
Utilizing Patient-Derived Epithelial Ovarian Cancer Tumor Organoids to Predict Carboplatin Resistance

 , , , and
, , , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Subjects
2.2. Tumor Organoid (TO) Development and Validation
2.3. Chemosensitivity Screens
2.4. Sequencing Methods
2.5. Gene Mutation and Gene Expression Bioinformatic Analysis
2.6. Statistical Analysis
3. Results
3.1. Subject Demographic and Treatment Charactistics
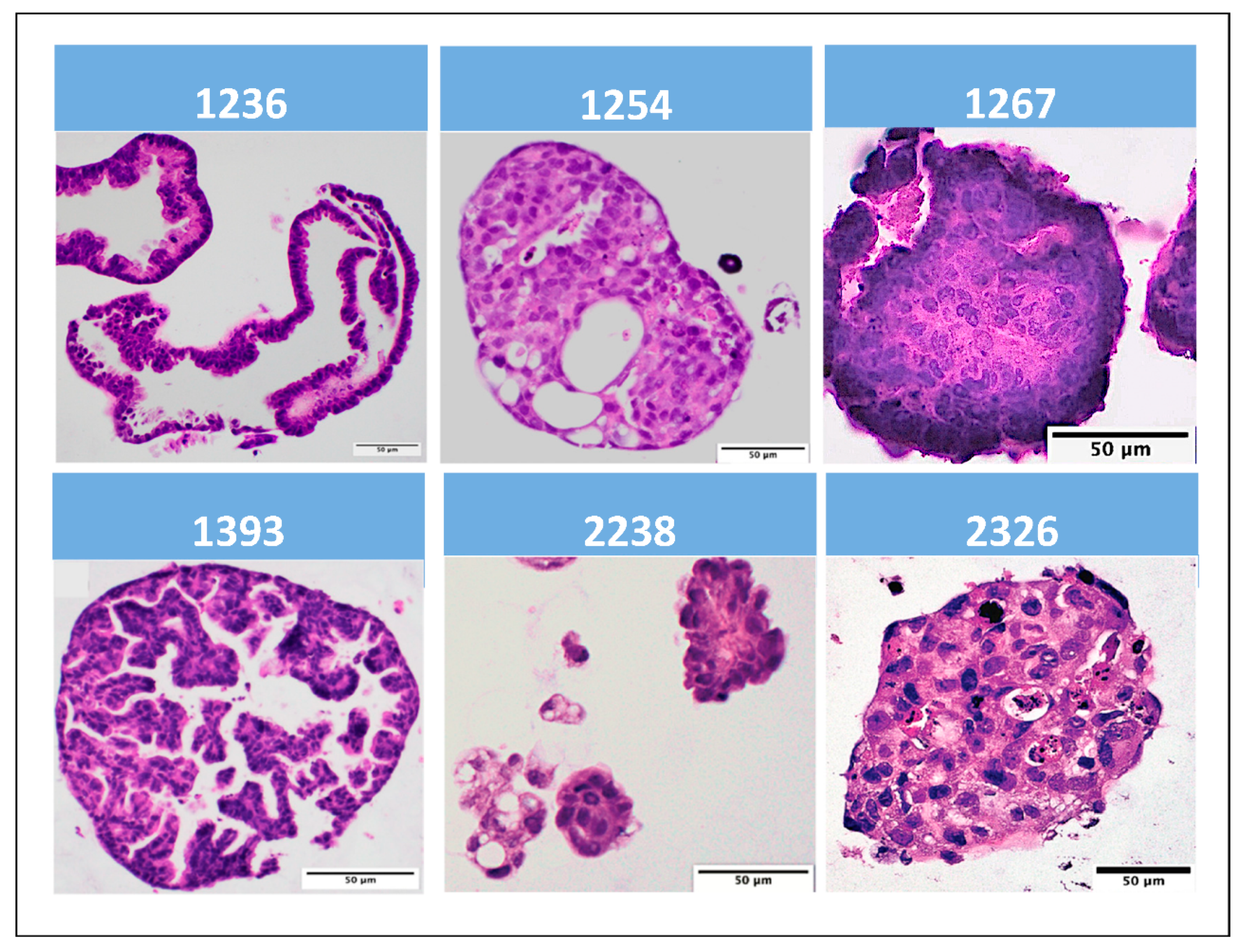
3.2. Tumor Organoid Validation
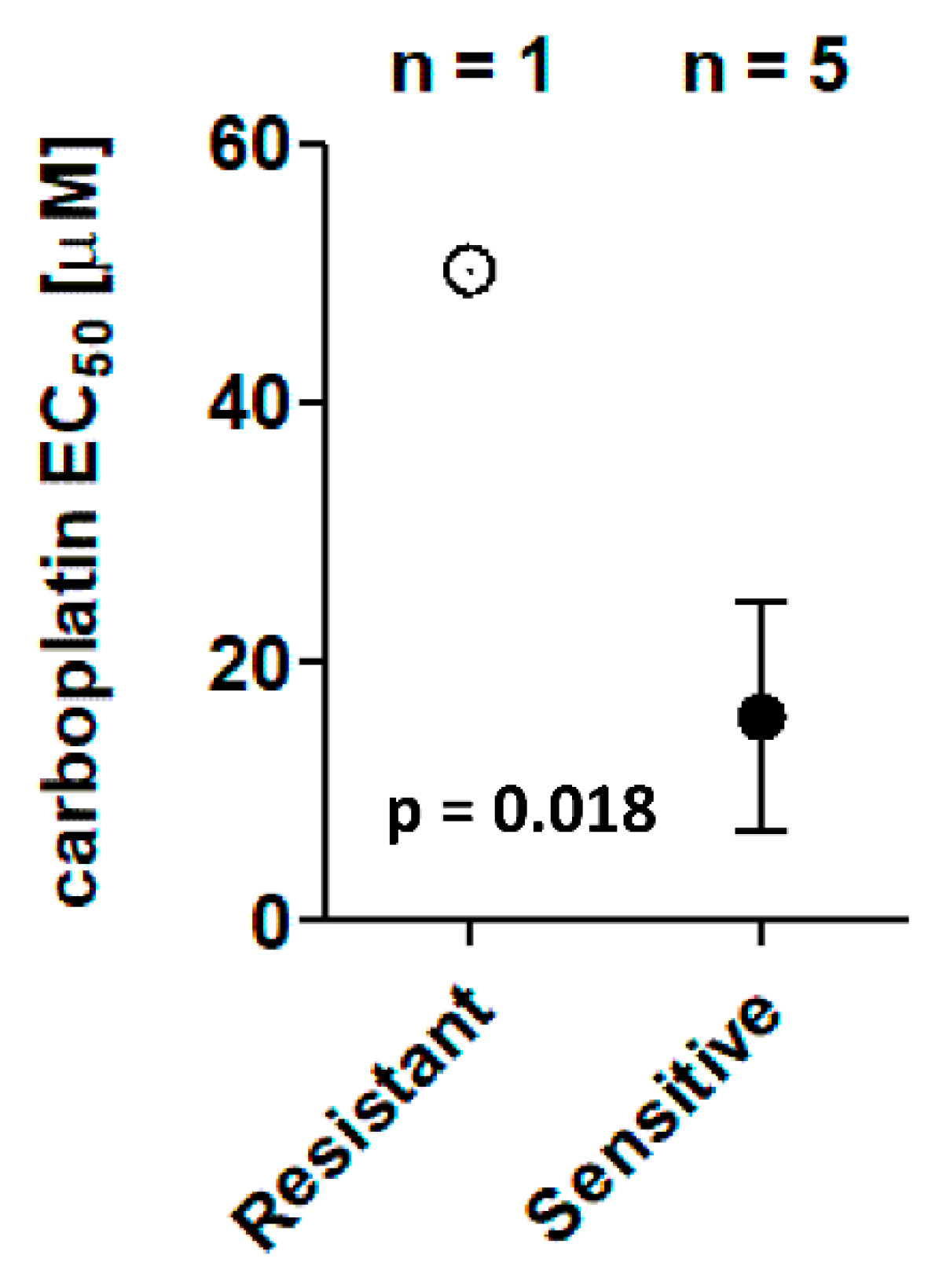
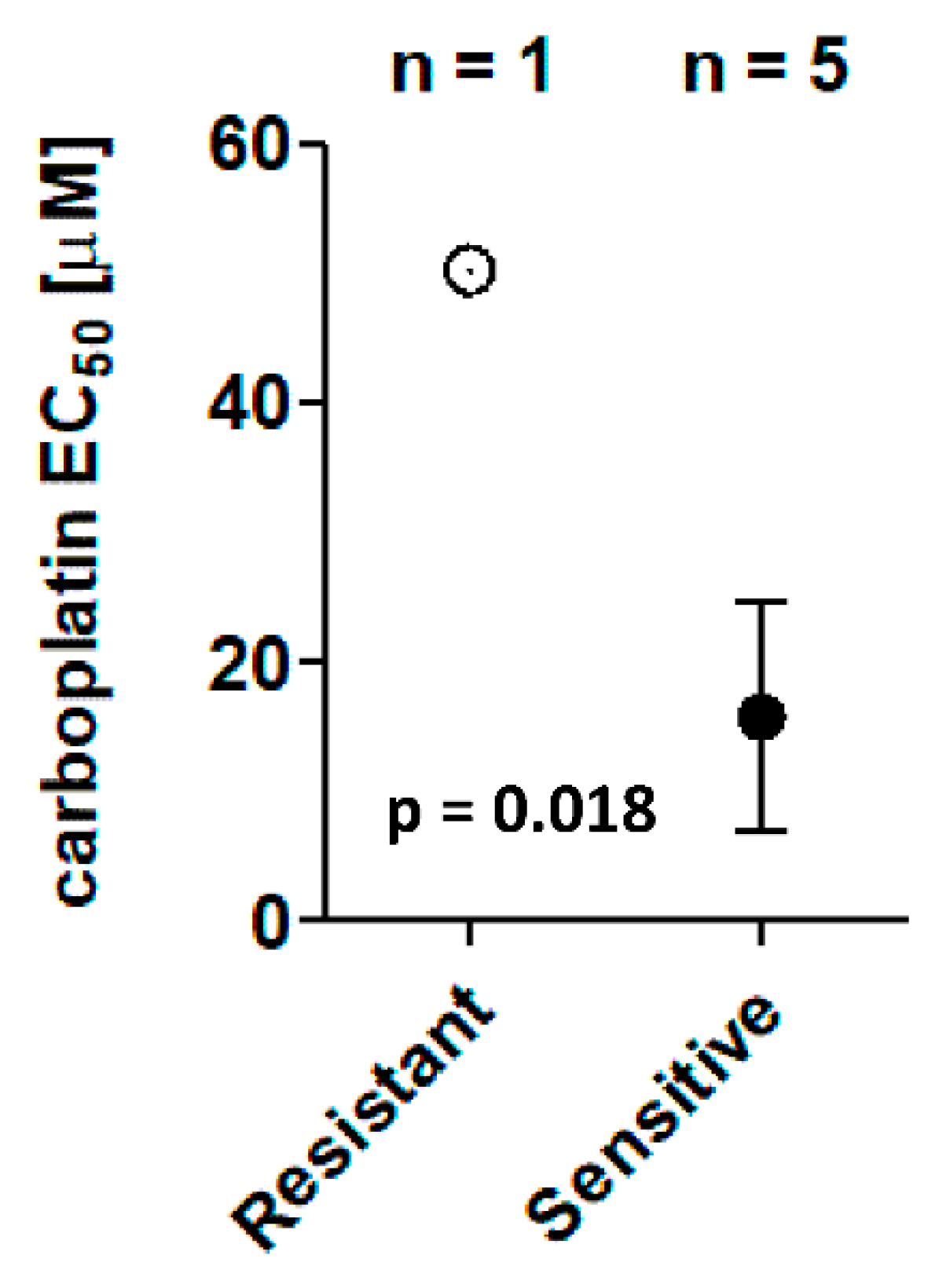
3.3. Chemosensitivity Screens
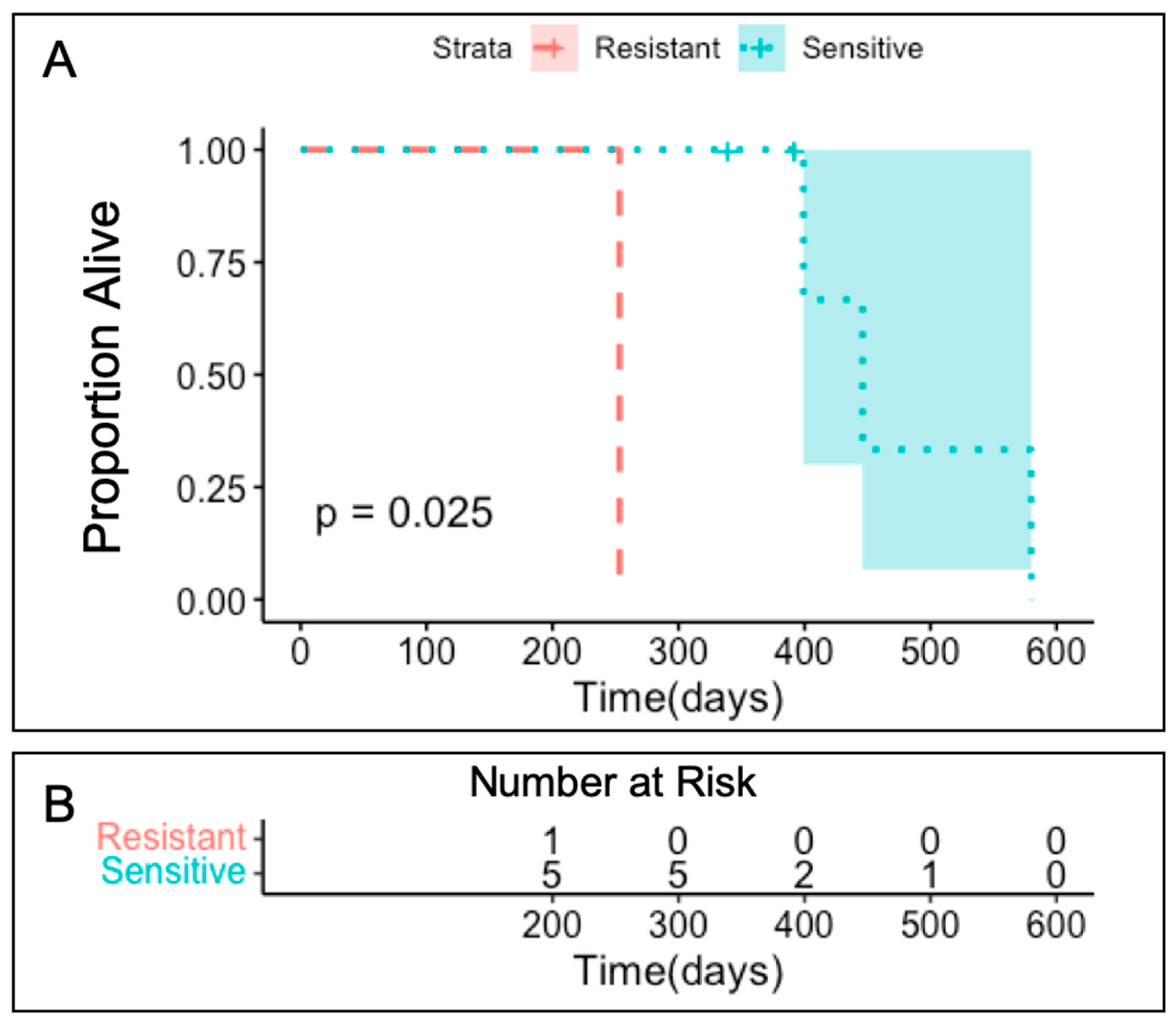
3.4. Clinical Outcomes
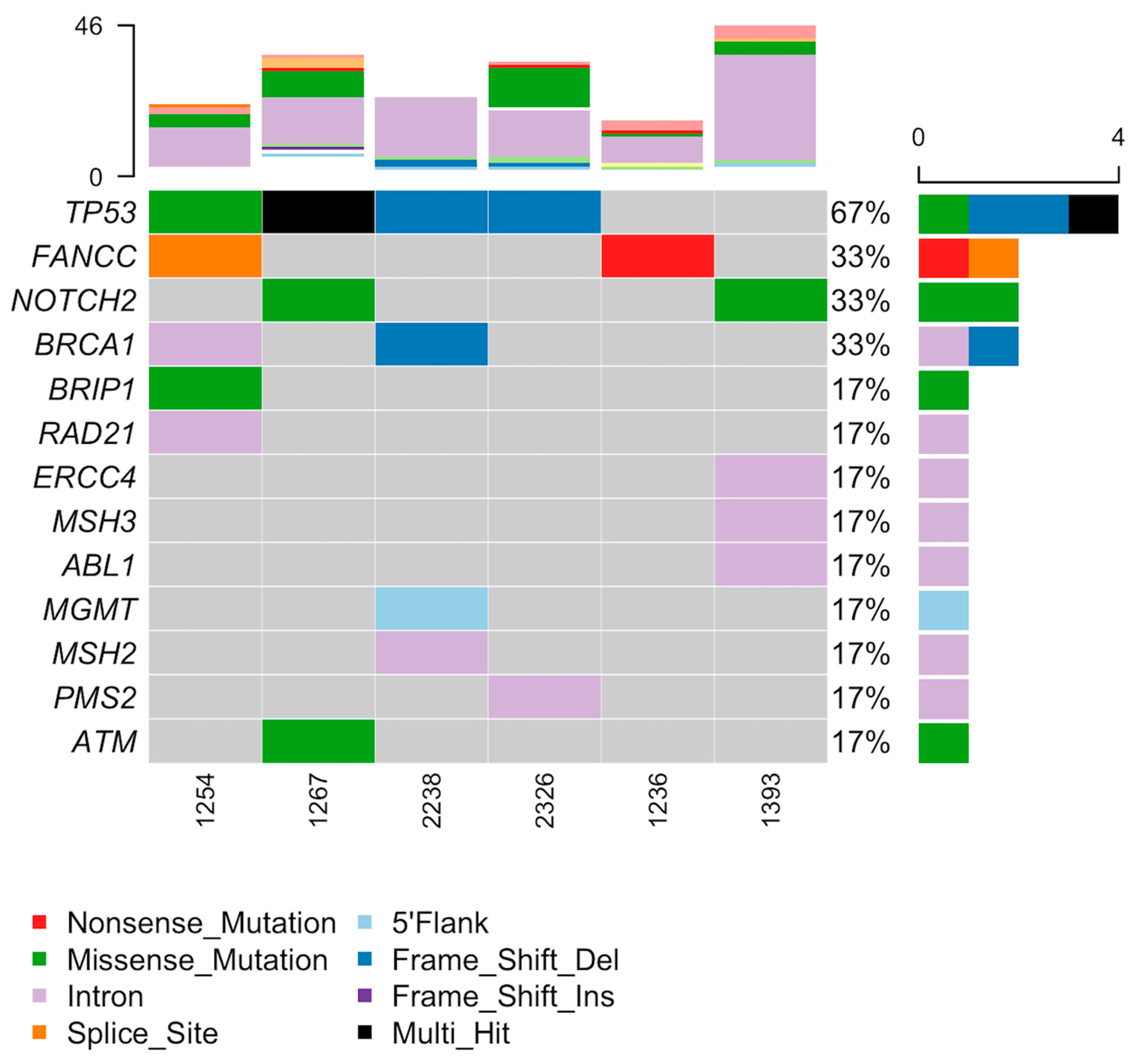
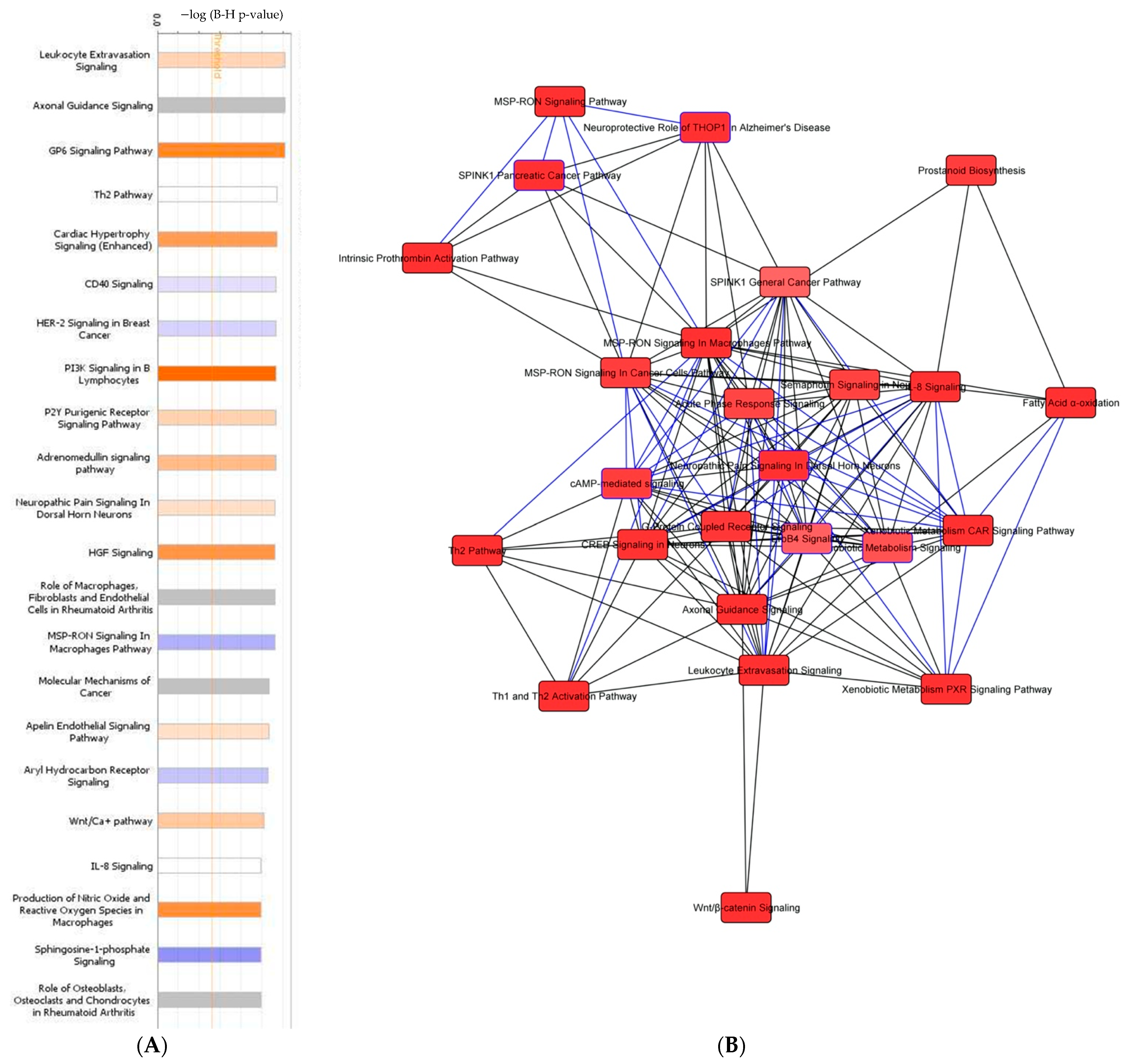
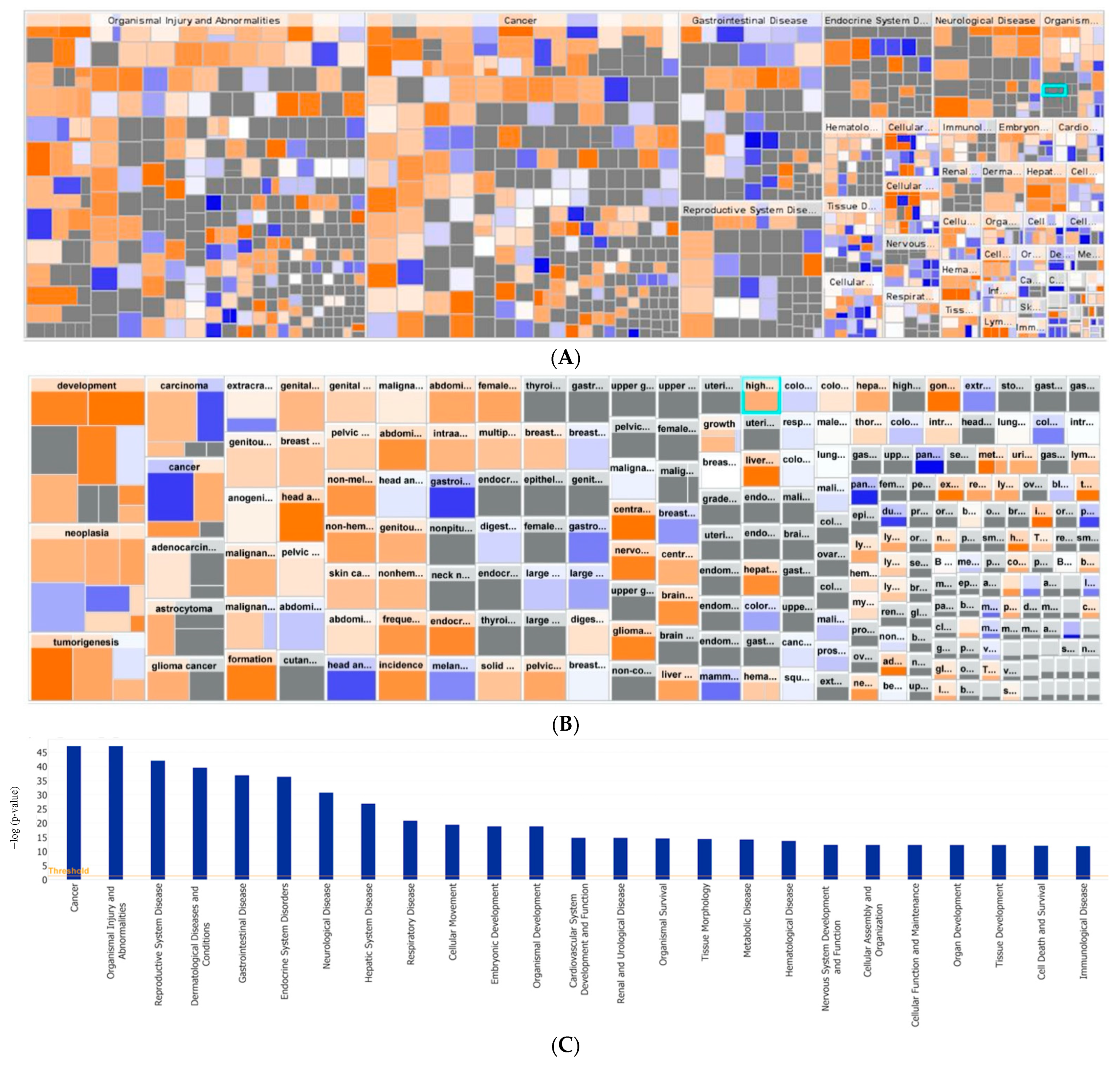
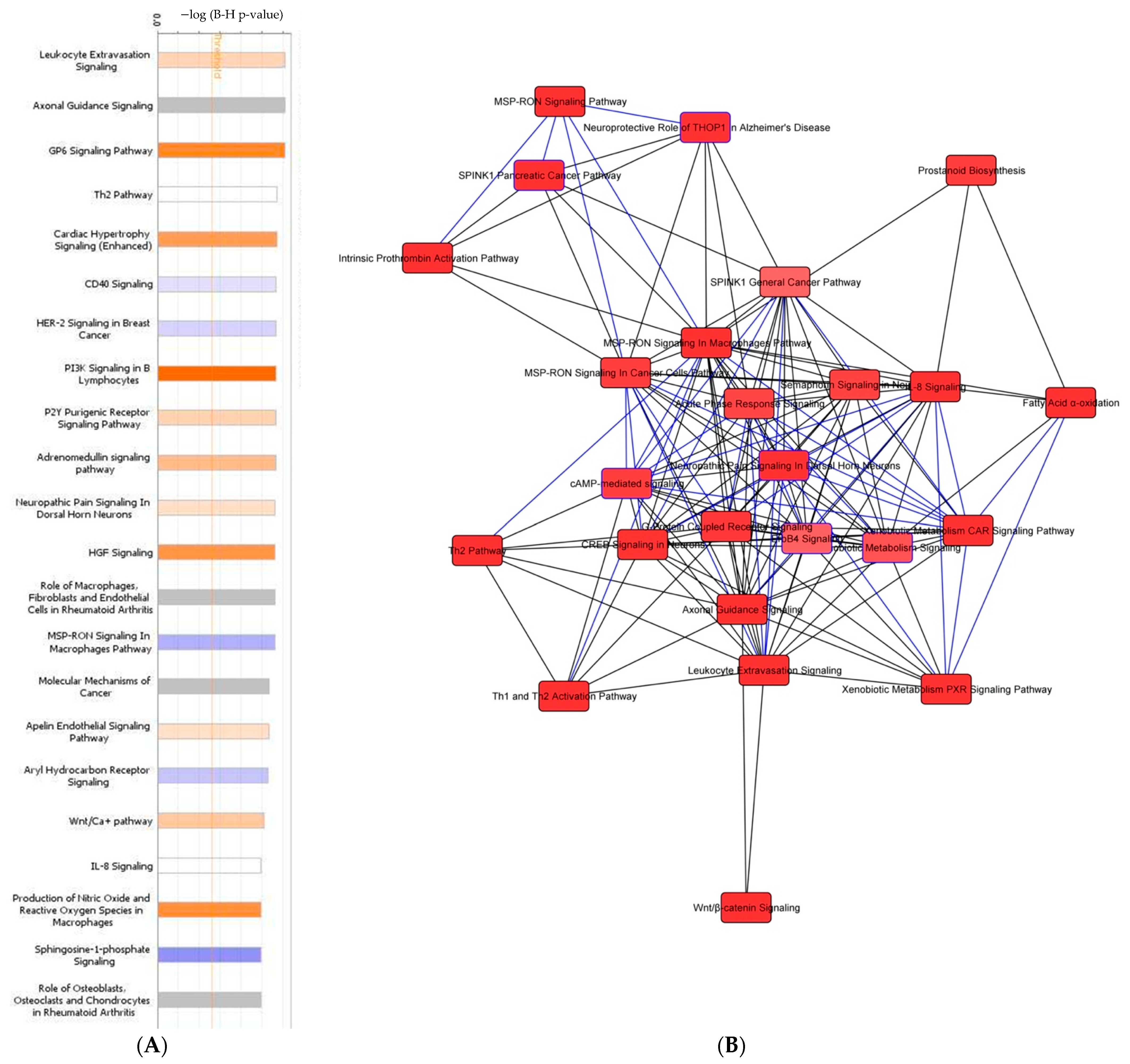
3.5. Tumor Organoid Mutation Analysis
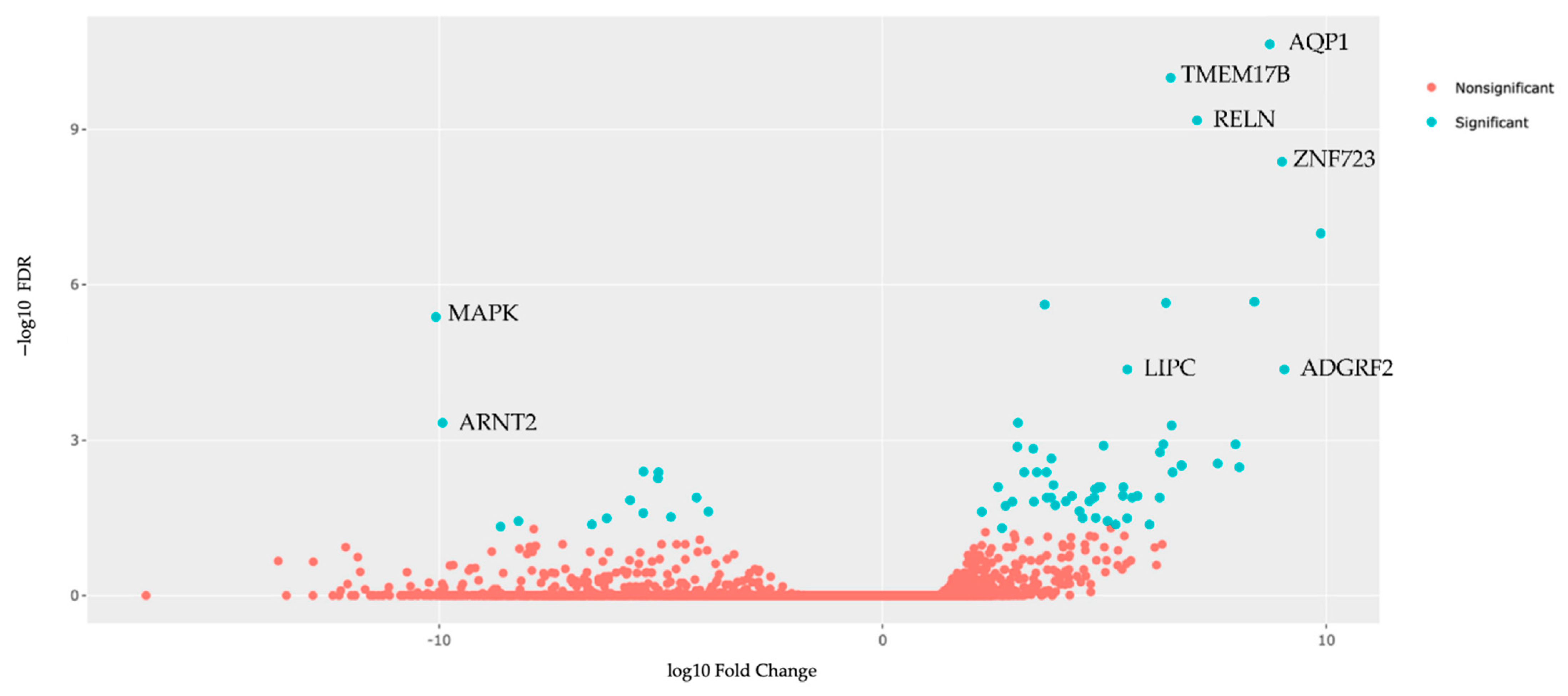
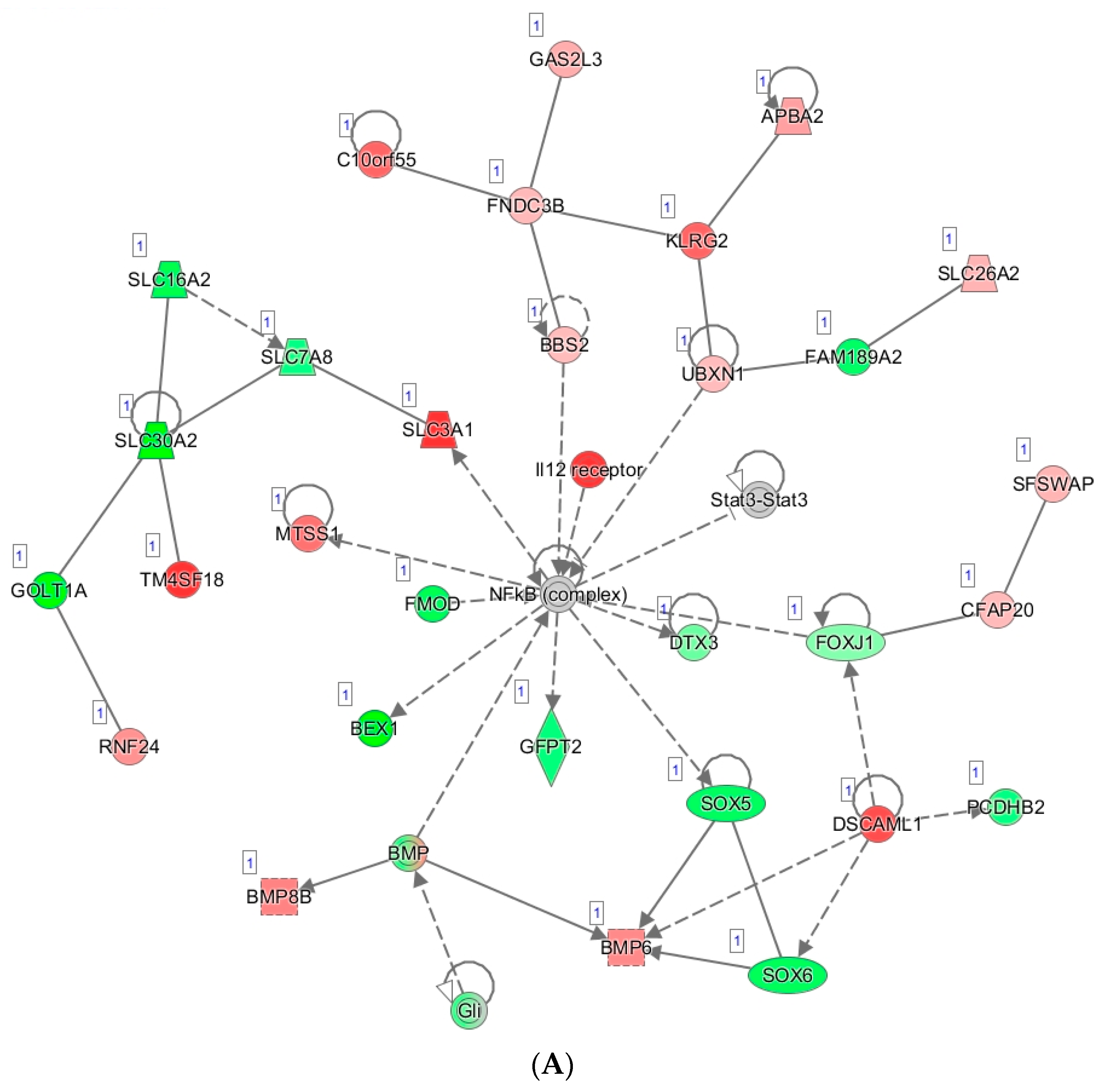
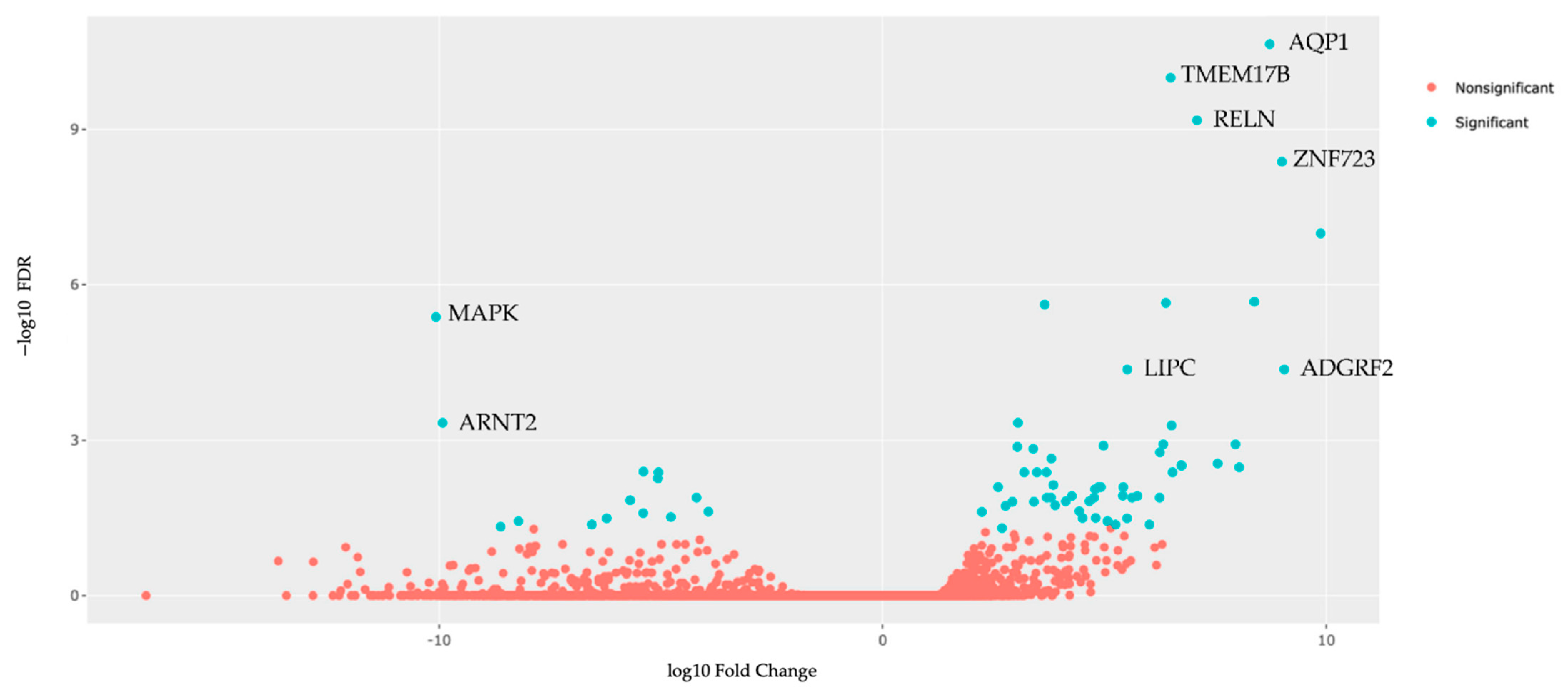
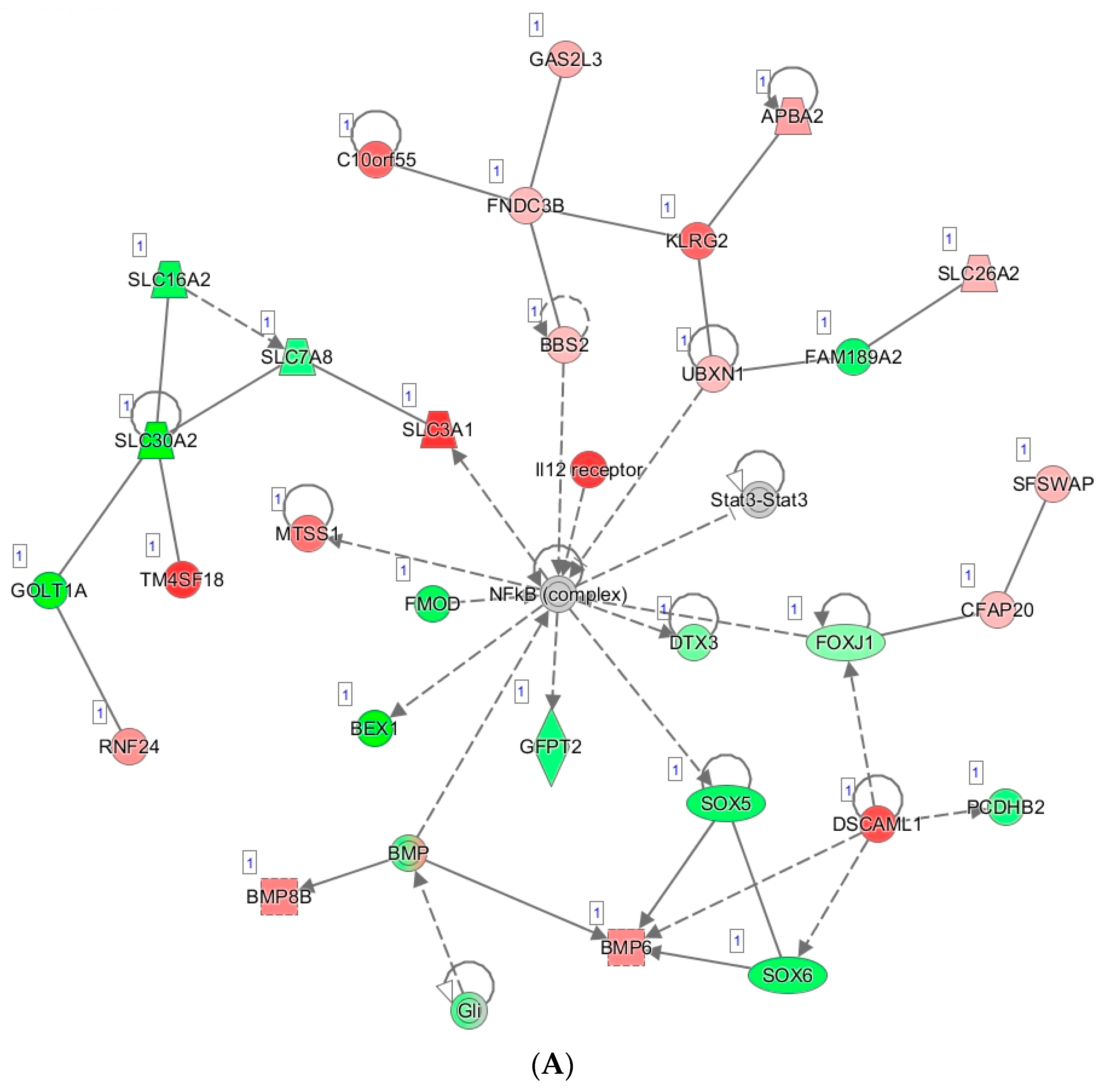
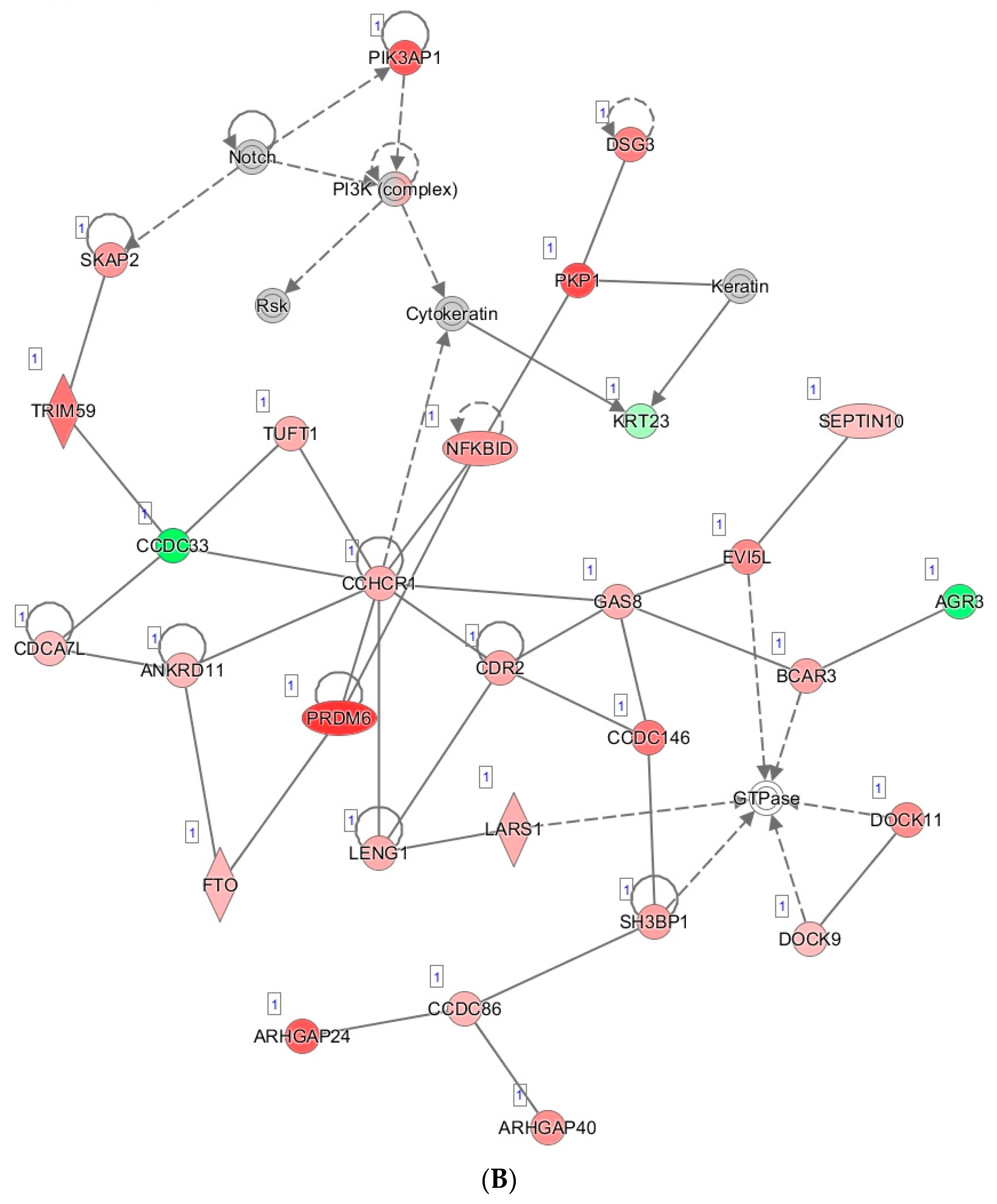
3.6. Tumor Organoid Gene Expression Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Armstrong, D.K.; Alvarez, R.D.; Bakkum-Gamez, J.N.; Barroilhet, L.; Behbakht, K.; Berchuck, A.; Chen, L.M.; Cristea, M.; DeRosa, M.; Eisenhauer, E.L.; et al. Ovarian Cancer, Version 2.2020, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2021, 19, 191–226. [Google Scholar] [CrossRef] [PubMed]
- Lheureux, S.; Braunstein, M.; Oza, A.M. Epithelial ovarian cancer: Evolution of management in the era of precision medicine. CA Cancer J. Clin. 2019, 69, 280–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matulonis, U.A.; Sood, A.K.; Fallowfield, L.; Howitt, B.E.; Sehouli, J.; Karlan, B.Y. Ovarian cancer. Nat. Rev. Dis. Primers 2016, 2, 16061. [Google Scholar] [CrossRef]
- Davis, A.; Tinker, A.V.; Friedlander, M. “Platinum resistant” ovarian cancer: What is it, who to treat and how to measure benefit? Gynecol. Oncol. 2014, 133, 624–631. [Google Scholar] [CrossRef]
