
The Histone Variant MacroH2A1 Impacts Circadian Gene Expression and Cell Phenotype in an In Vitro Model of Hepatocellular Carcinoma

 ,
,  ,
,  ,
, 
 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Primary Datasets
2.2. Cell Culture
2.3. Generation of Lentiviral Vectors
2.4. RNA-Seq
2.5. Analysis for Functional Categorization of the Most Significant Pathways Enriched by Deregulated Circadian Genes
2.6. Knockdown of PER1 by RNA Interference
2.7. Western Blotting
2.8. Cell Proliferation Assay
2.9. Cell Migration Assay
2.10. Flow Cytometry Analysis
2.10.1. Cell Cycle
2.10.2. Half Maximal Inhibitory Concentration (IC50) of Cisplatin
2.10.3. DNA Damage and Apoptosis Assay with and without Cisplatin
2.10.4. Mitochondria and Cellular Respiration Measurements
2.10.5. Statistical Analysis
3. Results
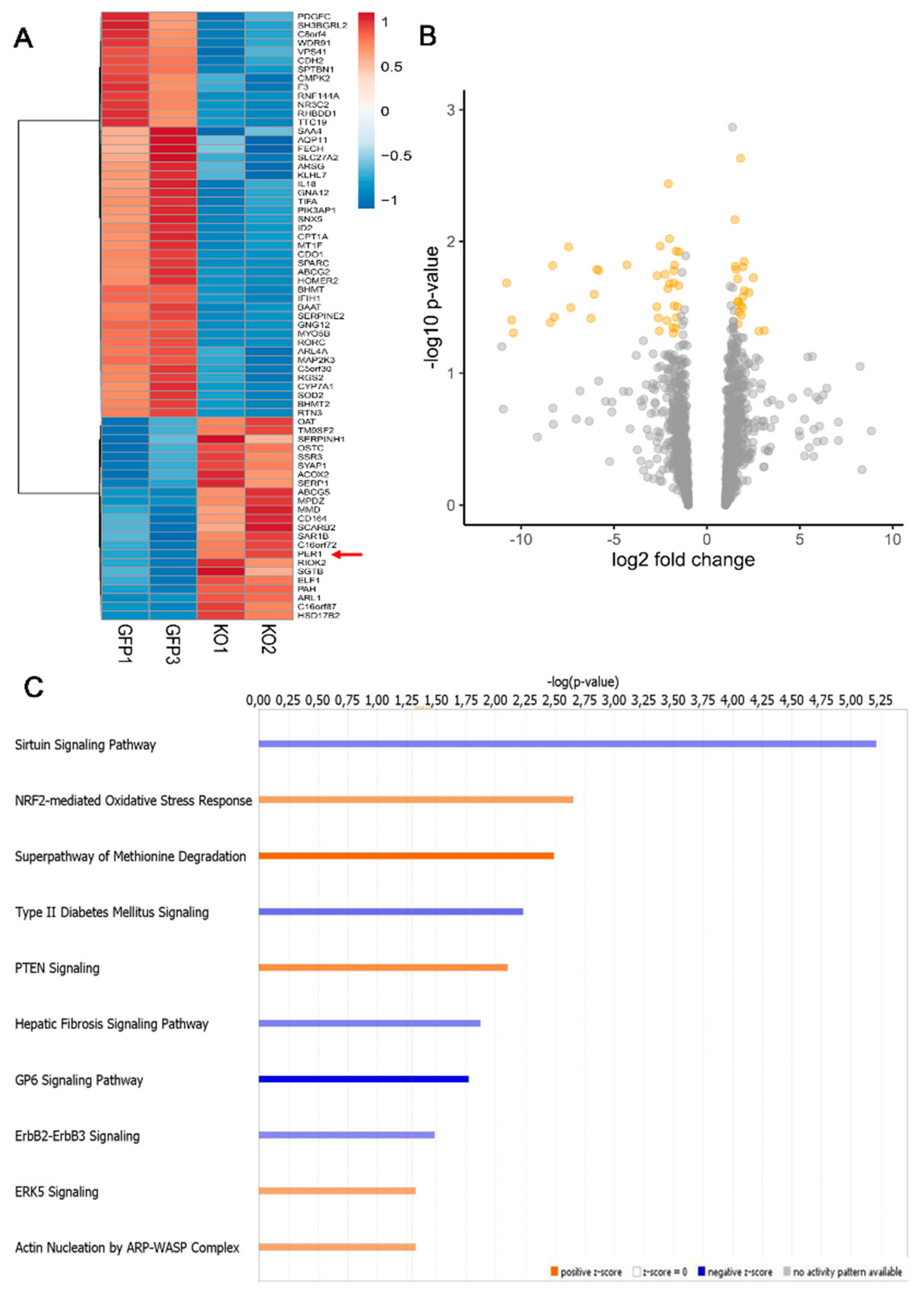
3.1. Loss of MacroH2A1 Histone Variants Changes the Expression of Circadian Genes in HCC Cells
3.2. Circadian Genes Deregulated upon MacroH2A1 Knockout Enrich Important Pathways in HCC Cells
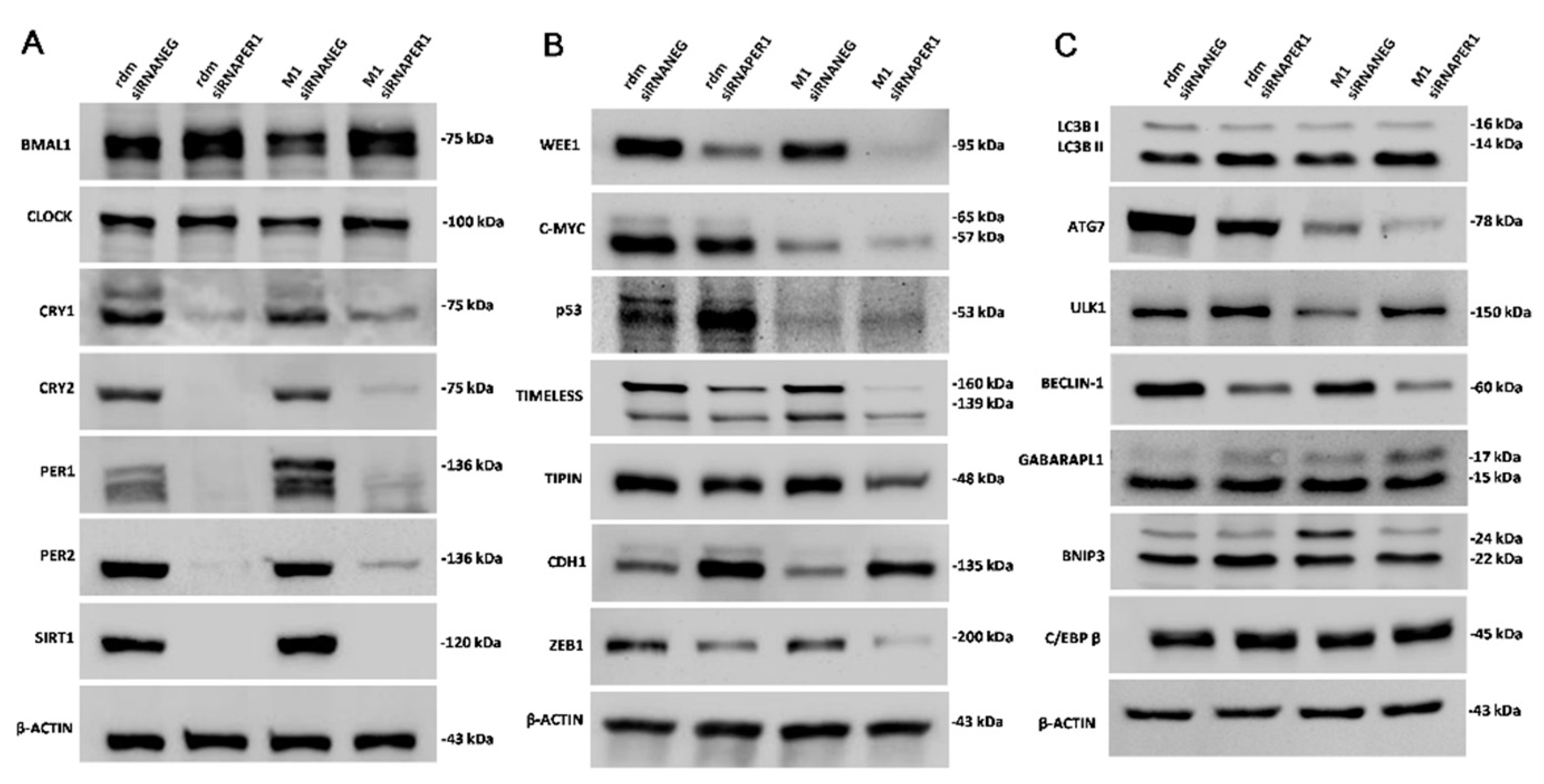
3.3. MacroH2A1 Histone Knockdown in HCC Alters the Expression of Proteins Entailed in Key Cell Functions
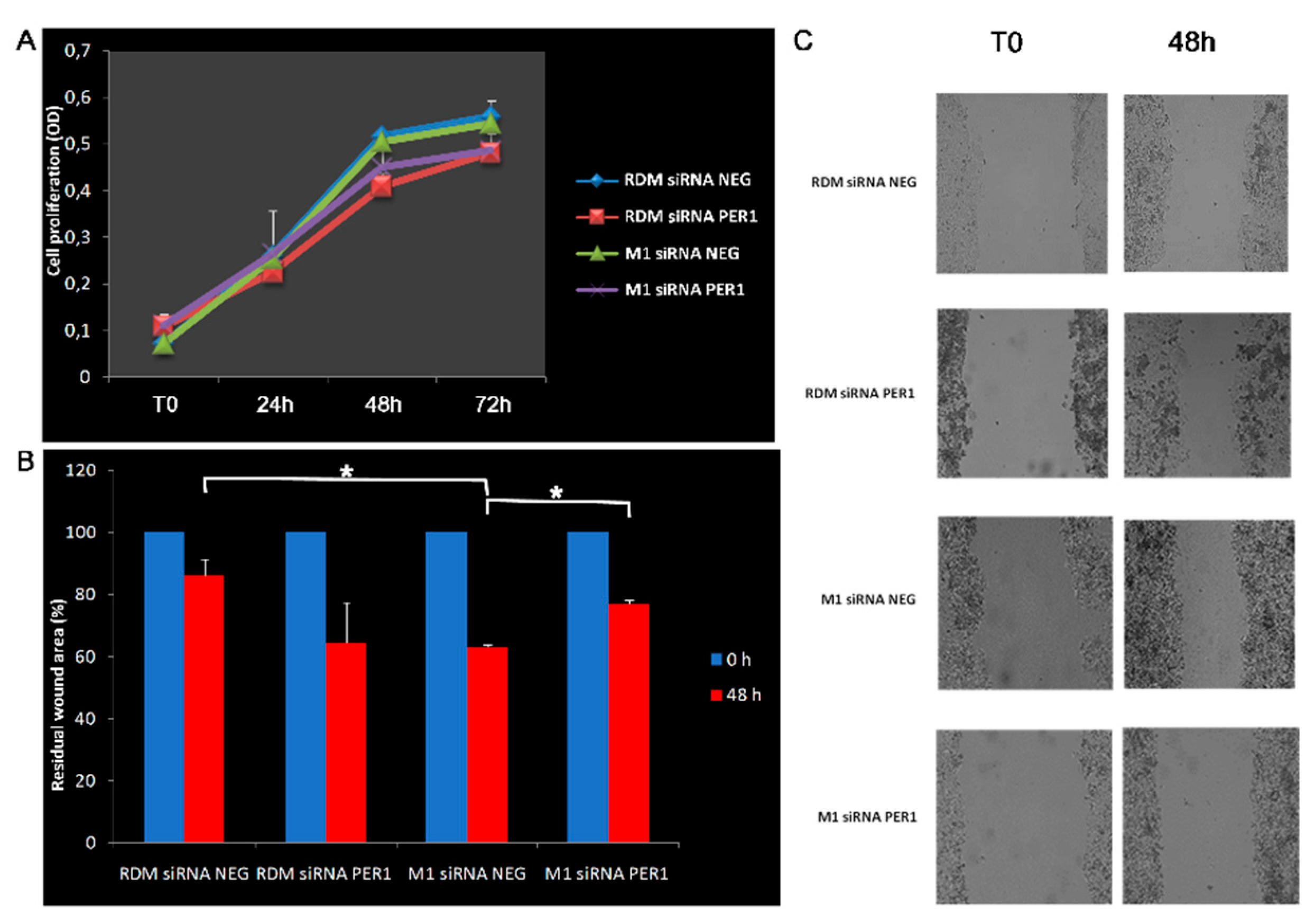
3.4. Knockdown of MacroH2A1 Histone Modulates Proliferative and Migratory Capacity of HCC Cells
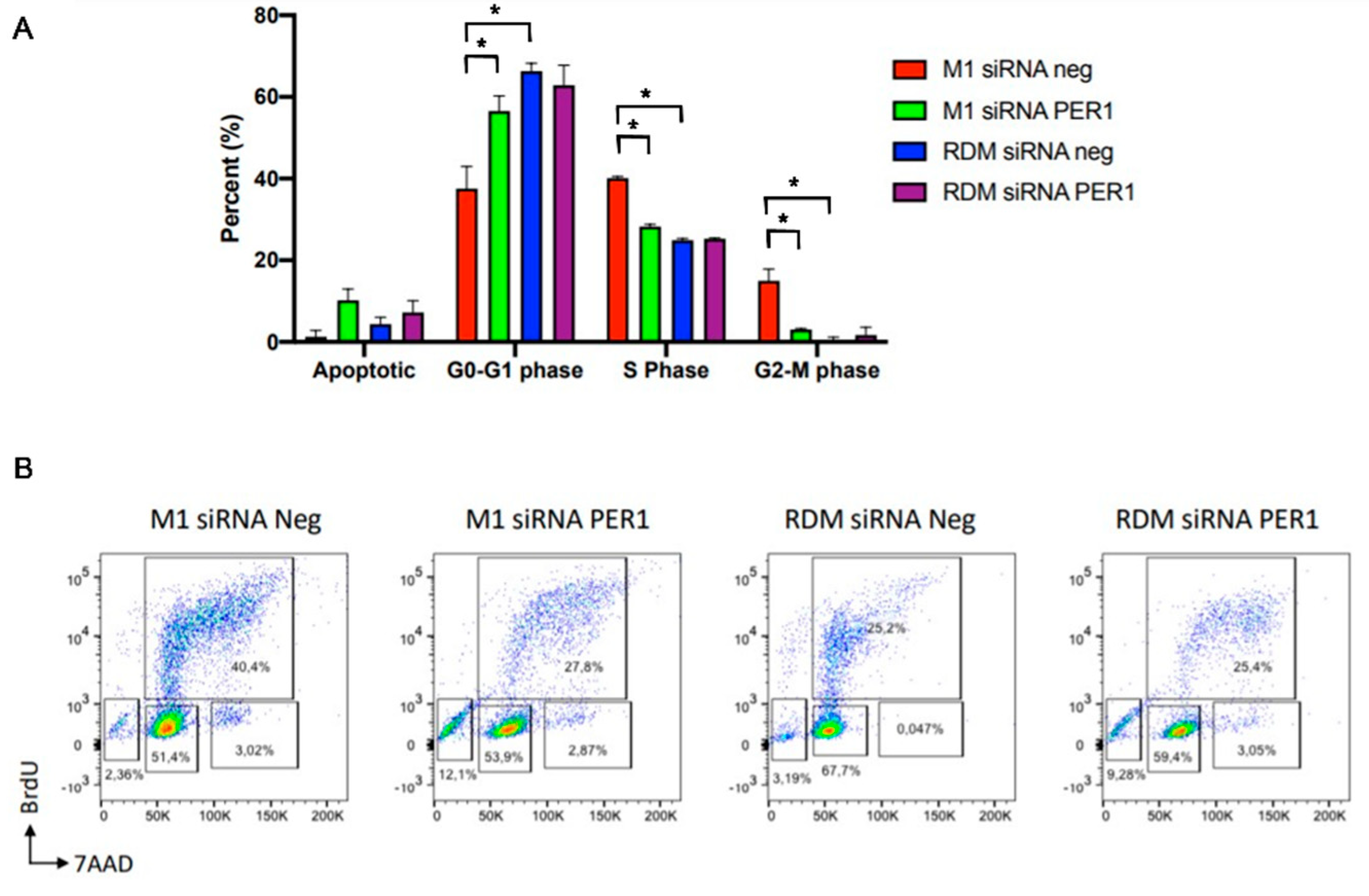
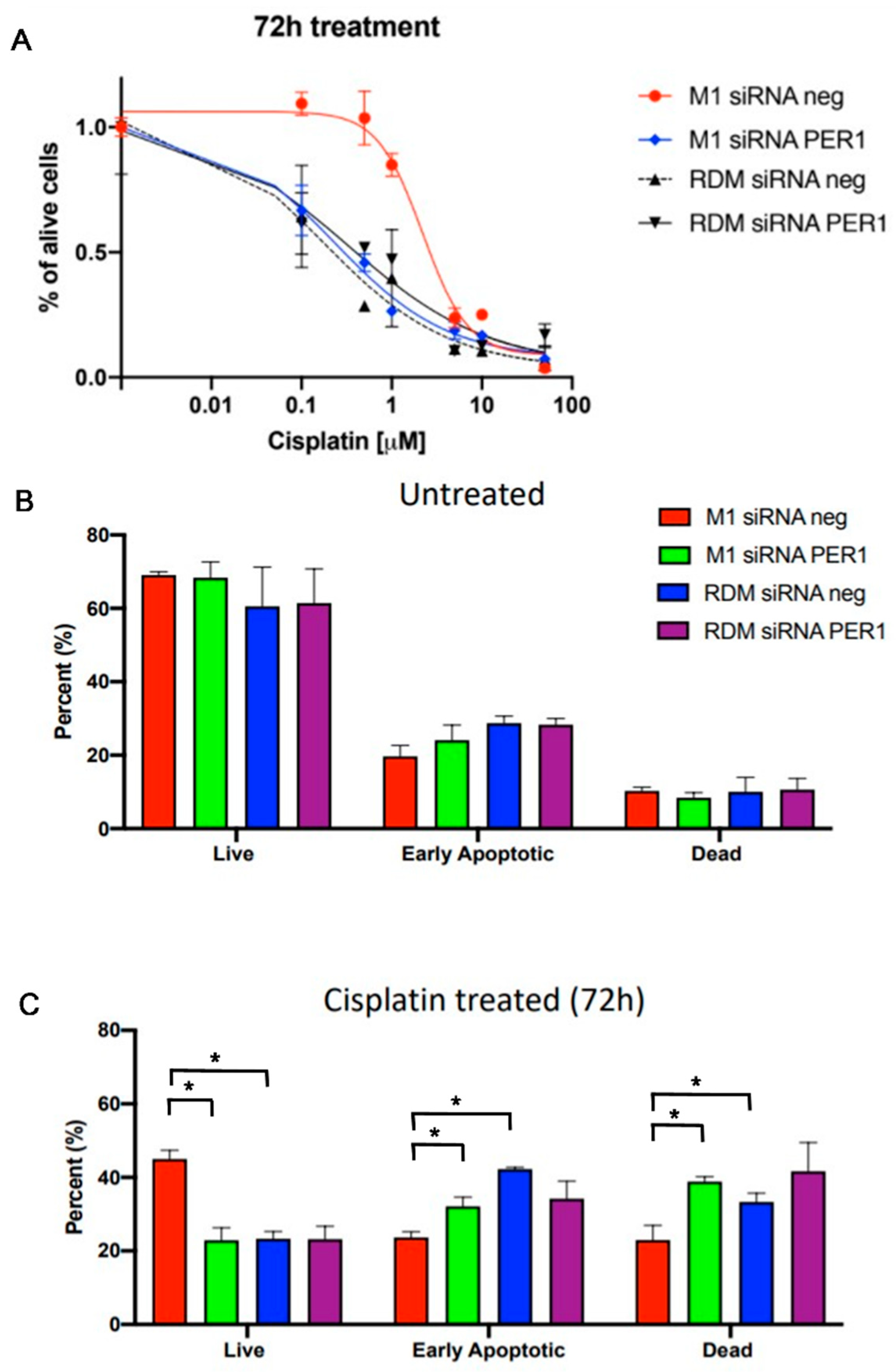
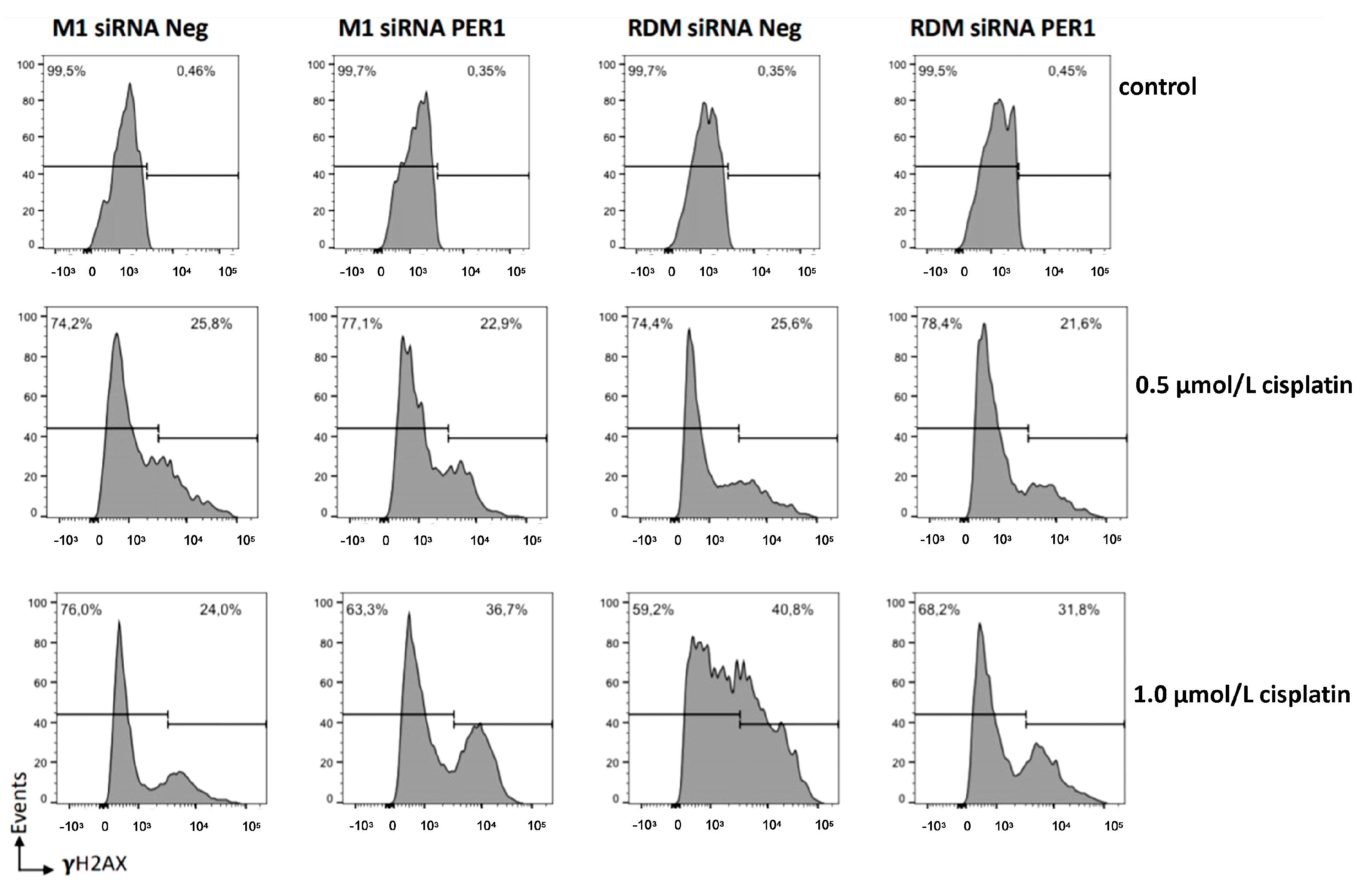
3.5. MacroH2A1 Histone Loss Impacts Cell Cycle, Anticancer Drug Sensitivity, DNA Damage/Apoptosis in HCC Cells
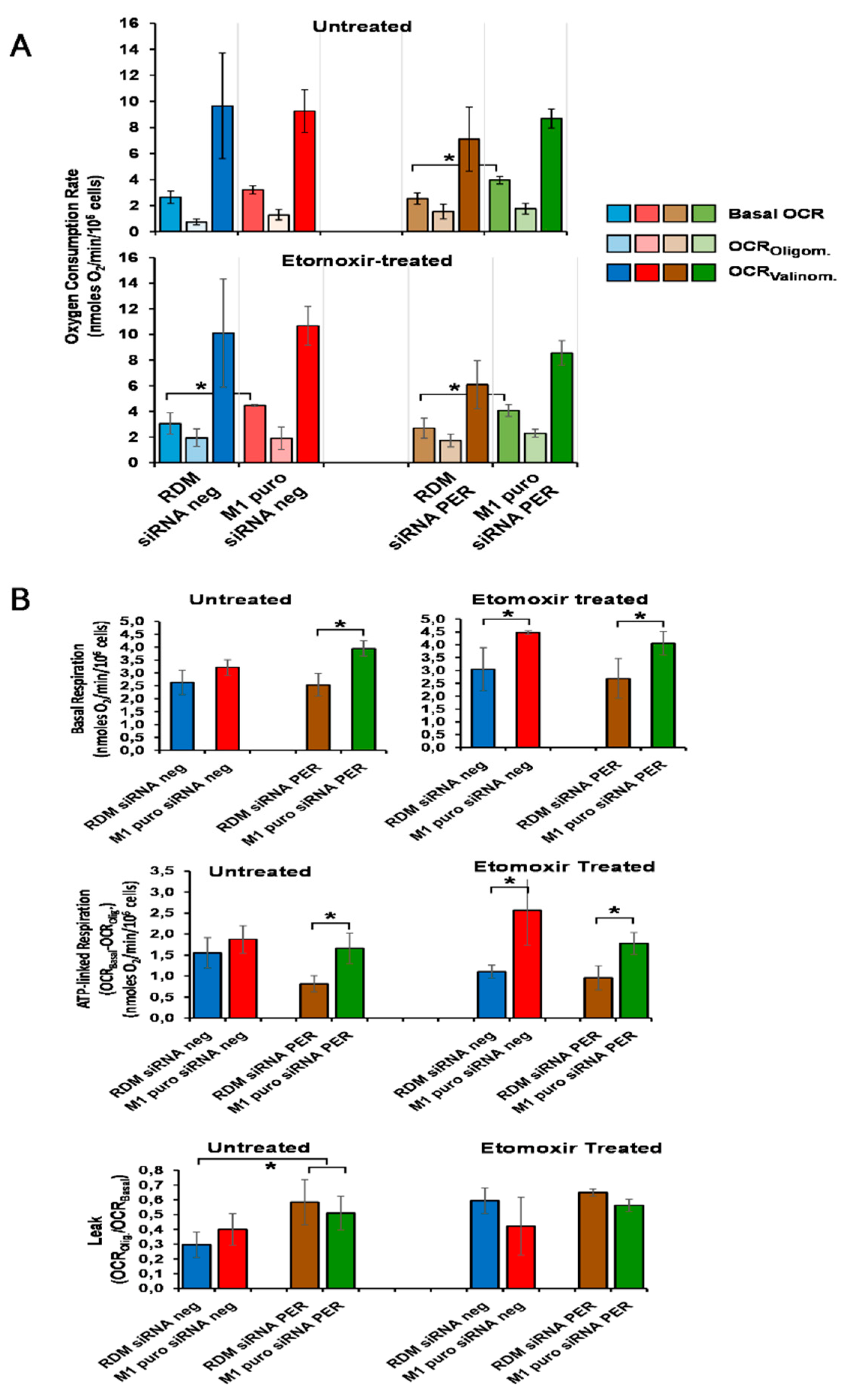
3.6. Loss of MacroH2A1 Decreases Mitochondrial Oxidative Phosphorylation Efficiency
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 2019, 144, 1941–1953. [Google Scholar] [CrossRef]
- Akinyemiju, T.; Abera, S.; Ahmed, M.; Alam, N.; Alemayohu, M.A.; Allen, C.; Al-Raddadi, R.; Alvis-Guzman, N.; Amoako, Y.; Artaman, A.; et al. The Burden of Primary Liver Cancer and Underlying Etiologies from 1990 to 2015 at the Global, Regional, and National Level: Results from the Global Burden of Disease Study 2015. JAMA Oncol. 2017, 3, 1683–1691. [Google Scholar] [PubMed]
- Mazzoccoli, G.; Miele, L.; Oben, J.; Grieco, A.; Vinciguerra, M. Biology, Epidemiology, Clinical Aspects of Hepatocellular Carcinoma and the Role of Sorafenib. Curr. Drug Targets 2016, 17, 783–799. [Google Scholar] [CrossRef] [PubMed]
- Mazzoccoli, G.; Tarquini, R.; Valoriani, A.; Oben, J.; Vinciguerra, M.; Marra, F. Management strategies for hepatocellular carcinoma: Old certainties and new realities. Clin. Exp. Med. 2016, 16, 243–256. [Google Scholar] [CrossRef]
- Anstee, Q.M.; Reeves, H.L.; Kotsiliti, E.; Govaere, O.; Heikenwalder, M. From NASH to HCC: Current concepts and future challenges. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 411–428. [Google Scholar] [CrossRef] [PubMed]
- Mazzoccoli, G.; Vinciguerra, M.; Oben, J.; Tarquini, R.; De Cosmo, S. Non-alcoholic fatty liver disease: The role of nuclear receptors and circadian rhythmicity. Liver Int. 2014, 34, 1133–1152. [Google Scholar] [CrossRef]
- Takahashi, J.S. Transcriptional architecture of the mammalian circadian clock. Nat. Rev. Genet. 2017, 18, 164–179. [Google Scholar] [CrossRef]
- Cox, K.H.; Takahashi, J.S. Circadian clock genes and the transcriptional architecture of the clock mechanism. J. Mol. Endocrinol. 2019, 63, R93–R102. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.H.; Lazar, M.A. Transcriptional Control of Circadian Rhythms and Metabolism: A Matter of Time and Space. Endocr. Rev. 2020, 41, 707–732. [Google Scholar] [CrossRef]
