
Role of Leptin in Non-Alcoholic Fatty Liver Disease

 , ,
, ,
Abstract
:1. Introduction
2. Non-Alcoholic Fatty Liver Disease (NAFLD): Characteristics and Signaling Pathways of Leptin Receptor
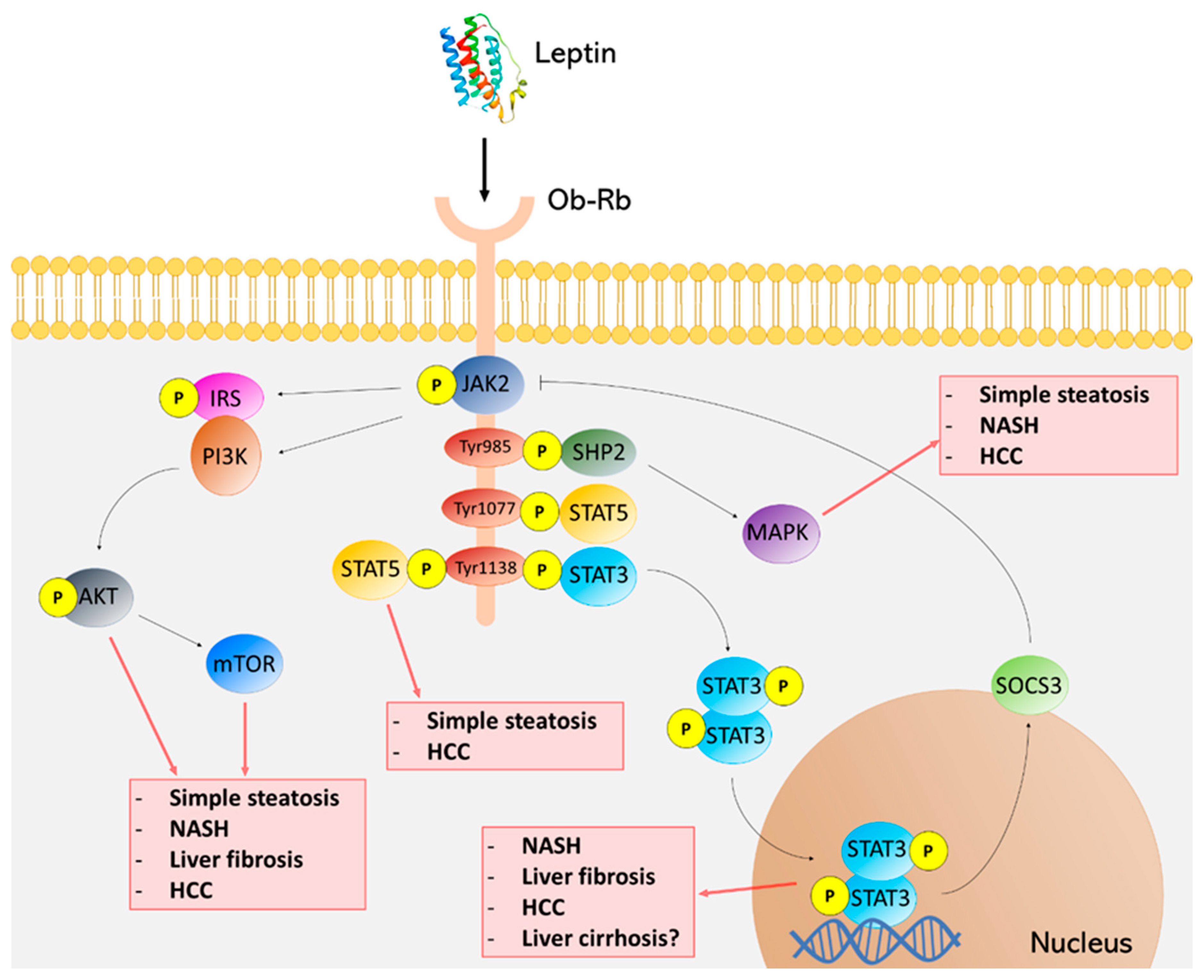
Leptin Receptor Signaling and NAFLD
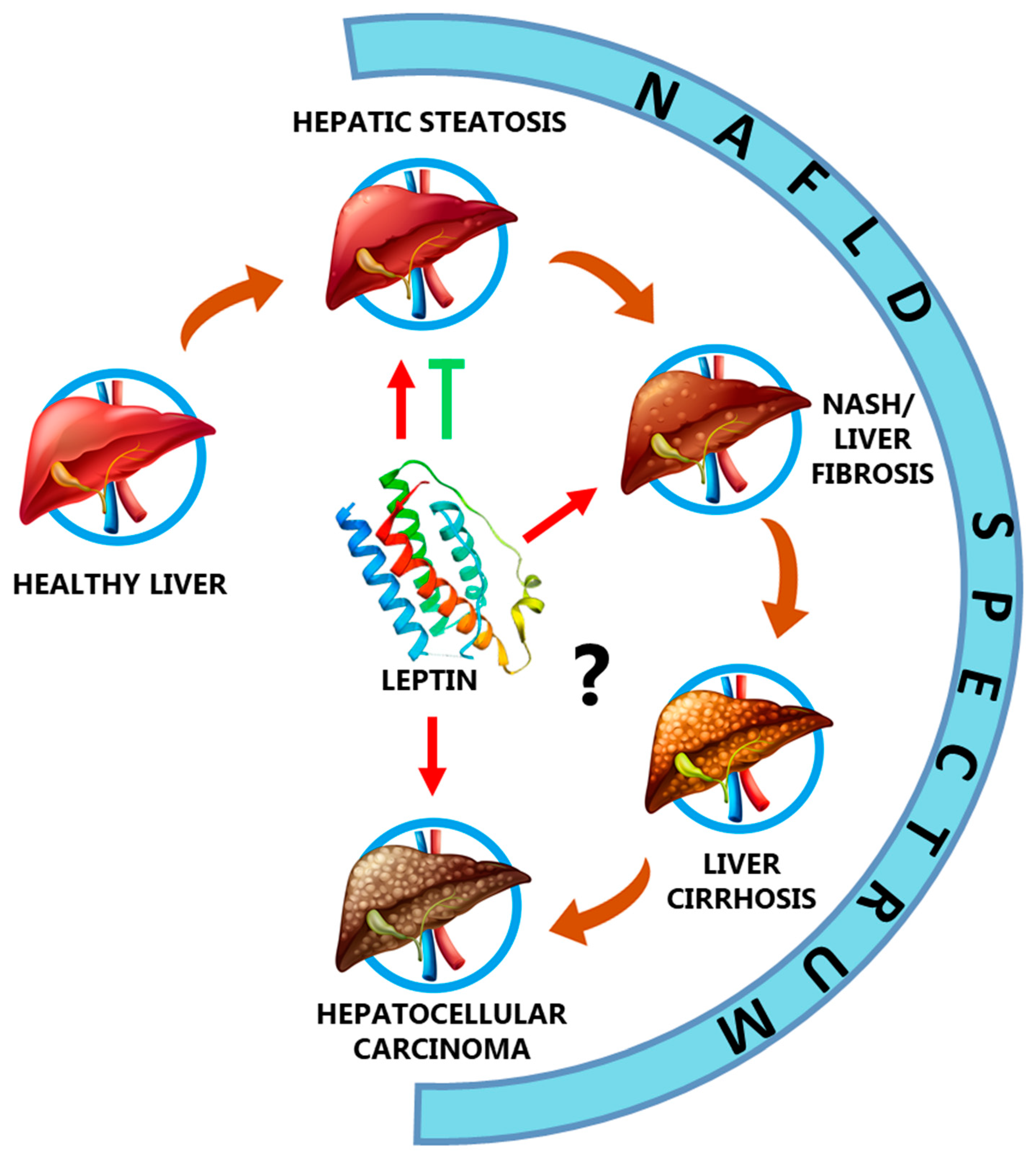
3. Leptin in the NAFLD Spectrum
3.1. Leptin and Hepatic Steatosis
3.2. Leptin, Non-Alcoholic Steatohepatitis (NASH), and Fibrosis
3.3. Leptin and Liver Cirrhosis
3.4. Leptin and Hepatocellular Carcinoma
4. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhang, Y.; Proenca, R.; Maffei, M. Positional cloning of the mouse obese gene and its human homologue. Nature 1994, 372, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Ingalls, A.M.; Dickie, M.M.; Snell, G.D. Obese, a new mutation in the house mouse. J. Hered. 1950, 41, 317–318. [Google Scholar] [CrossRef] [PubMed]
- Hummel, K.P.; Dickie, M.M.; Coleman, D.L. Diabetes, a new mutation in the mouse. Science 1966, 153, 1127–1128. [Google Scholar] [CrossRef]
- Denver, R.J.; Bonett, R.M.; Boorse, G.C. Evolution of leptin structure and function. Neuroendocrinology 2011, 94, 21–38. [Google Scholar] [CrossRef]
- Zhang, F.; Basinski, M.B.; Beals, J.M.; Briggs, S.L.; Churgay, L.M.; Clawson, D.K.; DiMarchi, R.D.; Furman, T.C.; Hale, J.E.; Hsiung, H.M.; et al. Crystal structure of the obese protein leptin-E100. Nature 1997, 387, 206–209. [Google Scholar] [CrossRef]
- Tartaglia, L.A.; Dembski, M.; Weng, X.; Deng, N.; Culpepper, J.; Devos, R.; Richards, G.J.; Campfield, L.A.; Clark, F.T.; Deeds, J.; et al. Identification and expression cloning of a leptin receptor OB-R. Cell 1995, 83, 1263–1271. [Google Scholar] [CrossRef] [Green Version]
- Tartaglia, L.A. The leptin receptor. J. Biol. Chem. 1997, 272, 6093–6096. [Google Scholar] [CrossRef] [Green Version]
- Myers, M.G., Jr. Leptin receptor signaling and the regulation of mammalian physiology. Recent Prog. Horm. Res. 2004, 59, 287–304. [Google Scholar] [CrossRef]
- Gorska, E.; Popko, K.; Stelmaszczyk-Emmel, A.; Ciepiela, O.; Kucharska, A.; Wasik, M. Leptin receptors. Eur. J. Med. Res. 2010, 15 (Suppl. 2), 50–54. [Google Scholar] [CrossRef] [Green Version]
- Park, H.-Y.; Ahima, R.S. Leptin signaling. F1000Prime Rep. 2014, 6, 73. [Google Scholar] [CrossRef]