- Barakat, R.R.; Markman, M.; Randall, M. Principles and Practice of Gynecologic Oncology, 5th ed.; Wolters Kluwer Health/Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2009; p. 1072. [Google Scholar]
- Karam, A.K.; Chiang, J.W.; Fung, E.; Nossov, V.; Karlan, B.Y. Extreme drug resistance assay results do not influence survival in women with epithelial ovarian cancer. Gynecol. Oncol. 2009, 114, 246–252. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, K.; Eno, M.L.; Im, D.D.; Rosenshein, N.B.; Sood, A.K. Clinical relevance of extent of extreme drug resistance in epithelial ovarian carcinoma. Gynecol. Oncol. 2010, 116, 61–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallion, H.; Christopherson, W.A.; Coleman, R.L.; DeMars, L.; Herzog, T.; Hosford, S.; Schellhas, H.; Wells, A.; Sevin, B.U. Progression-free interval in ovarian cancer and predictive value of an ex vivo chemoresponse assay. Int. J. Gynecol. Cancer 2006, 16, 194–201. [Google Scholar] [CrossRef]
- UnitedHealthcare. Chemosensitivity and Chemoresistance Assays in Cancer. In UnitedHealthcare Commercial Medical Policy: 2019T0533O; United HealthCare Services, Incorporated: Minnetonka, MN, USA, 2019. [Google Scholar]
- Maru, Y.; Tanaka, N.; Itami, M.; Hippo, Y. Efficient use of patient-derived organoids as a preclinical model for gynecologic tumors. Gynecol. Oncol. 2019, 154, 189–198. [Google Scholar] [CrossRef]
- Tiriac, H.; Belleau, P.; Engle, D.D.; Plenker, D.; Deschênes, A.; Somerville, T.D.D.; Froeling, F.E.M.; Burkhart, R.A.; Denroche, R.E.; Jang, G.H.; et al. Organoid Profiling Identifies Common Responders to Chemotherapy in Pancreatic Cancer. Cancer Discov. 2018, 8, 1112–1129. [Google Scholar] [CrossRef] [Green Version]
- Eisenhauer, E.A.; Therasse, P.; Bogaerts, J.; Schwartz, L.H.; Sargent, D.; Ford, R.; Dancey, J.; Arbuck, S.; Gwyther, S.; Mooney, M.; et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur. J. Cancer 2009, 45, 228–247. [Google Scholar] [CrossRef] [PubMed]
