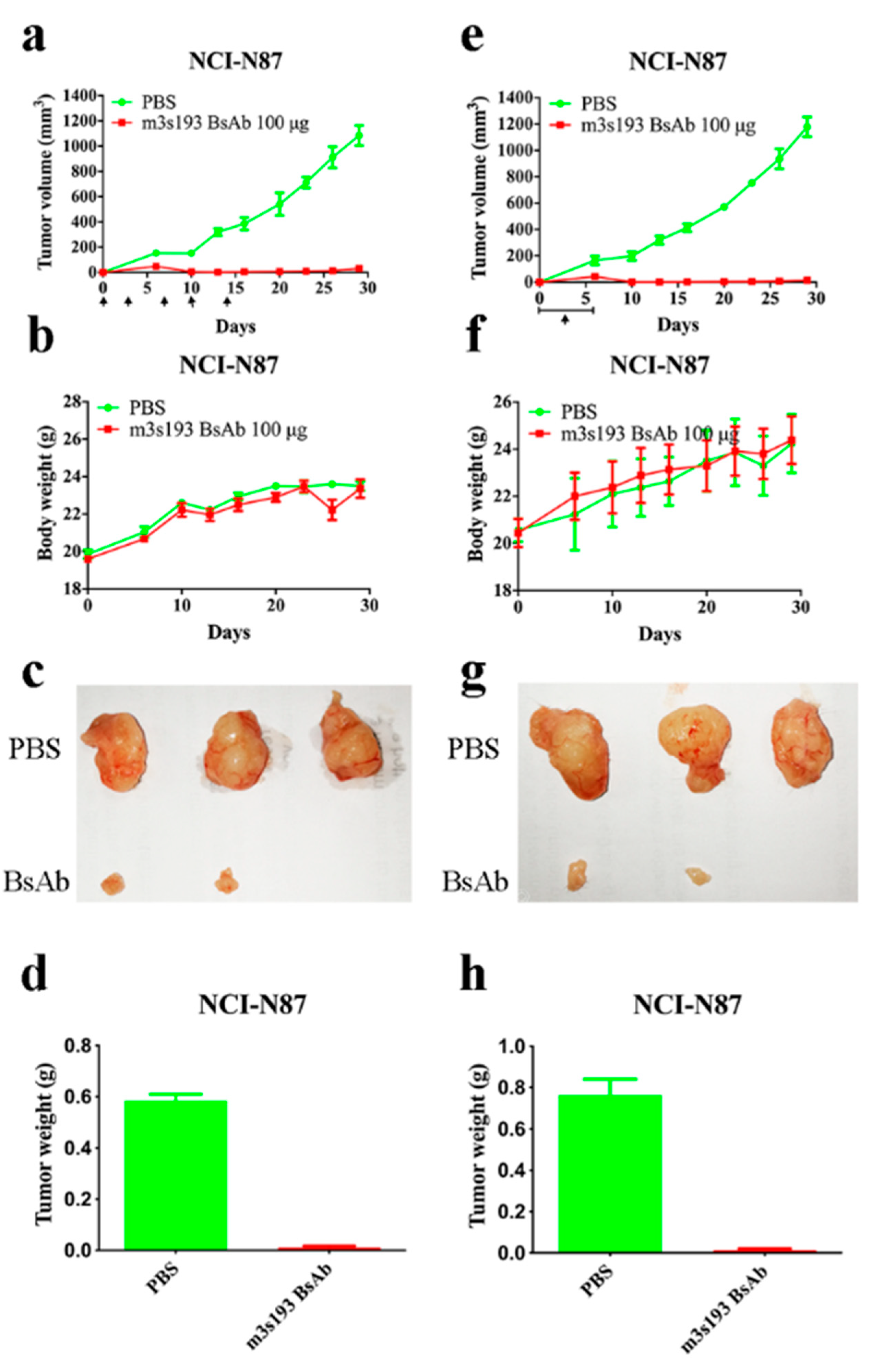
3.6.1. Tumor Inhibitory Activity of m3s193 BsAb
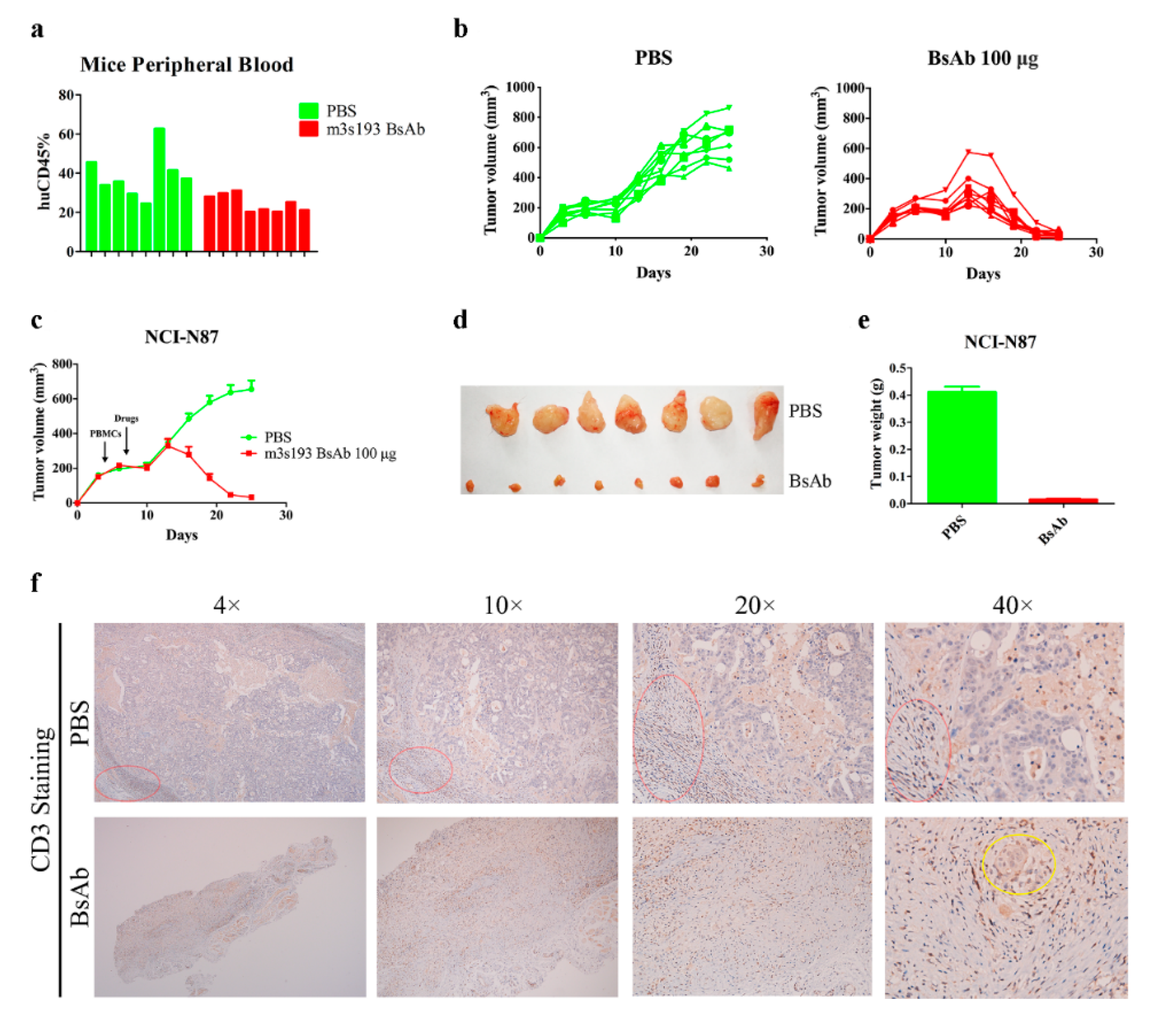
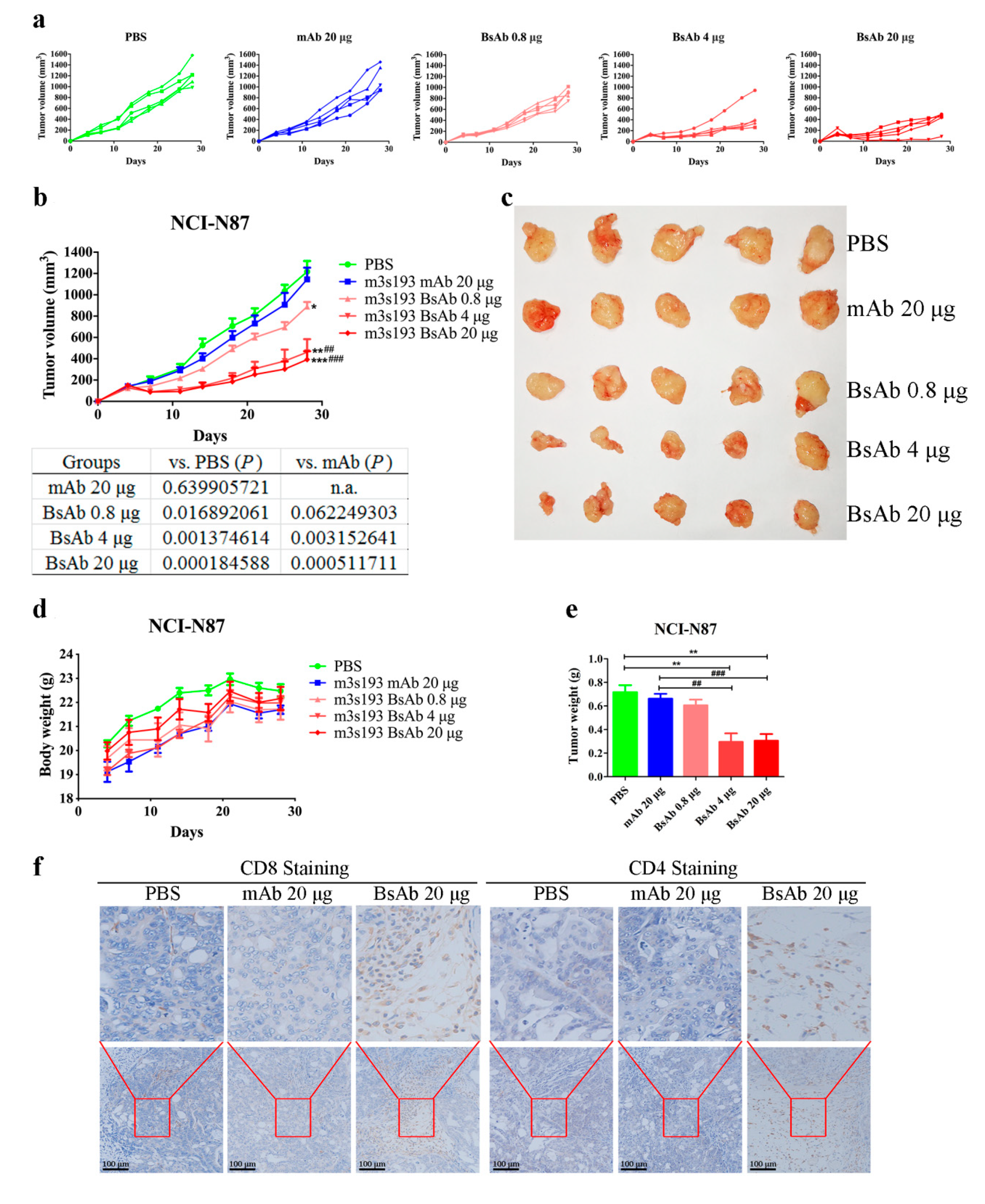
3.6.2. Remarkable Tumor Inhibitory Activity of m3s193 BsAb
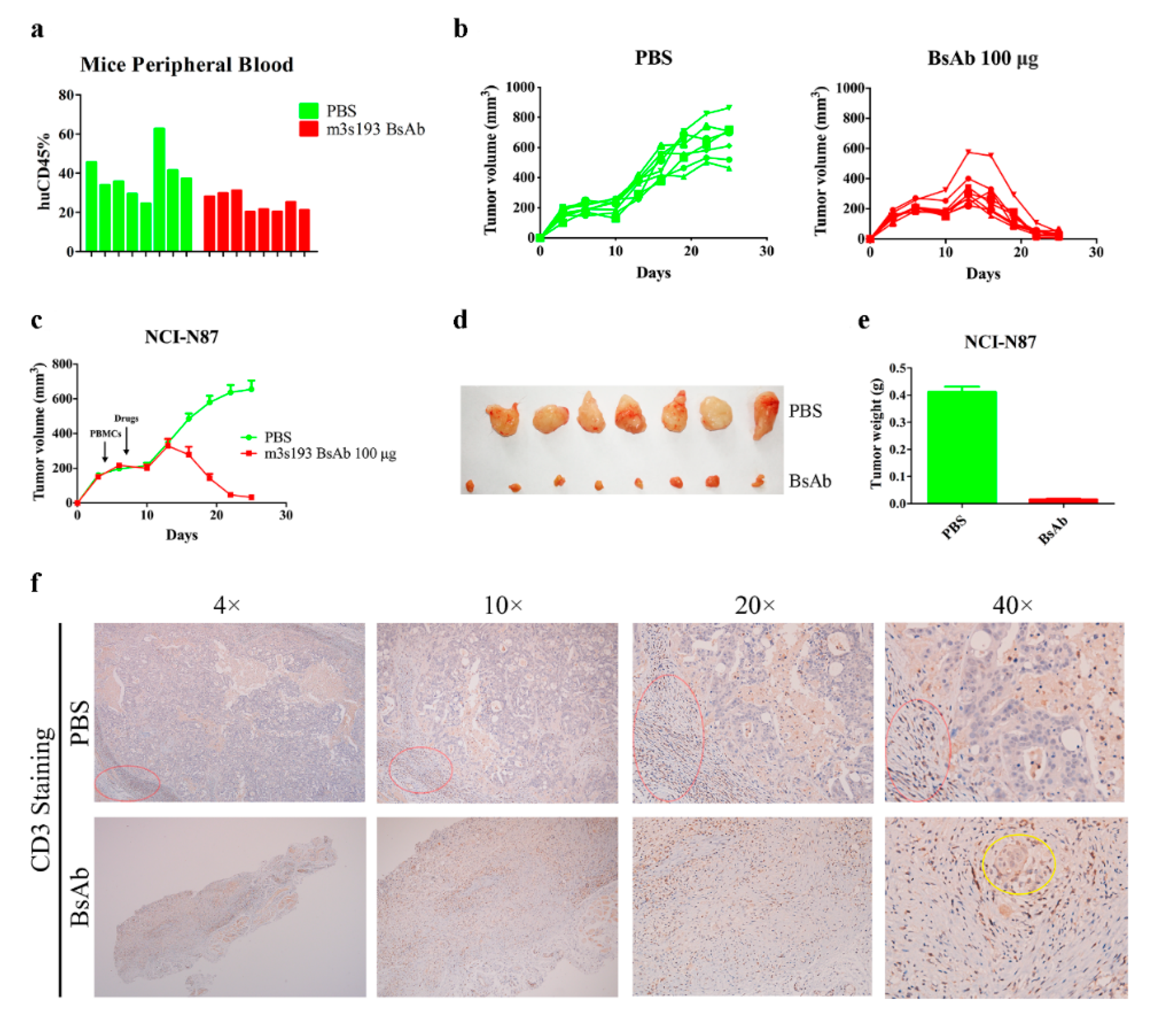
3.6.3. M3s193 BsAb Had Potent Anti-Tumor Activity in Therapeutic Study
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Labrijn, A.F.; Janmaat, M.L.; Reichert, J.M.; Parren, P.W. Bispecific antibodies: A mechanistic review of the pipeline. Nat. Rev. Drug Discov. 2019, 18, 585–608. [Google Scholar] [CrossRef]
- Haas, C.; Krinner, E.; Brischwein, K.; Hoffmann, P.; Lutterbüse, R.; Schlereth, B.; Kufer, P.; Baeuerle, P.A. Mode of cytotoxic action of T cell-engaging BiTE antibody MT110. Immunobiology 2009, 214, 441–453. [Google Scholar] [CrossRef] [PubMed]
- Stadler, C.R.; Bähr-Mahmud, H.; Plum, L.M.; Schmoldt, K.; Kölsch, A.C.; Türeci, Ö.; Sahin, U. Characterization of the first-in-class T-cell-engaging bispecific single-chain antibody for targeted immunotherapy of solid tumors expressing the oncofetal protein claudin 6. OncoImmunology 2015, 5, e1091555. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Cheung, N. T cell engaging bispecific antibody (T-BsAb): From technology to therapeutics. Pharmacol. Ther. 2018, 182, 161–175. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Lam, C.-Y.K.; Long, V.; Widjaja, L.; Yang, Y.; Li, H.; Jin, L.; Burke, S.; Gorlatov, S.; Brown, J.; et al. MGD011, A CD19 x CD3 Dual-Affinity Retargeting Bi-specific Molecule Incorporating Extended Circulating Half-life for the Treatment of B-Cell Malignancies. Clin. Cancer Res. 2017, 23, 1506–1518. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Albaitero, A.; Xu, H.; Guo, H.; Wang, L.; Wu, Z.; Tran, H.; Chandarlapaty, S.; Scaltriti, M.; Janjigian, Y.; De Stanchina, E.; et al. Overcoming resistance to HER2-targeted therapy with a novel HER2/CD3 bispecific antibody. OncoImmunology 2017, 6, e1267891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santich, B.H.; Park, J.A.; Tran, H.; Guo, H.-F.; Huse, M.; Cheung, N.-K.V. Interdomain spacing and spatial configuration drive the potency of IgG-[L]-scFv T cell bispecific antibodies. Sci. Transl. Med. 2020, 12, eaax1315. [Google Scholar] [CrossRef] [PubMed]