- Mazzoccoli, G.; De Cosmo, S.; Mazza, T. The Biological Clock: A Pivotal Hub in Non-alcoholic Fatty Liver Disease Pathogenesis. Front. Physiol. 2018, 9, 193. [Google Scholar] [CrossRef]
- Mazzoccoli, G.; Miele, L.; Marrone, G.; Mazza, T.; Vinciguerra, M.; Grieco, A. A Role for the Biological Clock in Liver Cancer. Cancers 2019, 11, 1778. [Google Scholar] [CrossRef]
- Podrini, C.; Borghesan, M.; Greco, A.; Pazienza, V.; Mazzoccoli, G.; Vinciguerra, M. Redox homeostasis and epigenetics in non-alcoholic fatty liver disease (NAFLD). Curr. Pharm. Des. 2013, 19, 2737–2746. [Google Scholar] [CrossRef]
- Lo Re, O.; Vinciguerra, M. Histone MacroH2A1: A Chromatin Point of Intersection between Fasting, Senescence and Cellular Regeneration. Genes 2017, 8, 367. [Google Scholar] [CrossRef] [PubMed]
- Pehrson, J.R.; Costanzi, C.; Dharia, C. Developmental and tissue expression patterns of histone macroH2A1 subtypes. J. Cell Biochem. 1997, 65, 107–113. [Google Scholar] [CrossRef]
- Costanzi, C.; Pehrson, J.R. Histone macroH2A1 is concentrated in the inactive X chromosome of female mammals. Nature 1998, 393, 599–601. [Google Scholar] [CrossRef] [PubMed]
- Pehrson, J.R.; Changolkar, L.N.; Costanzi, C.; Leu, N.A. Mice without macroH2A histone variants. Mol. Cell. Biol. 2014, 34, 4523–4533. [Google Scholar] [CrossRef] [PubMed]
- Podrini, C.; Koffas, A.; Chokshi, S.; Vinciguerra, M.; Lelliott, C.J.; White, J.K.; Adissu, H.A.; Williams, R.; Greco, A. MacroH2A1 isoforms are associated with epigenetic markers for activation of lipogenic genes in fat-induced steatosis. FASEB J. 2015, 29, 1676–1687. [Google Scholar] [CrossRef]
- Borghesan, M.; Fusilli, C.; Rappa, F.; Panebianco, C.; Rizzo, G.; Oben, J.A.; Mazzoccoli, G.; Faulkes, C.; Pata, I.; Agodi, A.; et al. DNA Hypomethylation and Histone Variant macroH2A1 Synergistically Attenuate Chemotherapy-Induced Senescence to Promote Hepatocellular Carcinoma Progression. Cancer Res. 2016, 76, 594–606. [Google Scholar] [CrossRef] [PubMed]
- Sheedfar, F.; Vermeer, M.; Pazienza, V.; Villarroya, J.; Rappa, F.; Cappello, F.; Mazzoccoli, G.; Villarroya, F.; van der Molen, H.; Hofker, M.H.; et al. Genetic ablation of macrohistone H2A1 leads to increased leanness, glucose tolerance and energy expenditure in mice fed a high-fat diet. Int. J. Obes. 2015, 39, 331–338. [Google Scholar] [CrossRef]
- Borghesan, M.; Mazzoccoli, G.; Sheedfar, F.; Oben, J.; Pazienza, V.; Vinciguerra, M. Histone variants and lipid metabolism. Biochem. Soc. Trans. 2014, 42, 1409–1413. [Google Scholar] [CrossRef]
- Pogribny, I.P.; Tryndyak, V.P.; Bagnyukova, T.V.; Melnyk, S.; Montgomery, B.; Ross, S.A.; Latendresse, J.R.; Rusyn, I.; Beland, F.A. Hepatic epigenetic phenotype predetermines individual susceptibility to hepatic steatosis in mice fed a lipogenic methyl-deficient diet. J. Hepatol. 2009, 51, 176–186. [Google Scholar] [CrossRef] [PubMed]
- Lo Re, O.; Douet, J.; Buschbeck, M.; Fusilli, C.; Pazienza, V.; Panebianco, C.; Castracani, C.C.; Mazza, T.; Li Volti, G.; Vinciguerra, M. Histone variant macroH2A1 rewires carbohydrate and lipid metabolism of hepatocellular carcinoma cells towards cancer stem cells. Epigenetics 2018, 13, 829–845. [Google Scholar] [CrossRef] [PubMed]
- Giallongo, S.; Lo Re, O.; Vinciguerra, M. Macro Histone Variants: Emerging Rheostats of Gastrointestinal Cancers. Cancers 2019, 11, 676. [Google Scholar] [CrossRef]
- Lo Re, O.; Fusilli, C.; Rappa, F.; Van Haele, M.; Douet, J.; Pindjakova, J.; Rocha, S.W.; Pata, I.; Valcikova, B.; Uldrijan, S.; et al. Induction of cancer cell stemness by depletion of macrohistone H2A1 in hepatocellular carcinoma. Hepatology 2018, 67, 636–650. [Google Scholar] [CrossRef] [PubMed]
