
Soluble Expression of Fc-Fused T Cell Receptors Allows Yielding Novel Bispecific T Cell Engagers

 , and
, and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Soluble Expression of TCRs and aCD3 in HEK293 Cells
2.3. Binding of NY-TCRmut to T2 Cells Loaded with Peptides
2.4. Flow Cytometry Analysis for Cell-Binding Assays
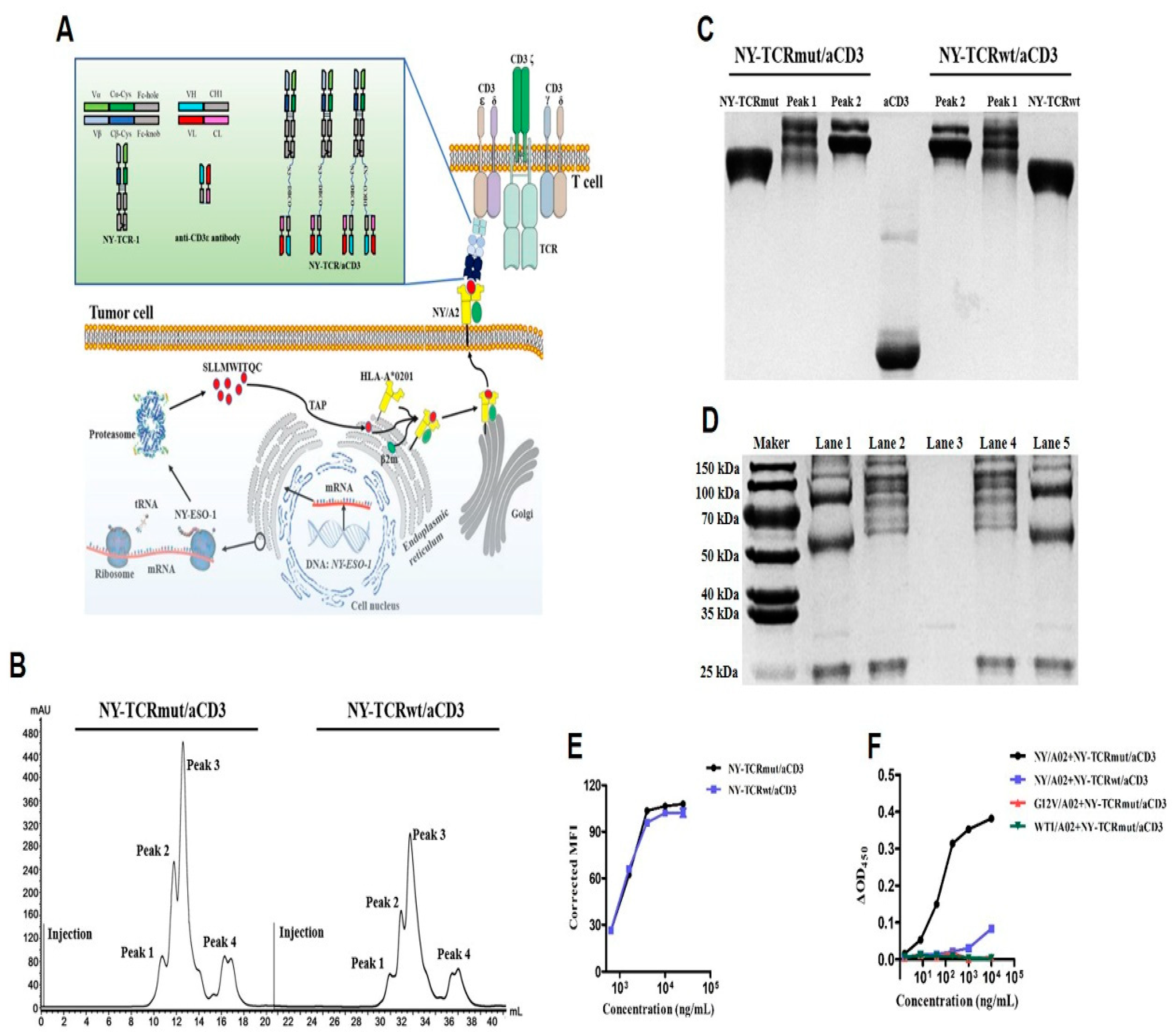
2.5. Generation of NY-TCR/aCD3
2.6. Enzyme-Linked Immunosorbent Assay (ELISA) for Affinity and Specificity Analysis of NY-TCR/aCD3
2.7. Preparation of Effector Cells
2.8. Evaluation of T Cell Activation Stimulated by NY-TCR/aCD3
2.9. Evaluation of the Anti-Tumor Activity of NY-TCR/aCD3 In Vitro
2.10. Evaluation of the Anti-Tumor Activity of NY-TCR/aCD3 In Vivo
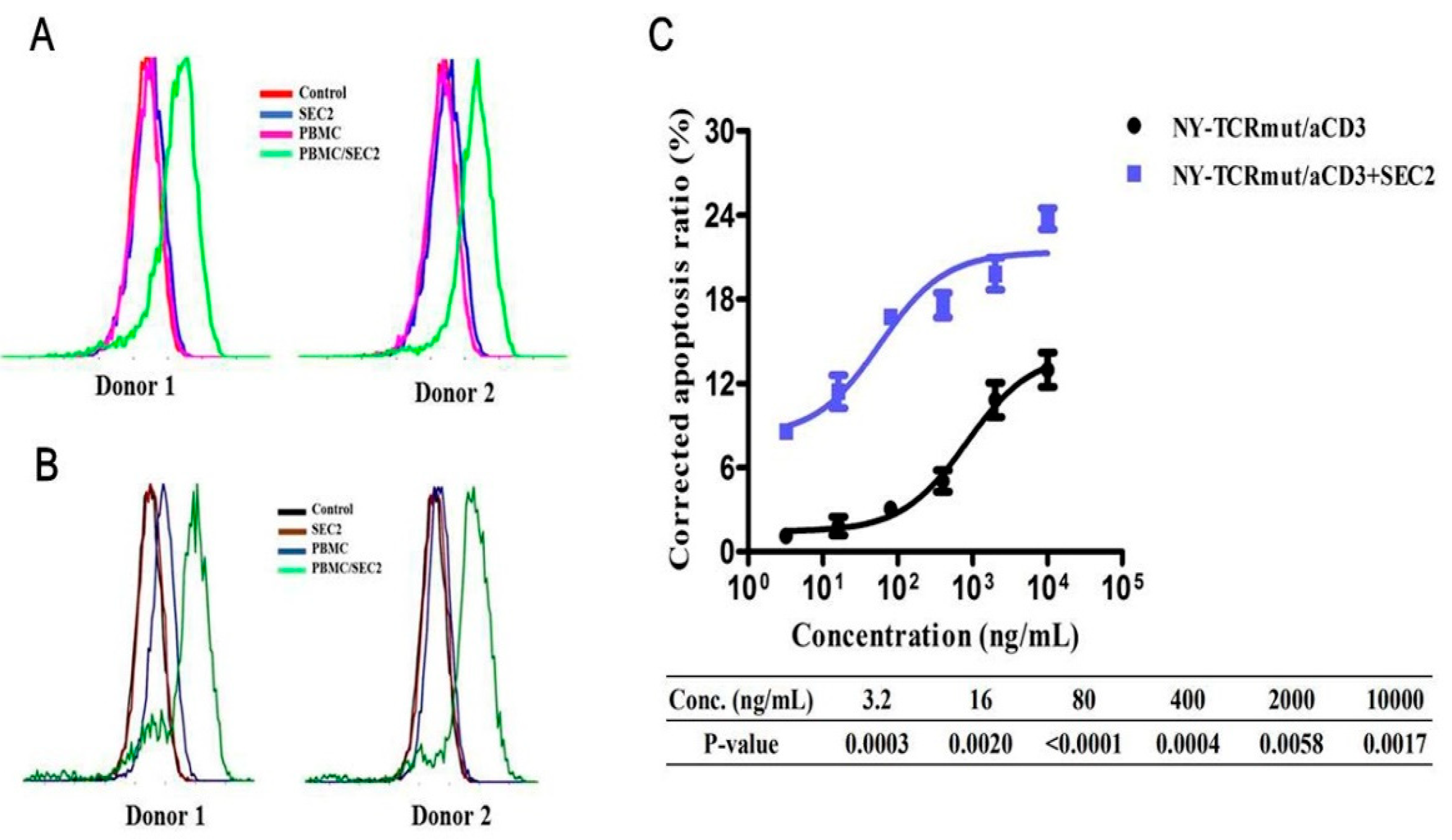
2.11. Analysis Expression Level of NY/A02 after Stimulated by SEC2
2.12. Evaluation of the Combined Anti-Tumor Activity of NY-TCRmut/aCD3 and SEC2
3. Results
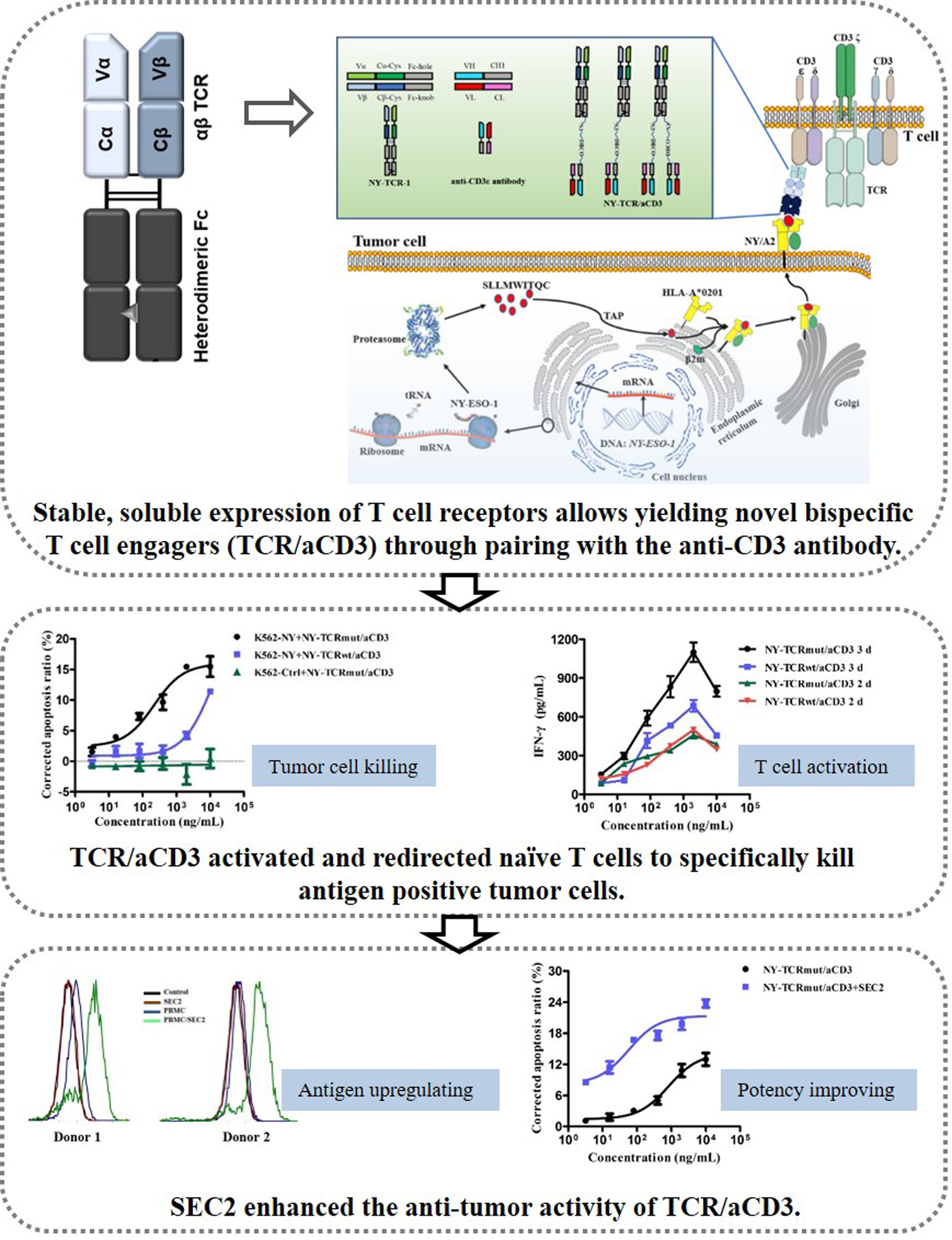
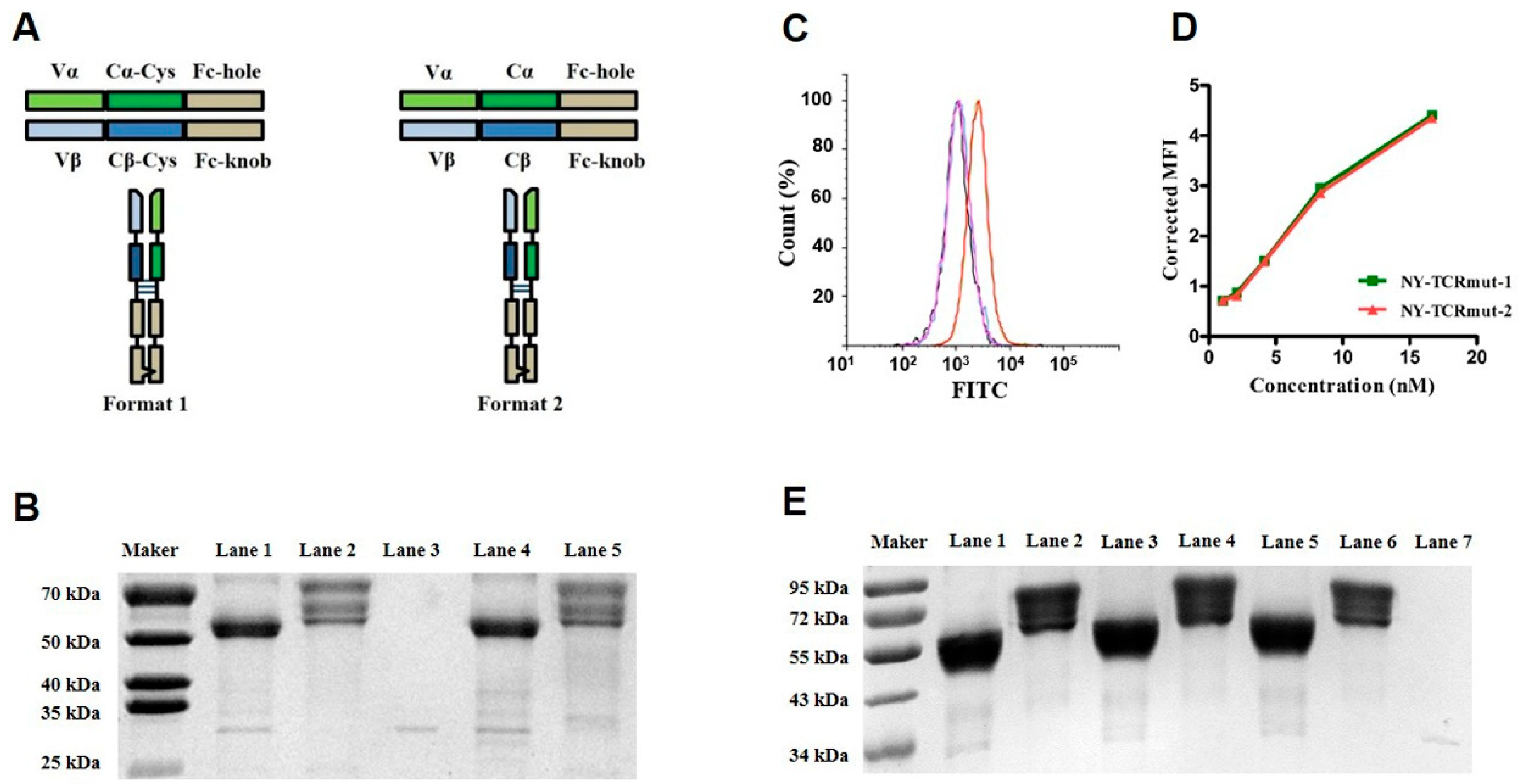
3.1. Soluble Expressions of TCRs and aCD3 in Mammalian Cells
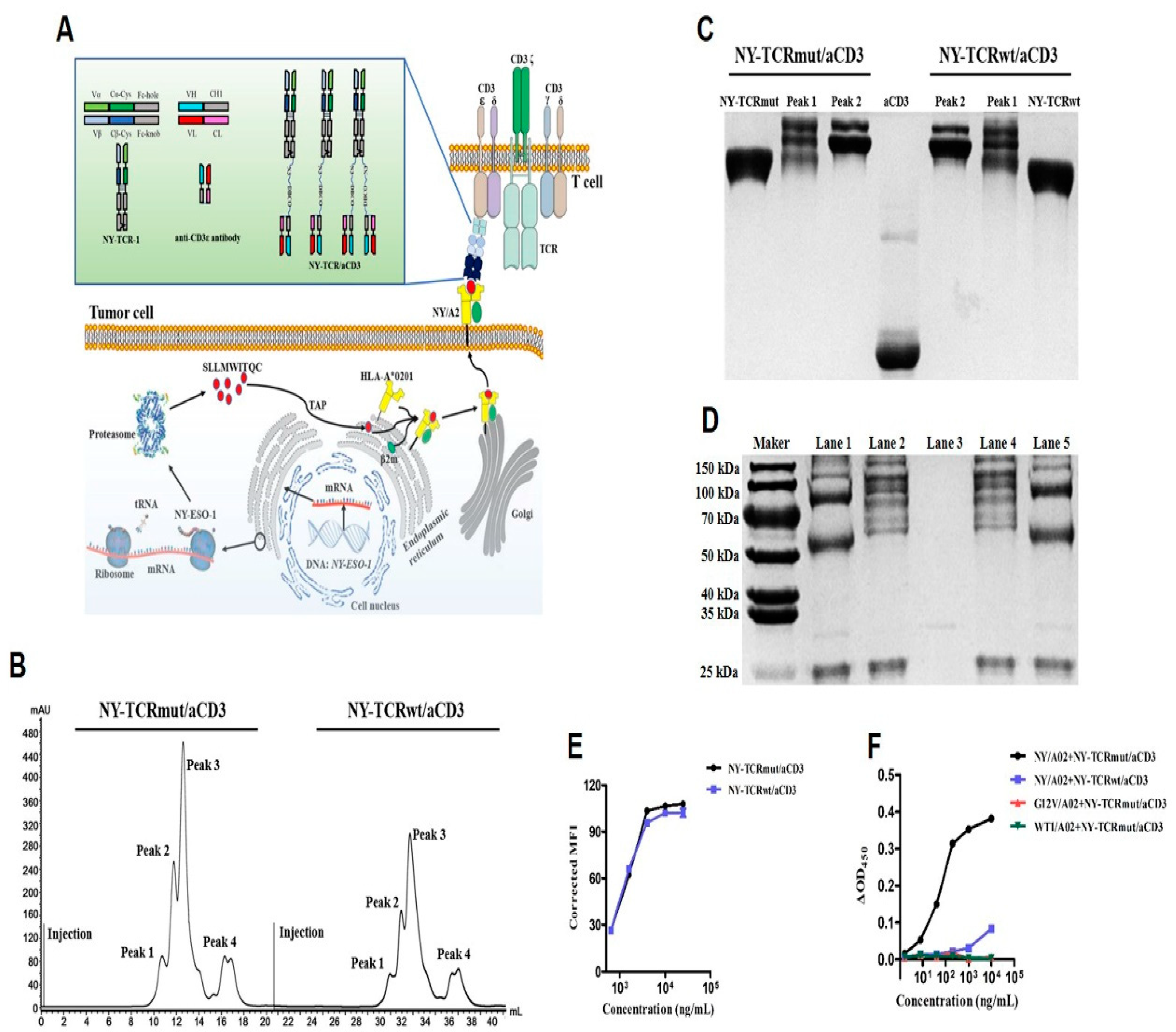
3.2. Generation of NY-TCR/aCD3
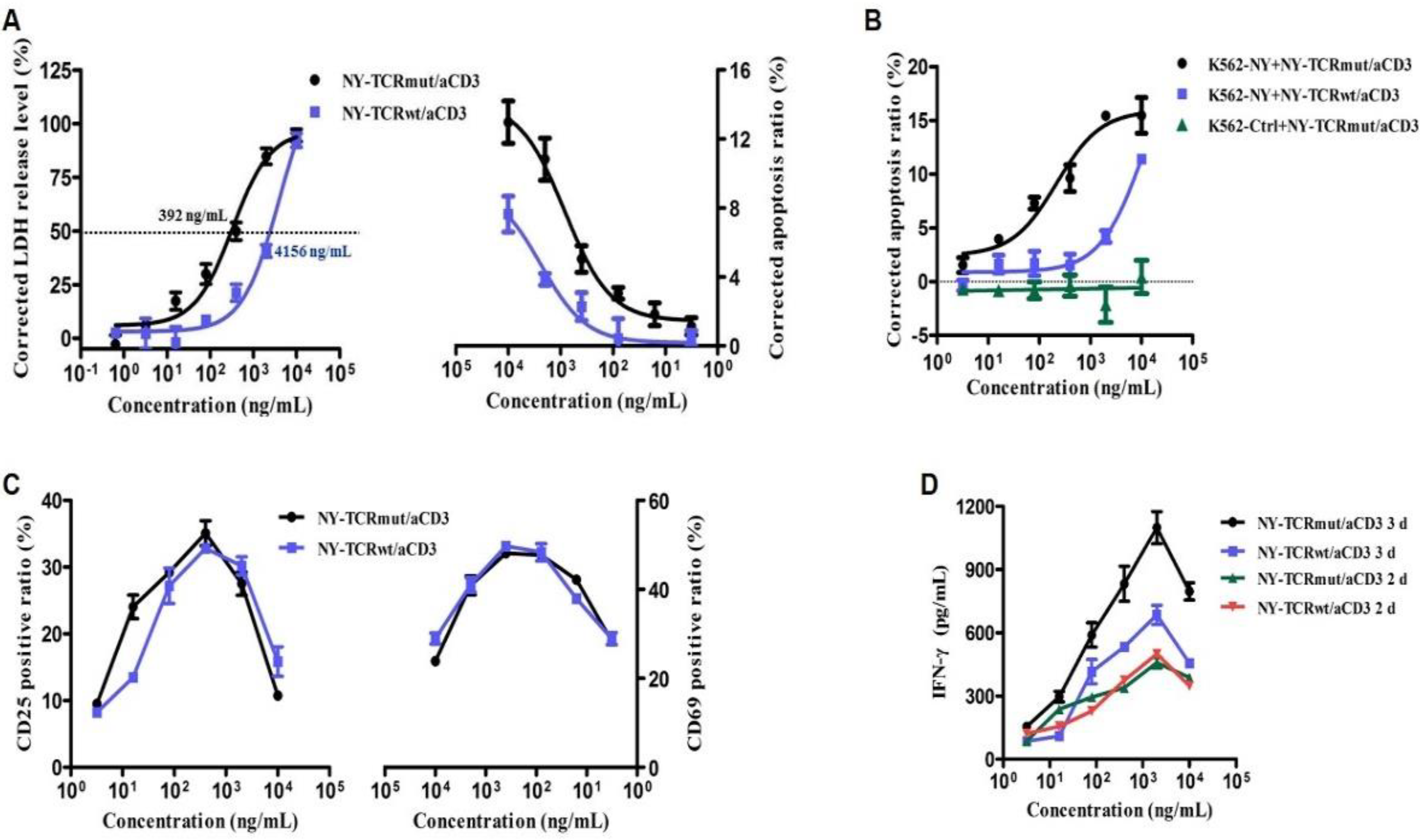
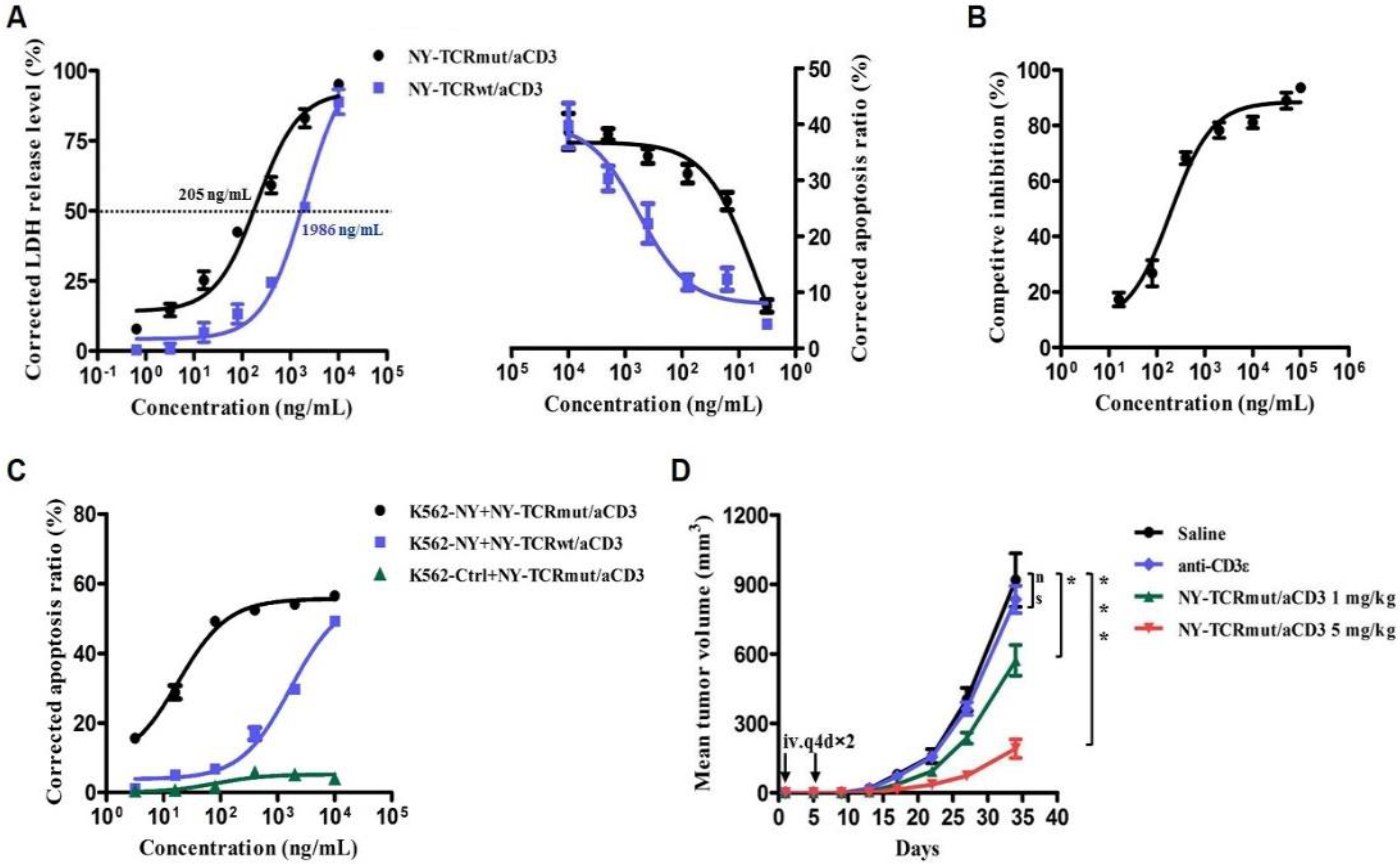
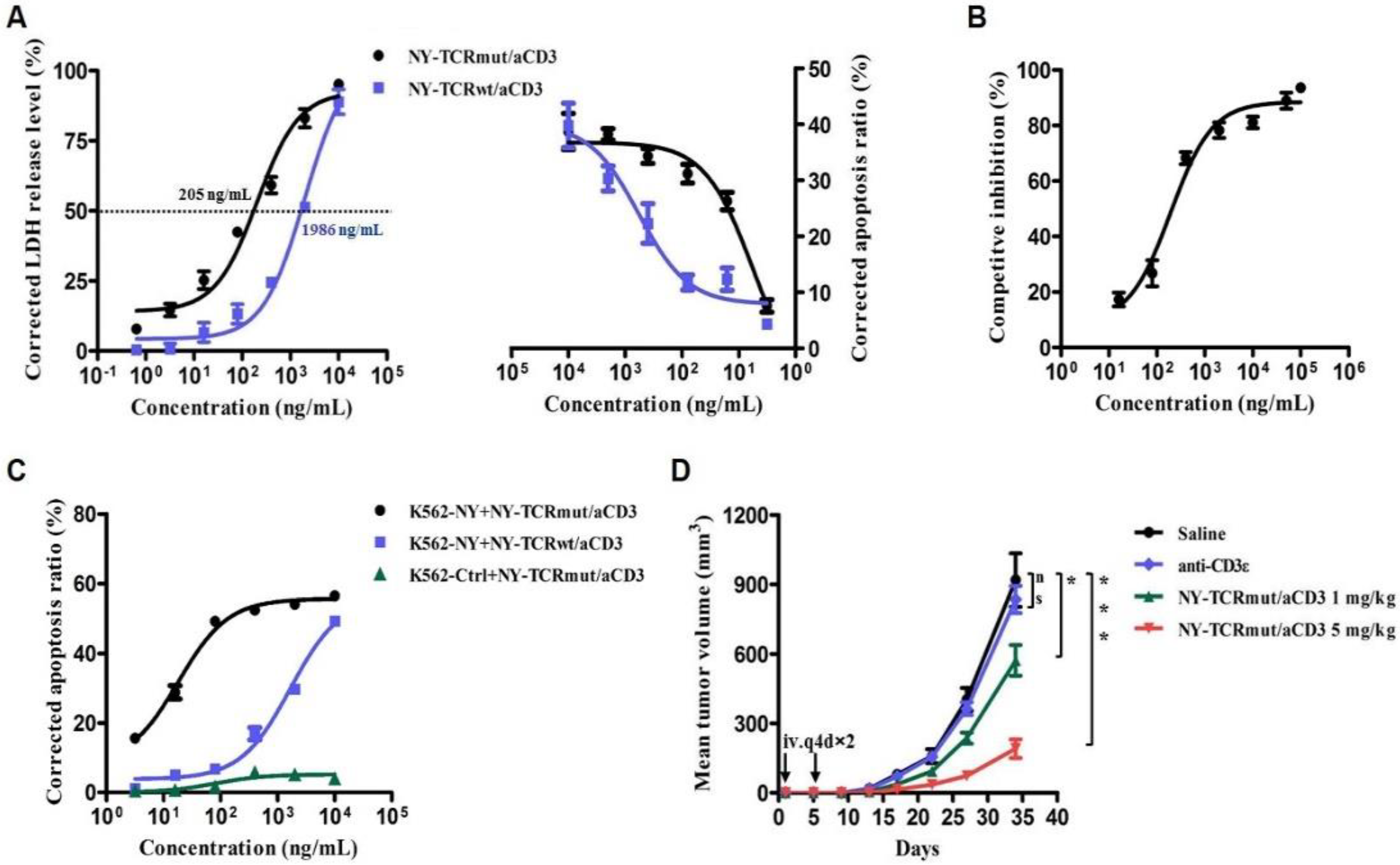
3.3. NY-TCR/aCD3 Activated and Redirected Naïve T Cells to Specifically Kill NY/A02+ Tumor Cells
3.4. NY-TCR/aCD3 Redirected ATC to Specifically Kill NY/A02+ Tumor Cells
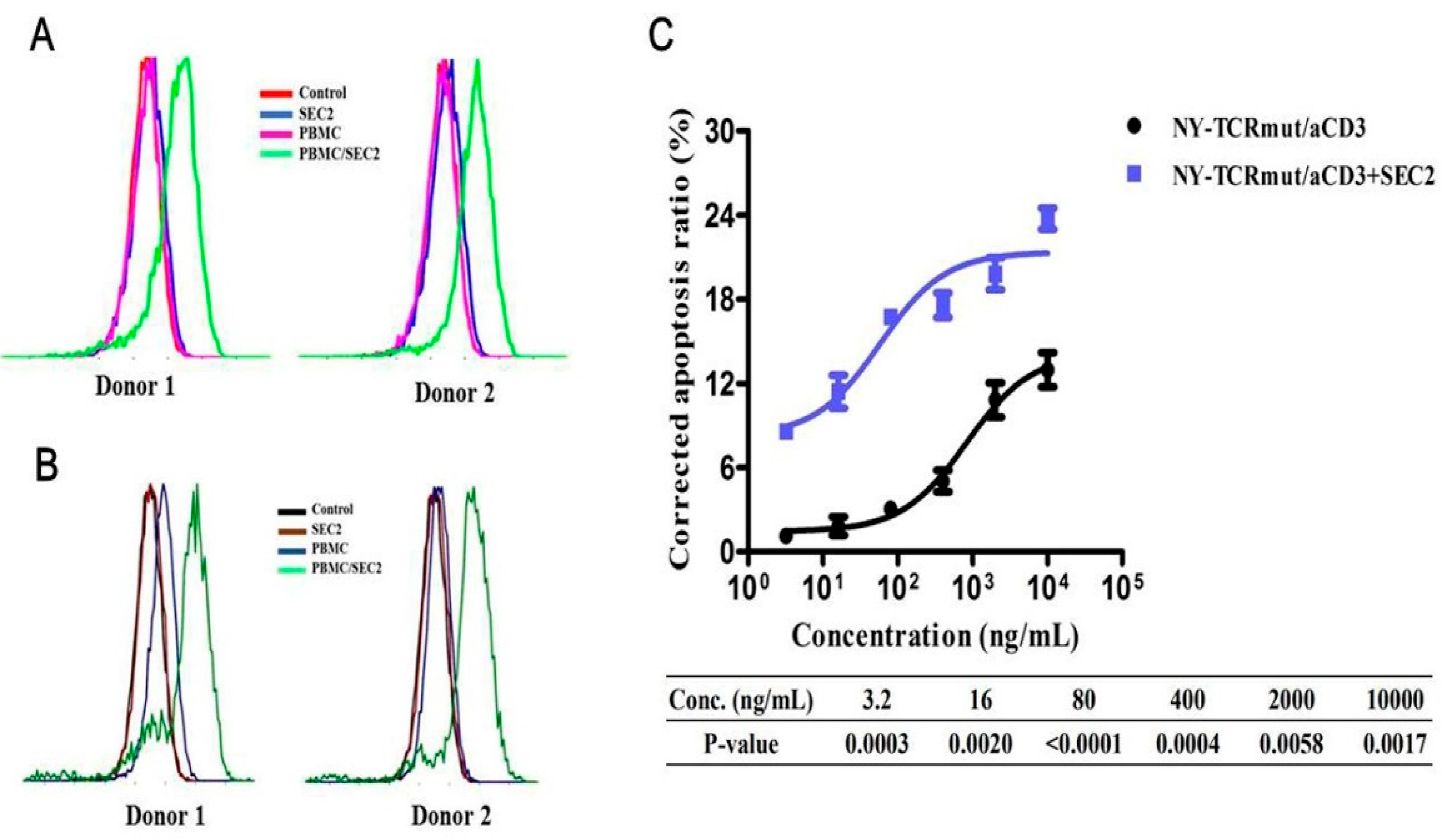
3.5. SEC2 Enhanced the Anti-Tumor Activity of NY-TCRmut/aCD3
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.-J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2019, 381, 1535–1546. [Google Scholar] [CrossRef] [Green Version]
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. CAR T cell immunotherapy for human cancer. Science 2018, 359, 1361–1365. [Google Scholar] [CrossRef] [Green Version]