- Hokke, C.H.; Neeleman, A.P.; Koeleman, C.A.; van den Eijnden, D.H. Identification of an alpha3-fucosyltransferase and a novel alpha2-fucosyltransferase activity in cercariae of the schistosome Trichobilharzia ocellata: Biosynthesis of the Fucalpha1-->2Fucalpha1-->3[Gal(NAc)beta1-->4]GlcNAc sequence. Glycobiology 1998, 8, 393–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitamura, K.; Stockert, E.; Garin-Chesa, P.; Welt, S.; Lloyd, K.O.; Armour, K.L.; Wallace, T.P.; Harris, W.J.; Carr, F.J.; Old, L.J. Specificity analysis of blood group Lewis-y (Le(y)) antibodies generatedagainst synthetic and natural Le(y) determinants. Proc. Natl. Acad. Sci. USA 1994, 91, 12957–12961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westwood, J.A.; Smyth, M.J.; Teng, M.W.L.; Moeller, M.; Trapani, J.A.; Scott, A.M.; Smyth, F.E.; Cartwright, G.A.; Power, B.E.; Hönemann, D.; et al. Adoptive transfer of T cells modified with a humanized chimeric receptor gene inhibits growth of Lewis-Y-expressing tumors in mice. Proc. Natl. Acad. Sci. USA 2005, 102, 19051–19056. [Google Scholar] [CrossRef] [Green Version]
- Scott, A.M.; Geleick, D.; Rubira, M.; Clarke, K.; Nice, E.C.; Smyth, F.E.; Stockert, E.; Richards, E.C.; Carr, F.J.; Harris, W.J.; et al. Construction, production, and characterization of humanized anti-Lewis Y monoclonal antibody 3S193 for targeted immunotherapy of solid tumors. Cancer Res. 2000, 60, 3254–3261. [Google Scholar] [PubMed]
- Dettke, M.; Pálfi, G.; Loibner, H. Activation-dependent expression of the blood group-related lewis Y antigen on peripheral blood granulocytes. J. Leukoc. Biol. 2000, 68, 511–514. [Google Scholar] [PubMed]
- Klinger, M.; Farhan, H.; Just, H.; Drobny, H.; Himmler, G.; Loibner, H.; Mudde, G.C.; Freissmuth, M.; Sexl, V. Antibodies directed against Lewis-Y antigen inhibit signaling of Lewis-Y modified ErbB receptors. Cancer Res. 2004, 64, 1087–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, M.; Zhu, L.; Zhuang, H.; Hao, Y.; Gao, S.; Liu, S.; Liu, Q.; Liu, D.; Liu, J.; Lin, B. Lewis Y antigen modified CD47 is an independent risk factor for poor prognosis and promotes early ovarian cancer metastasis. Am. J. Cancer Res. 2015, 5, 2777–2787. [Google Scholar]