- Rappa, F.; Greco, A.; Podrini, C.; Cappello, F.; Foti, M.; Bourgoin, L.; Peyrou, M.; Marino, A.; Scibetta, N.; Williams, R.; et al. Immunopositivity for histone macroH2A1 isoforms marks steatosis-associated hepatocellular carcinoma. PLoS ONE 2013, 8, e54458. [Google Scholar] [CrossRef]
- Lo Re, O.; Mazza, T.; Giallongo, S.; Sanna, P.; Rappa, F.; Vinh Luong, T.; Li Volti, G.; Drovakova, A.; Roskams, T.; Van Haele, M.; et al. Loss of histone macroH2A1 in hepatocellular carcinoma cells promotes paracrine-mediated chemoresistance and CD4(+)CD25(+)FoxP3(+) regulatory T cells activation. Theranostics 2020, 10, 910–924. [Google Scholar] [CrossRef]
- Pazienza, V.; Borghesan, M.; Mazza, T.; Sheedfar, F.; Panebianco, C.; Williams, R.; Mazzoccoli, G.; Andriulli, A.; Nakanishi, T.; Vinciguerra, M. SIRT1-metabolite binding histone macroH2A1.1 protects hepatocytes against lipid accumulation. Aging 2014, 6, 35–47. [Google Scholar] [CrossRef]
- Farcas, M.; Gavrea, A.A.; Gulei, D.; Ionescu, C.; Irimie, A.; Catana, C.S.; Berindan-Neagoe, I. SIRT1 in the Development and Treatment of Hepatocellular Carcinoma. Front. Nutr. 2019, 6, 148. [Google Scholar] [CrossRef] [PubMed]
- Garten, A.; Grohmann, T.; Kluckova, K.; Lavery, G.G.; Kiess, W.; Penke, M. Sorafenib-Induced Apoptosis in Hepatocellular Carcinoma Is Reversed by SIRT1. Int. J. Mol Sci. 2019, 20, 4048. [Google Scholar] [CrossRef] [PubMed]
- Portmann, S.; Fahrner, R.; Lechleiter, A.; Keogh, A.; Overney, S.; Laemmle, A.; Mikami, K.; Montani, M.; Tschan, M.P.; Candinas, D.; et al. Antitumor effect of SIRT1 inhibition in human HCC tumor models in vitro and in vivo. Mol. Cancer Ther. 2013, 12, 499–508. [Google Scholar] [CrossRef]
- Nakahata, Y.; Kaluzova, M.; Grimaldi, B.; Sahar, S.; Hirayama, J.; Chen, D.; Guarente, L.P.; Sassone-Corsi, P. The NAD+-dependent deacetylase SIRT1 modulates CLOCK-mediated chromatin remodeling and circadian control. Cell 2008, 134, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Nakahata, Y.; Sahar, S.; Astarita, G.; Kaluzova, M.; Sassone-Corsi, P. Circadian control of the NAD+ salvage pathway by CLOCK-SIRT1. Science 2009, 324, 654–657. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, K.M.; Yoshino, J.; Brace, C.S.; Abrassart, D.; Kobayashi, Y.; Marcheva, B.; Hong, H.K.; Chong, J.L.; Buhr, E.D.; Lee, C.; et al. Circadian clock feedback cycle through NAMPT-mediated NAD+ biosynthesis. Science 2009, 324, 651–654. [Google Scholar] [CrossRef]
- Hughes, M.E.; DiTacchio, L.; Hayes, K.R.; Vollmers, C.; Pulivarthy, S.; Baggs, J.E.; Panda, S.; Hogenesch, J.B. Harmonics of circadian gene transcription in mammals. PLoS Genet. 2009, 5, e1000442. [Google Scholar] [CrossRef]
- Cornel, K.M.C.; Bongers, M.Y.; Kruitwagen, R.P.F.M.; Romano, A. Local estrogen metabolism (intracrinology) in endometrial cancer: A systematic review. Mol. Cell Endocrinol. 2019, 489, 45–65. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Dai, C.; Huang, S.; Tang, J.; Chen, G.; Li, J.; Zhu, Z.; Zhu, X.; Zhou, S.; Gao, Y.; et al. Functional Silencing of HSD17B2 in Prostate Cancer Promotes Disease Progression. Clin. Cancer Res. 2019, 25, 1291–1301. [Google Scholar] [CrossRef]
- Kur, P.; Kolasa-Wołosiuk, A.; Misiakiewicz-Has, K.; Wiszniewska, B. Sex Hormone-Dependent Physiology and Diseases of Liver. Int. J. Environ. Res. Public Health 2020, 17, 2620. [Google Scholar] [CrossRef]