- Jackson, H.J.; Rafiq, S.; Brentjens, R.J. Driving CAR T-cells forward. Nat. Rev. Clin. Oncol. 2016, 13, 370–383. [Google Scholar] [CrossRef]
- Sahin, U.; Türeci, Ö. Personalized vaccines for cancer immunotherapy. Science 2018, 359, 1355–1360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Z.; Ott, P.A.; Wu, C.J. Towards personalized, tumour-specific, therapeutic vaccines for cancer. Nat. Rev. Immunol. 2018, 18, 168–182. [Google Scholar] [CrossRef]
- Das, S.; Johnson, D.B. Immune-related adverse events and anti-tumor efficacy of immune checkpoint inhibitors. J. Immunother. Cancer 2019, 7, 306. [Google Scholar] [CrossRef]
- Brudno, J.N.; Kochenderfer, J.N. Recent advances in CAR T-cell toxicity: Mechanisms, manifestations and management. Blood Rev. 2019, 34, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Shi, T.; Zhang, H.; Hu, J.; Song, Y.; Wei, J.; Ren, S.; Zhou, C. Tumor neoantigens: From basic research to clinical applications. J. Hematol. Oncol. 2019, 12, 93. [Google Scholar] [CrossRef] [Green Version]
- Melero, I.; Gaudernack, G.; Gerritsen, W.; Huber, C.; Parmiani, G.; Scholl, S.; Thatcher, N.; Wagstaff, J.; Zielinski, C.; Faulkner, I.; et al. Therapeutic vaccines for cancer: An overview of clinical trials. Nat. Rev. Clin. Oncol. 2014, 11, 509–524. [Google Scholar] [CrossRef] [PubMed]
- Shemesh, C.S.; Hsu, J.C.; Hosseini, I.; Shen, B.Q.; Rotte, A.; Twomey, P.; Girish, S.; Wu, B. Personalized Cancer Vaccines: Clinical Landscape, Challenges, and Opportunities. Mol. Ther. 2021, 29, 555–570. [Google Scholar] [CrossRef] [PubMed]
- Brameshuber, M.; Kellner, F.; Rossboth, B.K.; Ta, H.; Alge, K.; Sevcsik, E.; Göhring, J.; Axmann, M.; Baumgart, F.; Gascoigne, N.R.J.; et al. Monomeric TCRs drive T cell antigen recognition article. Nat. Immunol. 2018, 19, 487–496. [Google Scholar] [CrossRef]