- Gao, L.; Yan, L.; Lin, B.; Gao, J.; Liang, X.; Wang, Y.; Liu, J.; Zhang, S.; Masao, I. Enhancive effects of Lewis y antigen on CD44-mediated adhesion and spreading of human ovarian cancer cell line RMG-I. J. Exp. Clin. Cancer Res. 2011, 30, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Hu, Z.; Liu, J.; Liu, D.; Wang, Y.; Cai, M.; Zhang, D.; Tan, M.; Lin, B. Expression of CD147 and Lewis y antigen in ovarian cancer and their relationship to drug resistance. Med. Oncol. 2014, 31, 920. [Google Scholar] [CrossRef]
- Hellström, I.; Garrigues, H.J.; Garrigues, U.; Hellström, K.E. Highly tumor-reactive, internalizing, mouse monoclonal antibodies to Le(y)-related cell surface antigens. Cancer Res. 1990, 50, 2183–2190. [Google Scholar]
- Zhang, S.; Zhang, H.S.; Cordon-Cardo, C.; Reuter, V.E.; Singhal, A.K.; Lloyd, K.O.; Livingston, P.O. Selection of tumor antigens as targets for immune attack using immunohistochemistry: II. Blood group-related antigens. Int. J. Cancer 1997, 73, 50–56. [Google Scholar] [CrossRef]
- Madjd, Z.; Parsons, T.; Watson, N.F.; Spendlove, I.; Ellis, I.; Durrant, L.G. High expression of Lewisy/bantigens is associated with decreased survival in lymph node negative breast carcinomas. Breast Cancer Res. 2005, 7, R780–R787. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Liu, J.; Lin, B.; Wang, C.; Li, Q.; Liu, S.; Yan, L.; Zhang, S.; Iwamori, M. Study on the Expression and Clinical Significances of Lewis y Antigen and Integrin αv, β3 in Epithelial Ovarian Tumors. Int. J. Mol. Sci. 2011, 12, 3409–3421. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.-J.; Lin, B.; Hao, Y.-Y.; Li, F.-F.; Liu, D.-W.; Qi, Y.; Zhu, L.-C.; Zhang, S.-L.; Iwamori, M. Lewis(y) antigen stimulates the growth of ovarian cancer cells via regulation of the epidermal growth factor receptor pathway. Oncol. Rep. 2010, 23, 833–841. [Google Scholar] [PubMed]