- Deck, C.A.; Honeycutt, J.L.; Cheung, E.; Reynolds, H.M.; Borski, R.J. Assessing the Functional Role of Leptin in Energy Homeostasis and the Stress Response in Vertebrates. Front. Endocrinol. 2017, 8, 63. [Google Scholar] [CrossRef] [Green Version]
- Montserrat-de la Paz, S.; Pérez-Pérez, A.; Vilariño-García, T.; Jiménez-Cortegana, C.; Muriana, F.J.G.; Millán-Linares, M.C.; Sánchez-Margalet, V. Nutritional modulation of leptin expression and leptin action in obesity and obesity-associated complications. J. Nutr. Biochem. 2021, 89, 108561. [Google Scholar] [CrossRef]
- Pérez-Pérez, A.; Toro, A.; Vilariño-García, T.; Maymó, J.; Guadix, P.; Dueñas, J.L.; Fernández-Sánchez, M.; Varone, C.; Sánchez-Margalet, V. Leptin action in normal and pathological pregnancies. J. Cell. Mol. Med. 2018, 22, 716–727. [Google Scholar] [CrossRef] [PubMed]
- Reid, I.R.; Baldock, P.A.; Cornish, J. Effect of leptin on the skeleton. Endocr. Rev. 2018, 39, 938–959. [Google Scholar] [CrossRef]
- Navarini, L.; Margiotta, D.P.E.; Vadacca, M.; Afeltra, A. Leptin in autoimmune mechanisms of systemic rheumatic diseases. Cancer Lett. 2018, 423, 139–146. [Google Scholar] [CrossRef]
- Sánchez-Jiménez, F.; Pérez-Pérez, A.; De la Cruz-Merino, L.; Sánchez-Margalet, V. Obesity and Breast cancer: Role of leptin. Front. Oncol. 2019, 9, 596. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Pérez, A.; Sánchez-Jiménez, F.; Vilariño-García, T.; Sánchez-Margalet, V. Role of leptin in inflammation and vice versa. Int. J. Mol. Sci. 2020, 21, 5887. [Google Scholar] [CrossRef] [PubMed]
- Polyzos, S.A.; Kountouras, J.; Mantzoros, C.S. Leptin in nonalcoholic fatty liver disease: A narrative review. Metabolism 2015, 64, 60–78. [Google Scholar] [CrossRef]
- Arab, J.P.; Arrese, M.; Trauner, M. Recent Insights into the Pathogenesis of Nonalcoholic Fatty Liver Disease. Annu. Rev. Pathol. Mech. Dis. 2018, 13, 321–350. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease—Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [Green Version]
- Diehl, A.M.; Day, C. Cause, Pathogenesis, and Treatment of Nonalcoholic Steatohepatitis. N. Engl. J. Med. 2017, 377, 2063–2072. [Google Scholar] [CrossRef]
- Di Sessa, A.; Cirillo, G.; Guarino, S.; Marzuillo, P.; Miraglia-del Giudice, E. Pediatric non-alcoholic fatty liver disease: Current perspectives on diagnosis and management. Pediatric Health Med. Ther. 2019, 10, 89–97. [Google Scholar] [CrossRef] [Green Version]