- Hashimoto, C.; Miki, Y.; Tanaka, S.; Takagi, K.; Fue, M.; Doe, Z.; Li, B.; Yaegashi, N.; Suzuki, T.; Ito, K. 17β-Hydroxysteroid Dehydrogenase Type 2 Expression Is Induced by Androgen Signaling in Endometrial Cancer. Int. J. Mol. Sci. 2018, 19, 1139. [Google Scholar] [CrossRef]
- Zhang, L.S.; Yuan, F.; Guan, X.; Li, J.; Liu, X.L.; Sun, J.; Liu, B.; Ma, W.; Deng, F.M. Association of genetic polymorphisms in HSD17B1, HSD17B2 and SHBG genes with hepatocellular carcinoma risk. Pathol. Oncol. Res. 2014, 20, 661–666. [Google Scholar] [CrossRef]
- Falasca, M.; Maffucci, T. Emerging roles of phosphatidylinositol 3-monophosphate as a dynamic lipid second messenger. Arch. Physiol. Biochem. 2006, 112, 274–284. [Google Scholar] [CrossRef]
- Anquetil, T.; Payrastre, B.; Gratacap, M.P.; Viaud, J. The lipid products of phosphoinositide 3-kinase isoforms in cancer and thrombosis. Cancer Metastasis Rev. 2018, 37, 477–489. [Google Scholar] [CrossRef]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Tetzlaff, F.; Adam, M.G.; Feldner, A.; Moll, I.; Menuchin, A.; Rodriguez-Vita, J.; Sprinzak, D.; Fischer, A. MPDZ promotes DLL4-induced Notch signaling during angiogenesis. Elife 2018, 7, e32860. [Google Scholar] [CrossRef] [PubMed]
- Zein, A.A.; Kaur, R.; Hussein, T.O.K.; Graf, G.A.; Lee, J.Y. ABCG5/G8: A structural view to pathophysiology of the hepatobiliary cholesterol secretion. Biochem. Soc. Trans. 2019, 47, 1259–1268. [Google Scholar] [CrossRef] [PubMed]
- Tian, Q.; Sun, H.F.; Wang, W.J.; Li, Q.; Ding, J.; Di, W. miRNA-365b promotes hepatocellular carcinoma cell migration and invasion by downregulating SGTB. Future Oncol. 2019, 15, 2019–2028. [Google Scholar] [CrossRef]
- Zhang, J.; Lu, C.; He, S.; Wan, C.; Zhang, Y.; Cheng, C. Decreased expression of Small glutamine-rich tetratricopeptide repeat-containing protein (SGT) correlated with prognosis of Hepatocellular carcinoma. Neoplasma 2014, 61, 83–89. [Google Scholar] [CrossRef]
- Wu, G.; Ju, X.; Wang, Y.; Li, Z.; Gan, X. Up-regulation of SNHG6 activates SERPINH1 expression by competitive binding to miR-139-5p to promote hepatocellular carcinoma progression. Cell Cycle 2019, 18, 1849–1867. [Google Scholar] [CrossRef]
- Li, J.; Jin, C.; Zou, C.; Qiao, X.; Ma, P.; Hu, D.; Li, W.; Jin, J.; Jin, X.; Fan, P. GNG12 regulates PD-L1 expression by activating NF-κB signaling in pancreatic ductal adenocarcinoma. FEBS Open Bio 2020, 10, 278–287. [Google Scholar] [CrossRef]
- Liu, B.; Ricarte Filho, J.; Mallisetty, A.; Villani, C.; Kottorou, A.; Rodgers, K.; Chen, C.; Ito, T.; Holmes, K.; Gastala, N.; et al. Detection of Promoter DNA Methylation in Urine and Plasma Aids the Detection of Non-Small Cell Lung Cancer. Clin. Cancer Res. 2020, 26, 4339–4348. [Google Scholar] [CrossRef]
- Choi, J.I.; Cho, E.H.; Kim, S.B.; Kim, R.; Kwon, J.; Park, M.; Shin, H.J.; Ryu, H.S.; Park, S.H.; Lee, K.H. Promoter methylation of cysteine dioxygenase type 1: Gene silencing and tumorigenesis in hepatocellular carcinoma. Ann. Hepatobiliary Pancreat Surg. 2017, 21, 181–187. [Google Scholar] [CrossRef]
- Andresen, K.; Boberg, K.M.; Vedeld, H.M.; Honne, H.; Jebsen, P.; Hektoen, M.; Wadsworth, C.A.; Clausen, O.P.; Lundin, K.E.; Paulsen, V.; et al. Four DNA methylation biomarkers in biliary brush samples accurately identify the presence of cholangiocarcinoma. Hepatology 2015, 61, 1651–1659. [Google Scholar] [CrossRef]
- Vedeld, H.M.; Andresen, K.; Eilertsen, I.A.; Nesbakken, A.; Seruca, R.; Gladhaug, I.P.; Thiis-Evensen, E.; Rognum, T.O.; Boberg, K.M.; Lind, G.E. The novel colorectal cancer biomarkers CDO1, ZSCAN18 and ZNF331 are frequently methylated across gastrointestinal cancers. Int. J. Cancer 2015, 136, 844–853. [Google Scholar] [CrossRef] [PubMed]