- Liu, J.; Lin, B.; Hao, Y.; Qi, Y.; Zhu, L.; Li, F.; Liu, D.; Cong, J.; Zhang, S.; Iwamori, M. Lewis y antigen promotes the proliferation of ovarian carcinoma-derived RMG-I cells through the PI3K/Akt signaling pathway. J. Exp. Clin. Cancer Res. 2009, 28, 154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Liu, S.; Lin, B.; Yan, L.; Wang, Y.; Wang, C.; Zhang, S. Expression and correlation of Lewis y antigen and integrins ?5 and ?1 in ovarian serous and mucinous carcinoma. Int. J. Gynecol. Cancer 2010, 20, 1482–1489. [Google Scholar] [CrossRef]
- Gao, J.; Hu, Z.; Liu, D.; Liu, J.; Liu, C.; Hou, R.; Gao, S.; Zhang, D.; Zhang, S.; Lin, B. Expression of Lewis y antigen and integrin αv, β3 in ovarian cancer and their relationship with chemotherapeutic drug resistance. J. Exp. Clin. Cancer Res. 2013, 32, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Z.; Gao, S.; Gao, J.; Hou, R.; Liu, C.; Liu, J.; Li, B.; Liu, D.; Zhang, S.; Lin, B. Elevated Levels of Lewis Y and Integrin α5β1 Correlate with Chemotherapeutic Drug Resistance in Epithelial Ovarian Carcinoma. Int. J. Mol. Sci. 2012, 13, 15588–15600. [Google Scholar] [CrossRef] [Green Version]
- Baldus, S.E.; Mönig, S.P.; Zirbes, T.K.; Thakran, J.; Köthe, D.; Köppel, M.; Hanisch, F.-G.; Thiele, J.; Schneider, P.M.; Hölscher, A.H.; et al. Lewis(y) antigen (CD174) and apoptosis in gastric and colorectal carcinomas: Correlations with clinical and prognostic parameters. Histol. Histopathol. 2006, 21, 503–510. [Google Scholar] [PubMed]
- Clarke, K.; Lee, F.T.; Brechbiel, M.W.; Smyth, F.E.; Old, L.J.; Scott, A.M. Therapeutic efficacy of anti-Lewis(y) humanized 3S193 radioimmunotherapy in a breast cancer model: Enhanced activity when combined with taxol chemotherapy. Clin. Cancer Res. 2000, 6, 3621–3628. [Google Scholar] [PubMed]
- Aziz, F.; Yang, X.; Wang, X.; Yan, Q. Anti-LeY antibody enhances therapeutic efficacy of celecoxib against gastric cancer by downregulation of MAPKs/COX-2 signaling pathway: Correlation with clinical study. J. Cancer Res. Clin. Oncol. 2014, 141, 1221–1235. [Google Scholar] [CrossRef]
- Kawai, S.; Kato, S.; Imai, H.; Okada, Y.; Ishioka, C. Suppression of FUT1 attenuates cell proliferation in the HER2-overexpressing cancer cell line NCI-N87. Oncol. Rep. 2012, 29, 13–20. [Google Scholar] [CrossRef]