- Shiha, G.; Korenjak, M.; Eskridge, W.; Casanovas, T.; Velez-Moller, P.; Högström, S.; Richardson, B.; Munoz, C.; Siguroardóttir, S.; Coulibaly, A.; et al. Redefining fatty liver disease: An international patient perspective. Lancet Gastroenterol. Hepatol. 2020, 6, 73–79. [Google Scholar] [CrossRef]
- Lanuza, F.; Sapunar, J.; Hofmann, E. Management of non-alcoholic fatty liver disease. Rev. Med. Chil. 2018, 146, 894–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunt, E.M.; Wong, V.W.-S.; Nobili, V.; Day, C.P.; Sookoian, S.; Maher, J.J.; Bugianesi, E.; Sirlin, C.B.; Neuschwander-Tetri, B.A.; Rinella, M.E. Non-alcoholic fatty liver disease. Nat. Rev. Dis. Primers 2015, 1, 15080. [Google Scholar] [CrossRef] [PubMed]
- Flier, J.S. Obesity Wars: Molecular Progress Confronts an Expanding Epidemic. Cell 2004, 116, 337–350. [Google Scholar] [CrossRef] [Green Version]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef]
- Estes, C.; Anstee, Q.M.; Arias-Loste, M.T.; Bantel, H.; Bellentani, S.; Caballeria, J.; Colombo, M.; Craxi, A.; Crespo, J.; Day, C.P.; et al. Modeling NAFLD disease burden in China, France, Germany, Italy, Japan, Spain, United Kingdom, and United States for the period 2016–2030. J. Hepatol. 2018, 69, 896–904. [Google Scholar] [CrossRef]
- Jacobs, M.; Van Greevenbroek, M.M.J.; Van der Kallen, C.J.H.; Ferreira, I.; Feskens, E.J.M.; Jansen, E.H.J.M.; Schalkwijk, C.G.; Stehouwer, C. The association between the metabolic syndrome and alanine amino transferase is mediated by insulin resistance via related metabolic intermediates (the Cohort on Diabetes and Atherosclerosis Maastricht [CODAM] study). Metabolism 2011, 60, 969–975. [Google Scholar] [CrossRef]
- Polyzos, S.A.; Aronis, K.N.; Kountouras, J.; Raptis, D.D.; Vasiloglou, M.F.; Mantzoros, C.S. Circulating leptin in non-alcoholic fatty liver disease: A systematic review and meta-analysis. Diabetologia 2016, 59, 30–43. [Google Scholar] [CrossRef]
- Mendez-Sanchez, N.; Arrese, M.; Gadano, A.; Oliveira, C.P.; Fassio, E.; Arab, J.P.; Chávez-Tapia, N.C.; Dirchwolf, M.; Torre, A.; Ridruejo, E.; et al. The Latin American Association for the Study of the Liver (ALEH) position statement on the redefinition of fatty liver disease. Lancet Gastroenterol. Hepatol. 2021, 6, 65–72. [Google Scholar] [CrossRef]
- The Lancet Gastroenterology & Hepatology. Redefining non-alcoholic fatty liver disease: What’s in a name? Lancet Gastroenterol. Hepatol. 2020, 5, 419. [Google Scholar] [CrossRef]