- Darweesh, S.K.; Abd Alziz, R.A.; Omar, H.; Sabry, D.; Fathy, W. Secreted protein acidic and rich in cysteine gene variants: Impact on susceptibility and survival of hepatocellular carcinoma patients. J. Gastroenterol. Hepatol. 2019, 34, 1424–1431. [Google Scholar] [CrossRef]
- Deng, B.; Qu, L.; Li, J.; Fang, J.; Yang, S.; Cao, Z.; Mei, Z.; Sun, X. MiRNA-211 suppresses cell proliferation, migration and invasion by targeting SPARC in human hepatocellular carcinoma. Sci. Rep. 2016, 6, 26679. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yang, B.; Du, Z.; Bai, T.; Gao, Y.T.; Wang, Y.J.; Lou, C.; Wang, F.M.; Bai, Y. Aberrant methylation of SPARC in human hepatocellular carcinoma and its clinical implication. World J. Gastroenterol. 2012, 18, 2043–2052. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gadaleta, R.M.; Scialpi, N.; Peres, C.; Cariello, M.; Ko, B.; Luo, J.; Porru, E.; Roda, A.; Sabbà, C.; Moschetta, A. Suppression of Hepatic Bile Acid Synthesis by a non-tumorigenic FGF19 analogue Protects Mice from Fibrosis and Hepatocarcinogenesis. Sci. Rep. 2018, 8, 17210. [Google Scholar] [CrossRef]
- Niu, Y.; Zhang, J.; Tong, Y.; Li, J.; Liu, B. miR-145-5p restrained cell growth, invasion, migration and tumorigenesis via modulating RHBDD1 in colorectal cancer via the EGFR-associated signaling pathway. Int. J. Biochem. Cell Biol. 2019, 117, 105641. [Google Scholar] [CrossRef]
- Zhao, C.; Ling, X.; Li, X.; Hou, X.; Zhao, D. MicroRNA-138-5p inhibits cell migration, invasion and EMT in breast cancer by directly targeting RHBDD1. Breast Cancer 2019, 26, 817–825. [Google Scholar] [CrossRef]
- Song, W.; Liu, W.; Zhao, H.; Li, S.; Guan, X.; Ying, J.; Zhang, Y.; Miao, F.; Zhang, M.; Ren, X.; et al. Rhomboid domain containing 1 promotes colorectal cancer growth through activation of the EGFR signalling pathway. Nat. Commun. 2015, 6, 8022. [Google Scholar] [CrossRef]
- Han, Y.; Meng, F.; Venter, J.; Wu, N.; Wan, Y.; Standeford, H.; Francis, H.; Meininger, C.; Greene, J., Jr.; Trzeciakowski, J.P.; et al. miR-34a-dependent overexpression of Per1 decreases cholangiocarcinoma growth. J. Hepatol. 2016, 64, 1295–1304. [Google Scholar] [CrossRef]
- Gery, S.; Komatsu, N.; Baldjyan, L.; Yu, A.; Koo, D.; Koeffler, H.P. The circadian gene per1 plays an important role in cell growth and DNA damage control in human cancer cells. Mol. Cell. 2006, 22, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Wood, P.A.; Ansell, C.M.; Quiton, D.F.; Oh, E.Y.; Du-Quiton, J.; Hrushesky, W.J. The circadian clock gene Per1 suppresses cancer cell proliferation and tumor growth at specific times of day. Chronobiol. Int. 2009, 26, 1323–1339. [Google Scholar] [CrossRef] [PubMed]
- Nogueiras, R.; Habegger, K.M.; Chaudhary, N.; Finan, B.; Banks, A.S.; Dietrich, M.O.; Horvath, T.L.; Sinclair, D.A.; Pfluger, P.T.; Tschöp, M.H. Sirtuin 1 and sirtuin 3: Physiological modulators of metabolism. Physiol. Rev. 2012, 92, 1479–1514. [Google Scholar] [CrossRef] [PubMed]
- Asher, G.; Gatfield, D.; Stratmann, M.; Reinke, H.; Dibner, C.; Kreppel, F.; Mostoslavsky, R.; Alt, F.W.; Schibler, U. SIRT1 regulates circadian clock gene expression through PER2 deacetylation. Cell 2008, 134, 317–328. [Google Scholar] [CrossRef] [PubMed]
- Belden, W.J.; Dunlap, J.C. SIRT1 is a circadian deacetylase for core clock components. Cell 2008, 134, 212–214. [Google Scholar] [CrossRef]
- Gauger, M.A.; Sancar, A. Cryptochrome, circadian cycle, cell cycle checkpoints, and cancer. Cancer Res. 2005, 65, 6828–6834. [Google Scholar] [CrossRef]