- Hill, S.J.; Decker, B.; Roberts, E.A.; Horowitz, N.S.; Muto, M.G.; Worley, M.J., Jr.; Feltmate, C.M.; Nucci, M.R.; Swisher, E.M.; Nguyen, H.; et al. Prediction of DNA Repair Inhibitor Response in Short-Term Patient-Derived Ovarian Cancer Organoids. Cancer Discov. 2018, 8, 1404–1421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kessler, M.; Hoffmann, K.; Brinkmann, V.; Thieck, O.; Jackisch, S.; Toelle, B.; Berger, H.; Mollenkopf, H.J.; Mangler, M.; Sehouli, J.; et al. The Notch and Wnt pathways regulate stemness and differentiation in human fallopian tube organoids. Nat. Commun. 2015, 6, 8989. [Google Scholar] [CrossRef] [PubMed]
- Beaubier, N.; Tell, R.; Huether, R.; Bontrager, M.; Bush, S.; Parsons, J.; Shah, K.; Baker, T.; Selkov, G.; Taxter, T.; et al. Clinical validation of the Tempus xO assay. Oncotarget 2018, 9, 25826–25832. [Google Scholar] [CrossRef] [Green Version]
- Beaubier, N.; Tell, R.; Lau, D.; Parsons, J.R.; Bush, S.; Perera, J.; Sorrells, S.; Baker, T.; Chang, A.; Michuda, J.; et al. Clinical validation of the tempus xT next-generation targeted oncology sequencing assay. Oncotarget 2019, 10, 2384–2396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayakonda, A.; Lin, D.; Assenov, Y.; Plass, C.; Koeffler, P.H. Maftools: Efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 2018, 28, 1747–1756. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Liston, D.R.; Davis, M. Clinically Relevant Concentrations of Anticancer Drugs: A Guide for Nonclinical Studies. Clin. Cancer Res. 2017, 23, 3489–3498. [Google Scholar] [CrossRef] [Green Version]
- A Study in Ovarian Cancer Patients Evaluating Rucaparib and Nivolumab as Maintenance Treatment Following Response to Front-Line Platinum-Based Chemotherapy (ATHENA). Available online: https://clinicaltrials.gov/ct2/show/NCT03522246 (accessed on 1 June 2021).
- Frohnmayer, D.; Frohnmayer, L.; Guinan, E.; Kennedy, T.; Larsen, K. (Eds.) Fanconi Anemia: Guidelines for Diagnosis and Management, 4th ed.; Fanconi Anemia Research Fund: Eugene, OR, USA, 2014. [Google Scholar]
- Maruoka, M.; Suzuki, J.; Kawata, S.; Yoshida, K.; Hira, N.; Sato, S.; Goff, S.; Takeya, T.; Tani, K.; Shishido, T. Identification of B cell adaptor for PI3-kinase (BCPA) as an Abl interactor 1-regulated substrate of Abl kinases. FEBS Lett. 2005, 570, 2986–2990. [Google Scholar] [CrossRef] [Green Version]