- Polyzos, S.A.; Kountouras, J.; Zavos, C. Nonalcoholic fatty liver disease: The pathogenetic roles of insulin resistance and adipocytokines. Curr. Mol. Med. 2009, 9, 299–314. [Google Scholar] [CrossRef] [PubMed]
- Bray, G.A. Obesity, a disorder of nutrient partitioning: The MONA LISA hypothesis. J. Nutr. 1991, 121, 1146–11462. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Una, M.; López-Mancheno, Y.; Diéguez, C.; Fernández-Rojo, M.A.; Novelle, M.G. Unraveling the Role of Leptin in Liver Function and Its Relationship with Liver Diseases. Int. J. Mol. Sci. 2020, 21, 9368. [Google Scholar] [CrossRef]
- Hossain, I.A.; Akter, S.; Rahman, M.K.; Ali, L. Gender specific association of serum leptin and insulinemic indices with nonalcoholic fatty liver disease in prediabetic subjects. PLoS ONE 2015, 10, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Cernea, S.; Roiban, A.L.; Both, E.; Huţanu, A. Serum leptin and leptin resistance correlations with NAFLD in patients with type 2 diabetes. Diabetes Metab. Res. Rev. 2018, 34, 1–11. [Google Scholar] [CrossRef]
- Angulo, P.; Alba, L.M.; Petrovic, L.M.; Adams, L.A.; Lindor, K.D.; Jensen, M.D. Leptin, insulin resistance, and liver fibrosis in human nonalcoholic fatty liver disease. J. Hepatol. 2004, 41, 943–949. [Google Scholar] [CrossRef]
- Chitturri, S.; Farrell, G.; Frost, L.; Kriketos, A.; Lin, R.; Fung, C.; Liddle, C.; Samarasinghe, D.; George, J. Serum leptin in NASH correlates with hepatic steatosis but not fibrosis: A manifestation of lipotoxicity? Hepatology 2002, 36, 403–409. [Google Scholar] [CrossRef]
- Ataseven, H.; Bahcecioglu, I.H.; Kuzu, N.; Yalniz, M.; Celebi, S.; Erensoy, A.; Ustundag, B. The levels of ghrelin, leptin, TNF-alpha, and IL-6 in liver cirrhosis and hepatocellular carcinoma due to HBV and HDV infection. Mediat. Inflamm. 2006, 2006, 78380. [Google Scholar] [CrossRef] [Green Version]
- Naveau, S.; Perlemuter, G.; Chaillet, M.; Raynard, B.; Balian, A.; Beuzen, F.; Portier, A.; Galanaud, P.; Emilie, D.; Chaput, J.-C. Serum leptin in patients with alcoholic liver disease. Alcohol. Clin. Exp. Res. 2006, 30, 1422–1428. [Google Scholar] [CrossRef] [PubMed]
- Ockenga, J.; Tietge, U.J.F.; Böker, K.H.W.; Manns, M.P.; Brabant, G.; Bahr, M.J. Distinct roles of free leptin, bound leptin and soluble leptin receptor during the metabolic-inflammatory response in patients with liver cirrhosis. Aliment. Pharmacol. Ther. 2007, 25, 1301–1309. [Google Scholar] [CrossRef] [PubMed]