- Unsal-Kaçmaz, K.; Mullen, T.E.; Kaufmann, W.K.; Sancar, A. Coupling of human circadian and cell cycles by the timeless protein. Mol. Cell Biol. 2005, 25, 3109–3116. [Google Scholar] [CrossRef] [PubMed]
- Sancar, A.; Lindsey-Boltz, L.A.; Kang, T.H.; Reardon, J.T.; Lee, J.H.; Ozturk, N. Circadian clock control of the cellular response to DNA damage. FEBS Lett. 2010, 584, 2618–2625. [Google Scholar] [CrossRef]
- Mazzoccoli, G.; Laukkanen, M.O.; Vinciguerra, M.; Colangelo, T.; Colantuoni, V. A Timeless Link between Circadian Patterns and Disease. Trends Mol. Med. 2016, 22, 68–81. [Google Scholar] [CrossRef]
- Bellet, M.M.; Stincardini, C.; Costantini, C.; Gargaro, M.; Pieroni, S.; Castelli, M.; Piobbico, D.; Sassone-Corsi, P.; Della-Fazia, M.A.; Romani, L.; et al. The Circadian Protein PER1 Modulates the Cellular Response to Anticancer Treatments. Int. J. Mol. Sci. 2021, 22, 2974. [Google Scholar] [CrossRef]
- Ma, D.; Panda, S.; Lin, J.D. Temporal orchestration of circadian autophagy rhythm by C/EBPβ. EMBO J. 2011, 30, 4642–4651. [Google Scholar] [CrossRef] [PubMed]
- Pastore, N.; Vainshtein, A.; Herz, N.J.; Huynh, T.; Brunetti, L.; Klisch, T.J.; Mutarelli, M.; Annunziata, P.; Kinouchi, K.; Brunetti-Pierri, N.; et al. Nutrient-sensitive transcription factors TFEB and TFE3 couple autophagy and metabolism to the peripheral clock. EMBO J. 2019, 38, e101347. [Google Scholar] [CrossRef] [PubMed]
- Mulcahy Levy, J.M.; Thorburn, A. Autophagy in cancer: Moving from understanding mechanism to improving therapy responses in patients. Cell Death Differ. 2020, 27, 843–857. [Google Scholar] [CrossRef] [PubMed]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef] [PubMed]
- Keller, M.; Mazuch, J.; Abraham, U.; Eom, G.D.; Herzog, E.D.; Volk, H.D.; Kramer, A.; Maier, B. A circadian clock in macrophages controls inflammatory immune responses. Proc. Natl. Acad. Sci. USA 2009, 106, 21407–21412. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, S.; Li, X.; Li, B.; Li, Y.; Xia, K.; Yang, Y.; Aman, S.; Wang, M.; Wu, H. Circadian protein BMAL1 promotes breast cancer cell invasion and metastasis by up-regulating matrix metalloproteinase9 expression. Cancer Cell Int. 2019, 19, 182. [Google Scholar] [CrossRef] [PubMed]
- Ho, J.S.; Ma, W.; Mao, D.Y.; Benchimol, S. p53-Dependent transcriptional repression of c-myc is required for G1 cell cycle arrest. Mol. Cell Biol. 2005, 25, 7423–7431. [Google Scholar] [CrossRef]
- Lee, Y.; Lahens, N.F.; Zhang, S.; Bedont, J.; Field, J.M.; Sehgal, A. G1/S cell cycle regulators mediate effects of circadian dysregulation on tumor growth and provide targets for timed anticancer treatment. PLoS Biol. 2019, 17, e3000228. [Google Scholar] [CrossRef]
- Sulli, G.; Lam, M.T.Y.; Panda, S. Interplay between Circadian Clock and Cancer: New Frontiers for Cancer Treatment. Trends Cancer 2019, 5, 475–494. [Google Scholar] [CrossRef]
- Sancar, A.; Van Gelder, R.N. Clocks, cancer, and chronochemotherapy. Science 2021, 371, eabb0738. [Google Scholar] [CrossRef]
- Cao, Q.; Gery, S.; Dashti, A.; Yin, D.; Zhou, Y.; Gu, J.; Koeffler, H.P. A role for the clock gene per1 in prostate cancer. Cancer Res. 2009, 69, 7619–7625. [Google Scholar] [CrossRef]
- Cheng, H.Y.; Obrietan, K. Revealing a role of microRNAs in the regulation of the biological clock. Cell Cycle 2007, 6, 3034–3035. [Google Scholar] [CrossRef]
- Cassim, S.; Vučetić, M.; Ždralević, M.; Pouyssegur, J. Warburg and Beyond: The Power of Mitochondrial Metabolism to Collaborate or Replace Fermentative Glycolysis in Cancer. Cancers 2020, 12, 1119. [Google Scholar] [CrossRef]