- Nanki, Y.; Chioda, T.; Hirasawa, A.; Ookubo, A.; Itoh, M.; Ueno, M.; Akahane, T.; Kameyama, K.; Yamagami, W.; Kataoka, F.; et al. Patient-derived ovarian cancer organoids capture the genomic profiles of primary tumours applicable for drug sensitivity and resistance testing. Sci. Rep. 2020, 10, 12581. [Google Scholar] [CrossRef]
- Niraj, J.; Farkkila, A.; D’Andrea, A.D. The Fanconi Anemia Pathway in Cancer. Annu. Rev. Cancer Biol. 2019, 3, 457–478. [Google Scholar] [CrossRef]
- Xiu, M.X.; Liu, Y.M. The role of oncogenic Notch2 signaling in cancer: A novel therapeutic target. Am. J. Cancer Res. 2019, 9, 837–854. [Google Scholar] [PubMed]
- Moyer, C.L.; Ivanovich, J.; Gillespie, J.L.; Doberstein, R.; Radke, M.R.; Richardson, M.E.; Kaufmann, S.H.; Swisher, E.M.; Goodfellow, P.J. Rare BRIP1 Missense Alleles Confer Risk for Ovarian and Breast Cancer. Cancer Res. 2020, 80, 857–867. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-kB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, e17023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrington, B.S.; Annunziata, C.M. NF-κB Signaling in Ovarian Cancer. Cancers 2019, 11, 1182. [Google Scholar] [CrossRef] [Green Version]
- Lagunas, V.M.; Meléndez-Zajgla, J. Nuclear Factor-kappa B as a Resistance Factor to Platinum-Based Antineoplasic Drugs. Met. Based Drugs 2008, 2008, 576104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kan, Y.; Liu, J.; Li, F. High Expression of Nuclear Transcription Factor-κB is Associated with Cisplatin Resistance and Prognosis for Ovarian Cancer. Cancer Manag. Res. 2020, 12, 8241–8252. [Google Scholar] [CrossRef]
- Sonoda, H.; Oshikawa-Hori, S.; Ikeda, M. An Early Decrease in Release of Aquaporin-2 in Urinary Extracellular Vesicles After Cisplatin Treatment in Rats. Cells 2019, 8, 139. [Google Scholar] [CrossRef] [Green Version]
- Xuejun, C.; Weimin, C.; Xiaoyan, D.; Wei, Z.; Qiong, Z.; Jianhua, Y. Effects of aquaporins on chemosensitivity to cisplatin in ovarian cancer cells. Arch. Gynecol. Obstet. 2014, 290, 525–532. [Google Scholar] [CrossRef] [PubMed]