- Ertle, J.; Dechene, A.; Sowa, J.P.; Penndorf, V.; Herzer, K.; Kaiser, G.; Schlaak, J.F.; Gerken, G.; Syn, W.-K.; Canbay, A. Non-alcoholic fatty liver disease progresses to hepatocellular carcinoma in the absence of apparent cirrhosis. Int. J. Cancer 2011, 128, 2436–2443. [Google Scholar] [CrossRef]
- Robertson, S.A.; Leinninger, G.M.; Myers, M.G., Jr. Molecular and neural mediators of leptin action. Physiol. Behav. 2008, 94, 637–642. [Google Scholar] [CrossRef] [Green Version]
- Wauman, J.; Zabeau, L.; Tavernier, J. The leptin receptor complex: Heavier than expected? Front. Endocrinol. 2017, 8, 30. [Google Scholar] [CrossRef] [Green Version]
- Park, H.K.; Ahima, R.S. Physiology of leptin: Energy homeostasis, neuroendocrine function and metabolism. Metabolism 2015, 64, 24–34. [Google Scholar] [CrossRef] [Green Version]
- Adolph, T.E.; Grander, C.; Grabherr, F.; Tilg, H. Adipokines and non-alcoholic fatty liver disease: Multiple interactions. Int. J. Mol. Sci. 2017, 18, 1649. [Google Scholar] [CrossRef] [Green Version]
- Myers, M.G.; Cowley, M.A.; Münzberg, H. Mechanisms of leptin action and leptin resistance. Annu. Rev. Physiol. 2008, 70, 537–556. [Google Scholar] [CrossRef] [Green Version]
- Münzberg, H.; Morrison, C.D. Structure, production and signaling of leptin. Metabolism 2015, 64, 13–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polyzos, S.A.; Kountouras, J.; Zavos, C.; Deretzi, G. The potential adverse role of leptin resistance in nonalcoholic fatty liver disease: A hypothesis based on critical review of the literature. J. Clin. Gastroenterol. 2011, 45, 50–54. [Google Scholar] [CrossRef] [PubMed]
- German, J.; Kim, F.; Schwartz, G.J.; Havel, P.J.; Rhodes, C.J.; Schwartz, M.W.; Morton, G.J. Hypothalamic leptin signaling regulates hepatic insulin sensitivity via a neurocircuit involving the vagus nerve. Endocrinology 2009, 150, 4502–4511. [Google Scholar] [CrossRef] [Green Version]
- Utzschneider, K.M.; Kahn, S.E. Review: The role of insulin resistance in nonalcoholic fatty liver disease. J. Clin. Endocrinol. Metab. 2006, 91, 4753–4761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bugianesi, E.; Moscatiello, S.; Ciaravella, M.F.; Marchesini, G. Insulin resistance in nonalcoholic fatty liver disease. Curr. Pharm. Des. 2010, 16, 1941–1951. [Google Scholar] [CrossRef]