- Tran, E.; Robbins, P.F.; Lu, Y.-C.; Prickett, T.D.; Gartner, J.J.; Jia, L.; Pasetto, A.; Zheng, Z.; Ray, S.; Groh, E.M.; et al. T-Cell Transfer Therapy Targeting Mutant KRAS in Cancer. N. Engl. J. Med. 2016, 375, 2255–2262. [Google Scholar] [CrossRef] [Green Version]
- Rohaan, M.W.; Van Den Berg, J.H.; Kvistborg, P.; Haanen, J.B.A.G. Adoptive transfer of tumor-infiltrating lymphocytes in melanoma: A viable treatment option 11 Medical and Health Sciences 1107 Immunology 11 Medical and Health Sciences 1112 Oncology and Carcinogenesis. J. Immunother. Cancer 2018, 6, 102. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Cheung, N.V. T cell engaging bispecific antibody (T-BsAb): From technology to therapeutics. Pharmacol. Ther. 2018, 182, 161–175. [Google Scholar] [CrossRef]
- Liddy, N.; Bossi, G.; Adams, K.J.; Lissina, A.; Mahon, T.M.; Hassan, N.J.; Gavarret, J.; Bianchi, F.C.; Pumphrey, N.J.; Ladell, K.; et al. Monoclonal TCR-redirected tumor cell killing. Nat. Med. 2012, 18, 980–987. [Google Scholar] [CrossRef]
- Middleton, M.R.; McAlpine, C.; Woodcock, V.K.; Corrie, P.; Infante, J.R.; Steven, N.M.; Jeffry Evans, T.R.; Anthoney, A.; Shoushtari, A.N.; Hamid, O.; et al. Tebentafusp, a TCR/Anti-CD3 bispecific fusion protein targeting gp100, potently activated antitumor immune responses in patients with metastatic melanoma. Clin. Cancer Res. 2020, 26, 5869–5878. [Google Scholar] [CrossRef]
- Boulter, J.M.; Glick, M.; Todorov, P.T.; Baston, E.; Sami, M.; Rizkallah, P.; Jakobsen, B.K. Stable, soluble T-cell receptor molecules for crystallization and therapeutics. Protein Eng. 2003, 16, 707–711. [Google Scholar] [CrossRef] [PubMed]
- Shukla, A.A.; Wolfe, L.S.; Mostafa, S.S.; Norman, C. Evolving trends in mAb production processes. Bioeng. Transl. Med. 2017, 2, 58–69. [Google Scholar] [CrossRef]