- Li, J.M.; Yang, F.; Li, J.; Yuan, W.Q.; Wang, H.; Luo, Y.Q. Reelin Promotes Cisplatin Resistance by Induction of Epithelial-Mesenchymal Transition via p38/GSK3β/Snail Signaling in Non-Small Cell Lung Cancer. Med. Sci. Monit. 2020, 26, e925298. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Goubar, A.; Olaussen, K.A.; Vitale, I.; Senovilla, L.; Michels, J.; Robin, A.; Dorvault, N.; Besse, B.; Validire, P.; et al. Prognostic value of LIPC in non-small cell lung carcinoma. Cell Cycle 2013, 12, 647–654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, I.L.; Chou, C.Y.; Wu, Y.Y.; Wu, J.E.; Liang, C.H.; Tsai, Y.T.; Ke, J.Y.; Chen, Y.L.; Hsu, K.F.; Hong, T.M. Targeting FXYD2 by cardiac glycosides potently blocks tumor growth in ovarian clear cell carcinoma. Oncotarget 2016, 7, 62925–62938. [Google Scholar] [CrossRef]
- Yoon, J.-H.; Cho, S.-G. ADGRF4 Regulates Non-small Cell Lung Cancer Cell Invasiveness. Anticancer. Res. 2020, 40, 6835–6844. [Google Scholar] [CrossRef]
- Decker, C.E.; Yang, Z.; Rimer, R.; Park-Min, K.-Y.; Macaubas, C.; Mellins, E.D.; Novack, E.D.; Faccio, R. Role of Tmem178 in bone homeostasis. Proc. Natl. Acad. Sci. USA 2015, 112, 15654–15659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Rauch, J.; Kolch, W. Targeting MAPK Signaling in Cancer: Mechanisms of Drug Resistance and Sensitivity. Int. J. Mol. Sci. 2020, 21, 1102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, X.; Chen, H.; Zhou, J.; Steed, H.; Postovit, L.-M.; Fu, Y. Pharmacological Inhibition of p38 MAPK by SB203580 Increases Resistance to Carboplatin in A2780cp Cells and Promotes Growth in Primary Ovarian Cancer Cells. Int. J. Mol. Sci. 2018, 19, 2184. [Google Scholar] [CrossRef] [Green Version]
- Jo, U.; Murai, Y.; Chakka, S.; Chen, L.; Cheng, K.; Murai, J.; Saha, L.K.; Miller Jenkins, L.M.; Pommier, Y. SLFN11 promotes CDT1 degradation by CUL4 in response to replicative DNA damage, while its absence leads to synthetic lethality with ATR/CHK1 inhibitors. Proc. Natl. Acad. Sci. USA 2021, 118, e2015654118. [Google Scholar] [CrossRef] [PubMed]