- Zong, W.X.; Rabinowitz, J.D.; White, E. Mitochondria and Cancer. Mol Cell. 2016, 61, 667–676. [Google Scholar] [CrossRef]
- Porporato, P.E.; Filigheddu, N.; Pedro, J.M.B.; Kroemer, G.; Galluzzi, L. Mitochondrial metabolism and cancer. Cell Res. 2018, 28, 265–280. [Google Scholar] [CrossRef]
- Cela, O.; Scrima, R.; Pazienza, V.; Merla, G.; Benegiamo, G.; Augello, B.; Fugetto, S.; Menga, M.; Rubino, R.; Fuhr, L.; et al. Clock genes-dependent acetylation of complex I sets rhythmic activity of mitochondrial OxPhos. Biochim. Biophys. Acta 2016, 1863, 596–606. [Google Scholar] [CrossRef]
- Scrima, R.; Cela, O.; Merla, G.; Augello, B.; Rubino, R.; Quarato, G.; Fugetto, S.; Menga, M.; Fuhr, L.; Relógio, A.; et al. Clock-genes and mitochondrial respiratory activity: Evidence of a reciprocal interplay. Biochim. Biophys. Acta 2016, 1857, 1344–1351. [Google Scholar] [CrossRef] [PubMed]
- Scrima, R.; Cela, O.; Agriesti, F.; Piccoli, C.; Tataranni, T.; Pacelli, C.; Mazzoccoli, G.; Capitanio, N. Mitochondrial calcium drives clock gene-dependent activation of pyruvate dehydrogenase and of oxidative phosphorylation. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118815. [Google Scholar] [CrossRef] [PubMed]
- Sardon Puig, L.; Valera-Alberni, M.; Cantó, C.; Pillon, N.J. Circadian Rhythms and Mitochondria: Connecting the Dots. Front. Genet. 2018, 9, 452. [Google Scholar] [CrossRef] [PubMed]
- Matilainen, O.; Quirós, P.M.; Auwerx, J. Mitochondria and Epigenetics—Crosstalk in Homeostasis and Stress. Trends Cell Biol. 2017, 27, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Pacelli, C.; Adipietro, I.; Malerba, N.; Squeo, G.M.; Piccoli, C.; Amoresano, A.; Pinto, G.; Pucci, P.; Lee, J.E.; Ge, K.; et al. Loss of Function of the Gene Encoding the Histone Methyltransferase KMT2D Leads to Deregulation of Mitochondrial Respiration. Cells 2020, 9, 1685. [Google Scholar] [CrossRef]
- Holmström, K.M.; Baird, L.; Zhang, Y.; Hargreaves, I.; Chalasani, A.; Land, J.M.; Stanyer, L.; Yamamoto, M.; Dinkova-Kostova, A.T.; Abramov, A.Y. Nrf2 impacts cellular bioenergetics by controlling substrate availability for mitochondrial respiration. Biol. Open 2013, 2, 761–770. [Google Scholar] [CrossRef]
- Ghiraldini, F.G.; Filipescu, D.; Bernstein, E. Solid tumours hijack the histone variant network. Nat. Rev. Cancer 2021, 21, 257–275. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Ding, Y.; Zeng, L. The diagnostic and prognostic value of H2AFY in hepatocellular carcinoma. BMC Cancer 2021, 21, 418. [Google Scholar] [CrossRef] [PubMed]
- Yuan, P.; Yang, T.; Mu, J.; Zhao, J.; Yang, Y.; Yan, Z.; Hou, Y.; Chen, C.; Xing, J.; Zhang, H.; et al. Circadian clock gene NPAS2 promotes reprogramming of glucose metabolism in hepatocellular carcinoma cells. Cancer Lett. 2020, 469, 498–509. [Google Scholar] [CrossRef] [PubMed]
- Fekry, B.; Ribas-Latre, A.; Baumgartner, C.; Mohamed, A.M.T.; Kolonin, M.G.; Sladek, F.M.; Younes, M.; Eckel-Mahan, K.L. HNF4α-Deficient Fatty Liver Provides a Permissive Environment for Sex-Independent Hepatocellular Carcinoma. Cancer Res. 2019, 79, 5860–5873. [Google Scholar] [CrossRef] [PubMed]
- Fekry, B.; Ribas-Latre, A.; Baumgartner, C.; Deans, J.R.; Kwok, C.; Patel, P.; Fu, L.; Berdeaux, R.; Sun, K.; Kolonin, M.G.; et al. Incompatibility of the circadian protein BMAL1 and HNF4alpha in hepatocellular carcinoma. Nat. Commun. 2018, 9, 4349. [Google Scholar] [CrossRef]