- Smaletz, O.; Diz, M.D.; do Carmo, C.C.; Sabbaga, J.; Cunha-Junior, G.F.; Azevedo, S.J.; Maluf, F.C.; Barrios, C.H.; Costa, R.L.; Fontana, A.G.; et al. A phase II trial with anti-Lewis-Y monoclonal antibody (hu3S193) for the treatment of platinum resistant/refractory ovarian, fallopian tube and primary peritoneal carcinoma. Gynecol. Oncol. 2015, 138, 272–277. [Google Scholar] [CrossRef]
- Smaletz, O.; Ismael, G.; Del Pilar Estevez-Diz, M.; O Nascimento, I.L.O.; de Morais, A.L.G.; Cunha-Junior, G.F.; Azevedo, S.J.; Alves, V.A.; Moro, A.M.; Yeda, F.P.; et al. Phase II consolidation trial with anti-Lewis-Y monoclonal antibody (hu3S193) in platinum-sensitive ovarian cancer after a second remission. Int. J. Gynecol. Cancer 2021, 31, 562–568. [Google Scholar] [CrossRef]
- Han, L.; Chen, J.; Ding, K.; Zong, H.; Xie, Y.; Jiang, H.; Zhang, B.; Lu, H.; Yin, W.; Gilly, J.; et al. Efficient generation of bispecific IgG antibodies by split intein mediated protein trans-splicing system. Sci. Rep. 2017, 7, 8360. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Cheng, M.; Guo, H.; Chen, Y.; Huse, M.; Cheung, N.-K.V. Retargeting T Cells to GD2 Pentasaccharide on Human Tumors Using Bispecific Humanized Antibody. Cancer Immunol. Res. 2015, 3, 266–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Z.; Guo, H.-F.; Xu, H.; Cheung, N.-K.V. Development of a tetravalent anti-GPA33/anti-CD3 bispecific antibody for colorectal cancers. Mol. Cancer Ther. 2018, 17, 2164–2175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo, M.; Kim, H.S.; Tong, R.K.; Bainbridge, T.W.; Vernes, J.-M.; Zhang, Y.; Lin, Y.L.; Chung, S.; Dennis, M.S.; Zuchero, Y.J.Y.; et al. Effector-attenuating Substitutions That Maintain Antibody Stability and Reduce Toxicity in Mice. J. Biol. Chem. 2017, 292, 3900–3908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adair, J.R.; Athwal, D.S.; Bodmer, M.W.; Bright, S.M.; Collins, A.M.; Pulito, V.L.; Rao, P.E.; Reedman, R.; Rothermel, A.L.; Xu, D.; et al. Humanization of the murine anti-human CD3 monoclonal antibody OKT3. Hum. Antibodies 1994, 5, 41–47. [Google Scholar] [CrossRef]
- Xiao, X.; Ho, M.; Zhu, Z.; Pastan, I.; Dimitrov, D.S. Identification and characterization of fully human anti-CD22 monoclonal antibodies. mAbs 2009, 1, 297–303. [Google Scholar] [CrossRef] [Green Version]