- Roška, J.; Wachsmannová, L.; Hurbanová, L.; Šestáková, Z.; Mueller, T.; Jurkovičová, D.; Chovanec, M. Differential gene expression in cisplatin-resistant and -sensitive testicular germ cell tumor cell lines. Oncotarget 2020, 11, 4735–4753. [Google Scholar] [CrossRef]
- Martinez, V.; Kennedy, S.; Doolan, P.; Gammell, P.; Joyce, H.; Kenny, E.; Mehta, J.P.; Ryan, E.; O’Connor, R.; Crown, J.; et al. Drug metabolism-related genes as potential biomarkers: Analysis of expression in normal and tumour breast tissue. Breast Cancer Res. Treat. 2008, 110, 521–530. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Virshup, D. Wnt Signaling and Drug Resistance in Cancer. Mol. Pharmacol. 2020, 97, 72–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maenhoudt, N.; Defraye, C.; Boretto, M.; Jan, Z.; Heremans, R.; Boeckx, B.; Hermans, F.; Arijs, I.; Cox, B.; Van Nieuwenhuysen, E.; et al. Developing Organoids from Ovarian Cancer as Experimental and Preclinical Models. Stem Cell Rep. 2020, 14, 717–729. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Gotimer, K.; De Souza, C.; Tepper, C.G.; Karnezis, A.N.; Leiserowitz, G.S.; Chien, J.; Smith, L.H. Short-term organoid culture for drug sensitivity testing of high-grade serous carcinoma. Gynecol. Oncol. 2020, 157, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Bi, J.; Newtson, A.M.; Zhang, Y.; Devor, E.J.; Samuelson, M.I.; Thiel, K.W.; Leslie, K.K. Successful Patient-Derived Organoid Culture of Gynecologic Cancers for Disease Modeling and Drug Sensitivity Testing. Cancers 2021, 13, 2901. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Age | TNM Stage | FIGO Stage | Primary Site | Histology | Grade |
|---|---|---|---|---|---|---|
| UK1236 | 48 | ypT3cN0M1 | IIIC | Ovary | Serous | 3 |
| UK1254 | 49 | ypT3cNX | IIIC | Ovary | Serous | 3 |
| UK1267 | 55 | T2bN0 | IIB | Fallopian Tube | Serous | 3 |
| UK1393 | 46 | T3cNX | IIIC | Ovary 1 | Serous | 3 |
| UK2238 | 58 | T3aN1b | IIIA | Fallopian Tube | Serous | 3 |
| UK2326 | 62 | T3cNX | IIIC | Ovary | Serous | 3 |
| ID | Residual Disease (cm) | Neoadjuvant | Adjuvant | Maintenance |