- Froning, K.; Maguire, J.; Sereno, A.; Huang, F.; Chang, S.; Weichert, K.; Frommelt, A.J.; Dong, J.; Wu, X.; Austin, H.; et al. Computational stabilization of T cell receptors allows pairing with antibodies to form bispecifics. Nat. Commun. 2020, 11, 2330. [Google Scholar] [CrossRef] [PubMed]
- Mosquera, L.A.; Card, K.F.; Price-Schiavi, S.A.; Belmont, H.J.; Liu, B.; Builes, J.; Zhu, X.; Chavaillaz, P.-A.; Lee, H.; Jiao, J.; et al. In Vitro and In Vivo Characterization of a Novel Antibody-Like Single-Chain TCR Human IgG1 Fusion Protein. J. Immunol. 2005, 174, 4381–4388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Moysey, R.; Molloy, P.E.; Vuidepot, A.L.; Mahon, T.; Baston, E.; Dunn, S.; Liddy, N.; Jacob, J.; Jakobsen, B.K.; et al. Directed evolution of human T-cell receptors with picomolar affinities by phage display. Nat. Biotechnol. 2005, 23, 349–354. [Google Scholar] [CrossRef]
- Zhao, W.; Li, Y.; Liu, W.; Ding, D.; Xu, Y.; Pan, L.; Chen, S. Transcytosis, Antitumor Activity and Toxicity of Staphylococcal Enterotoxin C2 as an Oral Administration Protein Drug. Toxins 2016, 8, 185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo, W.; Parkhurst, M.; Robbins, P.F.; Tran, E.; Lu, Y.C.; Jia, L.; Gartner, J.J.; Pasetto, A.; Deniger, D.; Malekzadeh, P.; et al. Immunologic recognition of a shared p53 mutated neoantigen in a patient with metastatic colorectal cancer. Cancer Immunol. Res. 2019, 7, 534–543. [Google Scholar] [CrossRef]
- Ahmed, M.; Cheng, M.; Cheung, I.Y.; Cheung, N.-K. V Human derived dimerization tag enhances tumor killing potency of a T-cell engaging bispecific antibody. Oncoimmunology 2015, 4, e989776. [Google Scholar] [CrossRef] [Green Version]
- Pan, L.; Zhao, W.; Lai, J.; Ding, D.; Zhang, Q.; Yang, X.; Huang, M.; Jin, S.; Xu, Y.; Zeng, S.; et al. Sortase A-Generated Highly Potent Anti-CD20-MMAE Conjugates for Efficient Elimination of B-Lineage Lymphomas. Small 2017, 13, 1602267. [Google Scholar] [CrossRef] [PubMed]