- Ding, K.; Han, L.; Zong, H.; Chen, J.; Zhang, B.; Zhu, J. Production process reproducibility and product quality consistency of transient gene expression in HEK293 cells with anti-PD1 antibody as the model protein. Appl. Microbiol. Biotechnol. 2017, 101, 1889–1898. [Google Scholar] [CrossRef]
- Zhu, J. Mammalian cell protein expression for biopharmaceutical production. Biotechnol. Adv. 2012, 30, 1158–1170. [Google Scholar] [CrossRef]
- Zhu, J. Update on Production of Recombinant Therapeutic Protein: Transient Gene Expression; Smithers Rapra: Shawbury, UK, 2013. [Google Scholar]
- Fan, D.; Li, W.; Yang, Y.; Zhang, X.; Zhang, Q.; Yan, Y.; Yang, M.; Wang, J.; Xiong, D. Redirection of CD4+ and CD8+ T lymphocytes via an anti-CD3 × anti-CD19 bi-specific antibody combined with cytosine arabinoside and the efficient lysis of patient-derived B-ALL cells. J. Hematol. Oncol. 2015, 8, 108. [Google Scholar] [CrossRef] [Green Version]
- Mohammad, G.R.K.S.; Ghahremanloo, A.; Soltani, A.; Fathi, E.; Hashemy, S.I. Cytokines as potential combination agents with PD-1/PD-L1 blockade for cancer treatment. J. Cell. Physiol. 2020, 235, 5449–5460. [Google Scholar] [CrossRef] [PubMed]
- Conlon, K.C.; Miljković, M.; Waldmann, T.A. Cytokines in the Treatment of Cancer. J. Interf. Cytokine Res. 2019, 39, 6–21. [Google Scholar] [CrossRef]
- Benczik, M.; Gaffen, S.L. The Interleukin (IL)-2 Family Cytokines: Survival and Proliferation Signaling Pathways in T Lymphocytes. Immunol. Investig. 2004, 33, 109–142. [Google Scholar] [CrossRef]
- Sun, L.L.; Ellerman, D.; Mathieu, M.; Hristopoulos, M.; Chen, X.; Li, Y.; Yan, X.; Clark, R.; Reyes, A.; Stefanich, E.; et al. Anti-CD20/CD3 T cell–dependent bispecific antibody for the treatment of B cell malignancies. Sci. Transl. Med. 2015, 7, 287ra70. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- de Olza, M.O.; Rodrigo, B.N.; Zimmermann, S.; Coukos, G. Turning up the heat on non-immunoreactive tumours: Opportunities for clinical development. Lancet Oncol. 2020, 21, e419–e430. [Google Scholar] [CrossRef]
- Zhou, Y.; Zong, H.; Han, L.; Xie, Y.; Jiang, H.; Gilly, J.; Zhang, B.; Lu, H.; Chen, J.; Sun, R.; et al. A novel bispecific antibody targeting CD3 and prolactin receptor (PRLR) against PRLR-expression breast cancer. J. Exp. Clin. Cancer Res. 2020, 39, 87. [Google Scholar] [CrossRef] [PubMed]