|---|---|---|---|---|
| UK1236 | 0 | carboplatin and paclitaxel 1 × 1 cycle; carboplatin and abraxane × 2 cycles | carboplatin and abraxane × 3 cycles | none |
| UK1254 | <0.5 | carboplatin and paclitaxel × 3 cycles | carboplatin and paclitaxel × 3 cycles | GOG 3020: rucaparib v. placebo and nivolumab v. placebo |
| UK1267 | 0 | None | carboplatin and paclitaxel × 6 cycles | none |
| UK1393 | 0 | None | carboplatin and paclitaxel × 6 cycles; bevacizumab with cycles 2–6 | none |
| UK2238 | <0.5 | None | carboplatin and paclitaxel × 6 cycles | olaparib |
| UK2326 | <0.5 | None | carboplatin and paclitaxel × 6 cycles | none |
| ID | Carboplatin EC50 (µM) | PFS (Days) |
|---|---|---|
| UK2326 | 0.8 | 398 |
| UK1267 | 1.1 | 338 |
| UK2238 | 3.3 | 391 |
| UK1236 | 28.5 | 579 |
| UK1393 | 44.8 | 445 |
| UK1254 | 50.2 1 | 252 |
| A. Upregulated | |||||
| Gene | LogFC | p Value | QValue (FDR) | Pathway ID | Pathway Description |
| 1. AQP1 | 8.722968 | 1.46 × 10−15 | 2.26 × 10−11 | GO:0022857 | Transmembrane transport activity |
| 2. TMEM178B | 6.489275 | 1.30 × 10−14 | 1.01 × 10−10 | --- | --- |
| 3. RELN | 7.083244 | 1.29 × 10−13 | 6.68 × 10−10 | GO:0030154 | Cell dedifferentiation |
| 4. ZNF723 | 8.998623 | 1.08 × 10−12 | 4.20 × 10−9 | GO:0003700 | DNA Binding transcription factor activity |
| 5. HAVCR1 | 9.870356 | 3.29 × 10−11 | 1.02 × 10−7 | GO:00023676 | Immune system process |
| 6. FXYD2 | 8.374937 | 8.24 × 10−10 | 2.13 × 10−6 | GO:0030234 | Enzyme regulator activity |
| 7. TGM3 | 6.3814 | 1.01 × 10−9 | 2.24 × 10−6 | GO:0006464 | Cellular protein modification process |
| 8. OGFRL1 | 3.648762 | 1.25 × 10−9 | 2.41 × 10−6 | GO:0007165 | Signal transduction |
| 9. LIPC | 5.511889 | 2.91 × 10−8 | 4.31 × 10−5 | GO:0006629 | Lipid metabolic process |
| 10. ADGRF2 | 9.051376 | 3.06 × 10−8 | 4.31 × 10−5 | GO:0007165 | Signal transduction |
| B. Downregulated | |||||
| Gene | LogFC | p Value | QValue (FDR) | Pathway ID | Pathway Description |
| 1. MAPK1 | −10.0724 | 2.43 × 10−9 | 4.19 × 10−6 | GO:0030154 | Cell differentiation |
| 2. ARNT2 | −9.9226 | 3.89 × 10−7 | 0.000463 | GO:0006950 | Response to stress |