- Denkberg, G.; Cohen, C.J.; Segal, D.; Kirkin, A.F.; Reiter, Y. Recombinant human single-chain MHC-peptide complexes made from E. coli by in vitro refolding: Functional single-chain MHC-peptide complexes and tetramers with tumor associated antigens. Eur. J. Immunol. 2000, 30, 3522–3532. [Google Scholar] [CrossRef]
- La Gruta, N.L.; Gras, S.; Daley, S.R.; Thomas, P.G.; Rossjohn, J. Understanding the drivers of MHC restriction of T cell receptors. Nat. Rev. Immunol. 2018, 18, 467–478. [Google Scholar] [CrossRef] [PubMed]
- Daniels, M.A.; Hogquist, K.A.; Jameson, S.C. Sweet “n” sour: The impact of differential glycosylation on T cell responses. Nat. Immunol. 2002, 3, 903–910. [Google Scholar] [CrossRef]
- Zhou, Q.; Qiu, H. The Mechanistic Impact of N-Glycosylation on Stability, Pharmacokinetics, and Immunogenicity of Therapeutic Proteins. J. Pharm. Sci. 2019, 108, 1366–1377. [Google Scholar] [CrossRef]
- Wu, X.; Sereno, A.J.; Huang, F.; Zhang, K.; Batt, M.; Fitchett, J.R.; He, D.; Rick, H.L.; Conner, E.M.; Demarest, S.J. Protein design of IgG/TCR chimeras for the co-expression of Fab-like moieties within bispecific antibodies. MAbs 2015, 7, 364–376. [Google Scholar] [CrossRef] [Green Version]
- Bausch-Fluck, D.; Hofmann, A.; Bock, T.; Frei, A.P.; Cerciello, F.; Jacobs, A.; Moest, H.; Omasits, U.; Gundry, R.L.; Yoon, C.; et al. A Mass Spectrometric-Derived Cell Surface Protein Atlas. PLoS ONE 2015, 10, e0121314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sim, M.J.W.; Lu, J.; Spencer, M.; Hopkins, F.; Tran, E.; Rosenberg, S.A.; Long, E.O.; Sun, P.D. High-affinity oligoclonal TCRs define effective adoptive T cell therapy targeting mutant KRAS-G12D. Proc. Natl. Acad. Sci. USA 2020, 117, 12826–12835. [Google Scholar] [CrossRef]