- Dickopf, S.; Georges, G.J.; Brinkmann, U. Format and geometries matter: Structure-based design defines the functionality of bispecific antibodies. Comput. Struct. Biotechnol. J. 2020, 18, 1221–1227. [Google Scholar] [CrossRef] [PubMed]
- Choudhuri, K.; Wiseman, D.; Brown, M.H.; Gould, K.; Van Der Merwe, P.A. T-cell receptor triggering is critically dependent on the dimensions of its peptide-MHC ligand. Nature 2005, 436, 578–582. [Google Scholar] [CrossRef]
- Bacac, M.; Fauti, T.; Sam, J.; Colombetti, S.; Weinzierl, T.; Ouaret, D.; Bodmer, W.F.; Lehmann, S.; Hofer, T.; Hosse, R.J.; et al. A Novel Carcinoembryonic Antigen T-Cell Bispecific Antibody (CEA TCB) for the Treatment of Solid Tumors. Clin. Cancer Res. 2016, 22, 3286–3297. [Google Scholar] [CrossRef] [Green Version]
- Bacac, M.; Klein, C.; Umana, P. CEA TCB: A novel head-to-tail 2:1 T cell bispecific antibody for treatment of CEA-positive solid tumors. OncoImmunology 2016, 5, e1203498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varki, A.; Cummings, R.D.; Esko, J.D.; Stanley, P.; Hart, G.W.; Aebi, M.; Darvill, A.G.; Kinoshita, T.; Packer, N.H.; Prestegard, J.H.; et al. Essentials of Glycobiology; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2015. [Google Scholar]
- Labrada, M.; Dorvignit, D.; Hevia, G.; Rodríguez-Zhurbenko, N.; Hernández, A.M.; Vázquez, A.M.; Fernández, L.E. GM3(Neu5Gc) ganglioside: An evolution fixed neoantigen for cancer immunotherapy. Semin. Oncol. 2018, 45, 41–51. [Google Scholar] [CrossRef]
- Ladenstein, R.; Pötschger, U.; Valteau-Couanet, D.; Luksch, R.; Castel, V.; Yaniv, I.; Laureys, G.; Brock, P.; Michon, J.M.; Owens, C.; et al. Interleukin 2 with anti-GD2 antibody ch14.18/CHO (dinutuximab beta) in patients with high-risk neuroblastoma (HR-NBL1/SIOPEN): A multicentre, randomised, phase 3 trial. Lancet Oncol. 2018, 19, 1617–1629. [Google Scholar] [CrossRef]