- Sánchez, D.I.; González-Fernández, B.; Crespo, I.; San-Miguel, B.; Álvarez, M.; González-Gallego, J.; Tuñón, M.J. Melatonin modulates dysregulated circadian clocks in mice with diethylnitrosamine-induced hepatocellular carcinoma. J. Pineal Res. 2018, 65, e12506. [Google Scholar] [CrossRef]
- Yuan, P.; Li, J.; Zhou, F.; Huang, Q.; Zhang, J.; Guo, X.; Lyu, Z.; Zhang, H.; Xing, J. NPAS2 promotes cell survival of hepatocellular carcinoma by transactivating CDC25A. Cell Death Dis. 2017, 8, e2704. [Google Scholar] [CrossRef]
- Li, H.; Lu, Y.F.; Chen, H.; Liu, J. Dysregulation of metallothionein and circadian genes in human hepatocellular carcinoma. Chronobiol. Int. 2017, 34, 192–202. [Google Scholar] [CrossRef]
- Kettner, N.M.; Voicu, H.; Finegold, M.J.; Coarfa, C.; Sreekumar, A.; Putluri, N.; Katchy, C.A.; Lee, C.; Moore, D.D.; Fu, L. Circadian Homeostasis of Liver Metabolism Suppresses Hepatocarcinogenesis. Cancer Cell 2016, 30, 909–924. [Google Scholar] [CrossRef]
- Cui, M.; Zheng, M.; Sun, B.; Wang, Y.; Ye, L.; Zhang, X. A long noncoding RNA perturbs the circadian rhythm of hepatoma cells to facilitate hepatocarcinogenesis. Neoplasia 2015, 17, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Neumann, O.; Kesselmeier, M.; Geffers, R.; Pellegrino, R.; Radlwimmer, B.; Hoffmann, K.; Ehemann, V.; Schemmer, P.; Schirmacher, P.; Lorenzo Bermejo, J.; et al. Methylome analysis and integrative profiling of human HCCs identify novel protumorigenic factors. Hepatology 2012, 56, 1817–1827. [Google Scholar] [CrossRef] [PubMed]
- Sahar, S.; Sassone-Corsi, P. Metabolism and cancer: The circadian clock connection. Nat. Rev. Cancer 2009, 9, 886–896. [Google Scholar] [CrossRef]
- Chen, J. The Cell-Cycle Arrest and Apoptotic Functions of p53 in Tumor Initiation and Progression. Cold Spring Harb. Perspect. Med. 2016, 6, a026104. [Google Scholar] [CrossRef] [PubMed]
- Ashok Kumar, P.V.; Dakup, P.P.; Sarkar, S.; Modasia, J.B.; Motzner, M.S.; Gaddameedhi, S. It’s About Time: Advances in Understanding the Circadian Regulation of DNA Damage and Repair in Carcinogenesis and Cancer Treatment Outcomes. Yale J. Biol. Med. 2019, 92, 305–316. [Google Scholar] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carbone, A.; De Santis, E.; Cela, O.; Giambra, V.; Miele, L.; Marrone, G.; Grieco, A.; Buschbeck, M.; Capitanio, N.; Mazza, T.; et al. The Histone Variant MacroH2A1 Impacts Circadian Gene Expression and Cell Phenotype in an In Vitro Model of Hepatocellular Carcinoma. Biomedicines 2021, 9, 1057. https://doi.org/10.3390/biomedicines9081057
Carbone A, De Santis E, Cela O, Giambra V, Miele L, Marrone G, Grieco A, Buschbeck M, Capitanio N, Mazza T, et al. The Histone Variant MacroH2A1 Impacts Circadian Gene Expression and Cell Phenotype in an In Vitro Model of Hepatocellular Carcinoma. Biomedicines. 2021; 9(8):1057. https://doi.org/10.3390/biomedicines9081057
Chicago/Turabian StyleCarbone, Annalucia, Elisabetta De Santis, Olga Cela, Vincenzo Giambra, Luca Miele, Giuseppe Marrone, Antonio Grieco, Marcus Buschbeck, Nazzareno Capitanio, Tommaso Mazza, and et al. 2021. "The Histone Variant MacroH2A1 Impacts Circadian Gene Expression and Cell Phenotype in an In Vitro Model of Hepatocellular Carcinoma" Biomedicines 9, no. 8: 1057. https://doi.org/10.3390/biomedicines9081057
APA StyleCarbone, A., De Santis, E., Cela, O., Giambra, V., Miele, L., Marrone, G., Grieco, A., Buschbeck, M., Capitanio, N., Mazza, T., & Mazzoccoli, G. (2021). The Histone Variant MacroH2A1 Impacts Circadian Gene Expression and Cell Phenotype in an In Vitro Model of Hepatocellular Carcinoma. Biomedicines, 9(8), 1057. https://doi.org/10.3390/biomedicines9081057