- Akazawa, Y.; Saito, Y.; Yoshikawa, T.; Saito, K.; Nosaka, K.; Shimomura, M.; Mizuno, S.; Nakamoto, Y.; Nakatsura, T. Efficacy of immunotherapy targeting the neoantigen derived from epidermal growth factor receptor T790M/C797S mutation in non–small cell lung cancer. Cancer Sci. 2020, 111, 2736–2746. [Google Scholar] [CrossRef] [PubMed]
- Chheda, Z.S.; Kohanbash, G.; Okada, K.; Jahan, N.; Sidney, J.; Pecoraro, M.; Yang, X.; Carrera, D.A.; Downey, K.M.; Shrivastav, S.; et al. Novel and shared neoantigen derived from histone 3 variant H3.3K27M mutation for glioma T cell therapy. J. Exp. Med. 2018, 215, 141–157. [Google Scholar] [CrossRef]
- Wu, D.; Gallagher, D.T.; Gowthaman, R.; Pierce, B.G.; Mariuzza, R.A. Structural basis for oligoclonal T cell recognition of a shared p53 cancer neoantigen. Nat. Commun. 2020, 11, 2908. [Google Scholar] [CrossRef] [PubMed]
- Purbhoo, M.A.; Sutton, D.H.; Brewer, J.E.; Mullings, R.E.; Hill, M.E.; Mahon, T.M.; Karbach, J.; Jäger, E.; Cameron, B.J.; Lissin, N.; et al. Quantifying and Imaging NY-ESO-1/LAGE-1-Derived Epitopes on Tumor Cells Using High Affinity T Cell Receptors. J. Immunol. 2006, 176, 7308–7316. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Xie, S.; Yang, X.; Cueva, J.C.; Hou, X.; Tang, Z.; Yao, H.; Mo, F.; Yin, S.; Liu, A.; et al. Opportunities and challenges for antibodies against intracellular antigens. Theranostics 2019, 9, 7792–7806. [Google Scholar] [CrossRef]
- Chang, A.Y.; Gejman, R.S.; Brea, E.J.; Oh, C.Y.; Mathias, M.D.; Pankov, D.; Casey, E.; Dao, T.; Scheinberg, D.A. Opportunities and challenges for TCR mimic antibodies in cancer therapy. Expert Opin. Biol. Ther. 2016, 16, 979–987. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.N.; Harris, D.T.; Kranz, D.M. T Cell Receptor Engineering and Analysis Using the Yeast Display Platform. Methods Mol. Biol. 2015, 1319, 95–141. [Google Scholar] [PubMed] [Green Version]