| 3. STRA6 | −5.39401 | 6.80 × 10−6 | 0.00405 | GO:0006629 | Lipid metabolic process |
| 4. RBP1 | −5.05496 | 8.05 × 10−6 | 0.004168 | GO:0006629 | Lipid metabolic process |
| 5. ANTXR1 | −5.06802 | 1.13 × 10−5 | 0.005453 | GO:0007010 | Cytoskeleton organization |
| 6. LTBP1 | −4.19728 | 3.59 × 10−5 | 0.01285 | GO:0006464 | Cellular protein modification process |
| 7. AXIN2 | −5.6952 | 4.47 × 10−5 | 0.01442 | GO:0008283 | Cell population proliferation |
| 8. SLFN11 | −3.93117 | 8.66 × 10−5 | 0.02396 | GO:0006950 | Cell response to stress |
| 9. PHACTR1 | −5.39943 | 9.58 × 10−5 | 0.025592 | GO:0007010 | Cytoskeleton organization |
| 10. LYPD1 | −4.77696 | 0.000116 | 0.030426 | GO:0007267 | Cell–cell signaling |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gorski, J.W.; Zhang, Z.; McCorkle, J.R.; DeJohn, J.M.; Wang, C.; Miller, R.W.; Gallion, H.H.; Dietrich, C.S.; Ueland, F.R.; Kolesar, J.M. Utilizing Patient-Derived Epithelial Ovarian Cancer Tumor Organoids to Predict Carboplatin Resistance. Biomedicines 2021, 9, 1021. https://doi.org/10.3390/biomedicines9081021
Gorski JW, Zhang Z, McCorkle JR, DeJohn JM, Wang C, Miller RW, Gallion HH, Dietrich CS, Ueland FR, Kolesar JM. Utilizing Patient-Derived Epithelial Ovarian Cancer Tumor Organoids to Predict Carboplatin Resistance. Biomedicines. 2021; 9(8):1021. https://doi.org/10.3390/biomedicines9081021
Chicago/Turabian StyleGorski, Justin W., Zhuwei Zhang, J. Robert McCorkle, Jodi M. DeJohn, Chi Wang, Rachel W. Miller, Holly H. Gallion, Charles S. Dietrich, Frederick R. Ueland, and Jill M. Kolesar. 2021. "Utilizing Patient-Derived Epithelial Ovarian Cancer Tumor Organoids to Predict Carboplatin Resistance" Biomedicines 9, no. 8: 1021. https://doi.org/10.3390/biomedicines9081021
APA StyleGorski, J. W., Zhang, Z., McCorkle, J. R., DeJohn, J. M., Wang, C., Miller, R. W., Gallion, H. H., Dietrich, C. S., Ueland, F. R., & Kolesar, J. M. (2021). Utilizing Patient-Derived Epithelial Ovarian Cancer Tumor Organoids to Predict Carboplatin Resistance. Biomedicines, 9(8), 1021. https://doi.org/10.3390/biomedicines9081021