- Cavdarli, S.; Dewald, J.H.; Yamakawa, N.; Guérardel, Y.; Terme, M.; Le Doussal, J.-M.; Delannoy, P.; Groux-Degroote, S. Identification of 9-O-acetyl-N-acetylneuraminic acid (Neu5,9Ac2) as main O-acetylated sialic acid species of GD2 in breast cancer cells. Glycoconj. J. 2019, 36, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Posey, A.D., Jr.; Schwab, R.D.; Boesteanu, A.C.; Steentoft, C.; Mandel, U.; Engels, B.; Stone, J.D.; Madsen, T.D.; Schreiber, K.; Haines, K.M.; et al. Engineered CAR T Cells Targeting the Cancer-Associated Tn-Glycoform of the Membrane Mucin MUC1 Control Adenocarcinoma. Immunity 2016, 44, 1444–1454. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Structure | Fc Mutation | Target | SEC-HPLC Purity (%) |
|---|---|---|---|---|
| m3s193 mAb | IgG1 | No | Lewis Y | 92.63% |
| m3s193 BsAb | IgG-[L]-scfv | LALA-PG | Lewis Y and CD3 | 98.44% |
| M971 BsAb | IgG-[L]-scfv | LALA-PG | CD22 and CD3 | 98.79% |
| Cells | MCF-7 | T47D | MDA-MB-231 | NCI-N87 | NCI-N87 | |
|---|---|---|---|---|---|---|
| Proteins | ||||||
| m3s193 mAb | 6.689 | 62.52 | 2.296E + 15 | 162.5 | ||
| m3s193 BsAb | 162.5 | 35.89 | 23.98 | 9.315 | 2.673 | |
| M971 BsAb | ND* | |||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, J.; Pan, Z.; Han, L.; Zhou, Y.; Zong, H.; Wang, L.; Sun, R.; Jiang, H.; Xie, Y.; Yuan, Y.; et al. A Novel Bispecific Antibody Targeting CD3 and Lewis Y with Potent Therapeutic Efficacy against Gastric Cancer. Biomedicines 2021, 9, 1059. https://doi.org/10.3390/biomedicines9081059
Chen J, Pan Z, Han L, Zhou Y, Zong H, Wang L, Sun R, Jiang H, Xie Y, Yuan Y, et al. A Novel Bispecific Antibody Targeting CD3 and Lewis Y with Potent Therapeutic Efficacy against Gastric Cancer. Biomedicines. 2021; 9(8):1059. https://doi.org/10.3390/biomedicines9081059
Chicago/Turabian StyleChen, Jie, Zhidi Pan, Lei Han, Yuexian Zhou, Huifang Zong, Lei Wang, Rui Sun, Hua Jiang, Yueqing Xie, Yunsheng Yuan, and et al. 2021. "A Novel Bispecific Antibody Targeting CD3 and Lewis Y with Potent Therapeutic Efficacy against Gastric Cancer" Biomedicines 9, no. 8: 1059. https://doi.org/10.3390/biomedicines9081059
APA StyleChen, J., Pan, Z., Han, L., Zhou, Y., Zong, H., Wang, L., Sun, R., Jiang, H., Xie, Y., Yuan, Y., Wu, M., Bian, Y., Zhang, B., & Zhu, J. (2021). A Novel Bispecific Antibody Targeting CD3 and Lewis Y with Potent Therapeutic Efficacy against Gastric Cancer. Biomedicines, 9(8), 1059. https://doi.org/10.3390/biomedicines9081059