- Pierce, B.G.; Hellman, L.M.; Hossain, M.; Singh, N.K.; Vander Kooi, C.W.; Weng, Z.; Baker, B.M. Computational Design of the Affinity and Specificity of a Therapeutic T Cell Receptor. PLoS Comput. Biol. 2014, 10, e1003478. [Google Scholar] [CrossRef]
- Holland, C.J.; Crean, R.M.; Pentier, J.M.; de Wet, B.; Lloyd, A.; Srikannathasan, V.; Lissin, N.; Lloyd, K.A.; Blicher, T.H.; Conroy, P.J.; et al. Specificity of bispecific T cell receptors and antibodies targeting peptide-HLA. J. Clin. Investg. 2020, 130, 2673–2688. [Google Scholar] [CrossRef] [Green Version]
- Huppa, J.B.; Davis, M.M. T-cell-antigen recognition and the immunological synapse. Nat. Rev. Immunol. 2003, 3, 973–983. [Google Scholar] [CrossRef]
- Esensten, J.H.; Helou, Y.A.; Chopra, G.; Weiss, A.; Bluestone, J.A. CD28 Costimulation: From Mechanism to Therapy. Immunity 2016, 44, 973–988. [Google Scholar] [CrossRef] [Green Version]
- Skokos, D.; Waite, J.C.; Haber, L.; Crawford, A.; Hermann, A.; Ullman, E.; Slim, R.; Godin, S.; Ajithdoss, D.; Ye, X.; et al. A class of costimulatory CD28-bispecific antibodies that enhance the antitumor activity of CD3-bispecific antibodies. Sci. Transl. Med. 2020, 12, eaaw7888. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Seung, E.; Xu, L.; Rao, E.; Lord, D.M.; Wei, R.R.; Cortez-Retamozo, V.; Ospina, B.; Posternak, V.; Ulinski, G.; et al. Trispecific antibodies enhance the therapeutic efficacy of tumor-directed T cells through T cell receptor co-stimulation. Nat. Cancer 2020, 1, 86–98. [Google Scholar] [CrossRef]
- Sun, H.Y.; Xue, Q.; Pan, Y.Q.; Ding, D.; Chen, J.; Chen, S.Q. Preparation and application of antibody against staphylococcal enterotoxin C2. Acta. Pharm. Sin. 2008, 43, 801–805. [Google Scholar]
- Zhou, F. Molecular mechanisms of IFN-γ to up-regulate MHC class I antigen processing and presentation. Int. Rev. Immunol. 2009, 28, 239–260. [Google Scholar] [CrossRef]
- Yang, Y.M.; Shang, D.S.; Zhao, W.D.; Fang, W.G.; Chen, Y.H. Microglial TNF-α-Dependent Elevation of MHC Class I Expression on Brain Endothelium Induced by Amyloid-Beta Promotes T Cell Transendothelial Migration. Neurochem. Res. 2013, 38, 2295–2304. [Google Scholar] [CrossRef]
- Von Scheidt, B.; Wang, M.; Oliver, A.J.; Chan, J.D.; Jana, M.K.; Ali, A.I.; Clow, F.; Fraser, J.D.; Quinn, K.M.; Darcy, P.K.; et al. Enterotoxins can support CAR T cells against solid tumors. Proc. Natl. Acad. Sci. USA 2019, 116, 25229–25235. [Google Scholar] [CrossRef]
- Yamamoto, T.N.; Kishton, R.J.; Restifo, N.P. Developing neoantigen-targeted T cell–based treatments for solid tumors. Nat. Med. 2019, 25, 1488–1499. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, W.-B.; Shen, Y.; Liu, W.-H.; Li, Y.-M.; Jin, S.-J.; Xu, Y.-C.; Pan, L.-Q.; Zhou, Z.; Chen, S.-Q. Soluble Expression of Fc-Fused T Cell Receptors Allows Yielding Novel Bispecific T Cell Engagers. Biomedicines 2021, 9, 790. https://doi.org/10.3390/biomedicines9070790
Zhao W-B, Shen Y, Liu W-H, Li Y-M, Jin S-J, Xu Y-C, Pan L-Q, Zhou Z, Chen S-Q. Soluble Expression of Fc-Fused T Cell Receptors Allows Yielding Novel Bispecific T Cell Engagers. Biomedicines. 2021; 9(7):790. https://doi.org/10.3390/biomedicines9070790
Chicago/Turabian StyleZhao, Wen-Bin, Ying Shen, Wen-Hui Liu, Yi-Ming Li, Shi-Jie Jin, Ying-Chun Xu, Li-Qiang Pan, Zhan Zhou, and Shu-Qing Chen. 2021. "Soluble Expression of Fc-Fused T Cell Receptors Allows Yielding Novel Bispecific T Cell Engagers" Biomedicines 9, no. 7: 790. https://doi.org/10.3390/biomedicines9070790
APA StyleZhao, W.-B., Shen, Y., Liu, W.-H., Li, Y.-M., Jin, S.-J., Xu, Y.-C., Pan, L.-Q., Zhou, Z., & Chen, S.-Q. (2021). Soluble Expression of Fc-Fused T Cell Receptors Allows Yielding Novel Bispecific T Cell Engagers. Biomedicines, 9(7), 790. https://doi.org/10.3390/biomedicines9070790