- Khan, R.S.; Bril, F.; Cusi, K.; Newsome, P.N. Modulation of Insulin Resistance in Nonalcoholic Fatty Liver Disease. Hepatology 2019, 70, 711–724. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Chen, G.; Liu, W.; Yang, X.; Gao, J.; Huang, L.; Guan, H.; Li, Z.; Zheng, Z.; Li, M.; et al. Intramuscular injection of exogenous leptin induces adiposity, glucose intolerance and fatty liver by repressing the JAK2-STAT3/PI3K pathway in a rat model. Gen. Comp. Endocrinol. 2017, 252, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Sahin-Efe, A.; Upadhyay, J.; Ko, B.J.; Dincer, F.; Park, K.H.; Migdal, A.; Vokonas, P.; Mantzoros, C. Irisin and leptin concentrations in relation to obesity, and developing type 2 diabetes: A cross sectional and a prospective case-control study nested in the Normative Aging Study. Metabolism 2018, 79, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Polyzos, S.A.; Kountouras, J.; Zavos, C.; Stergiopoulos, C. Adipocytokines in insulin resistance and non-alcoholic fatty liver disease: The two sides of the same coin. Med. Hypotheses. 2010, 74, 1089–1090. [Google Scholar] [CrossRef]
- Polyzos, S.A.; Perakakis, N.; Mantzoros, C.S. Fatty liver in lipodystrophy: A review with a focus on therapeutic perspectives of adiponectin and/or leptin replacement. Metabolism 2019, 96, 66–82. [Google Scholar] [CrossRef]
- Chan, J.L.; Lutz, K.; Cochran, E.; Huang, W.; Peters, Y.; Weyer, C.; Gorden, P. Clinical effects of long-term metreleptin treatment in patients with lipodystrophy. Endocr. Pract. 2011, 17, 922–932. [Google Scholar] [CrossRef] [Green Version]
- Polyzos, S.A.; Mantzoros, C.S. Leptin in Health and Disease: Facts and Expectations at its Twentieth Anniversary. Metabolism 2015, 64, 5–12. [Google Scholar] [CrossRef]
- Boutari, C.; Mantzoros, C.S. Adiponectin and leptin in the diagnosis and therapy of NAFLD. Metabolism 2020, 103, 154028. [Google Scholar] [CrossRef]
- DePaoli, A.M. Leptin in common obesity and associated disorders of metabolism. J. Endocrinol. 2014, 223, T71–T81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; He, W.; Tsai, P.-J.; Chen, P.-H.; Ye, M.; Guo, J.; Su, Z. Mutual interaction between endoplasmic reticulum and mitochondria in nonalcoholic fatty liver disease. Lipids Health Dis. 2020, 19, 72. [Google Scholar] [CrossRef] [Green Version]
- Hirsova, P.; Gores, G.J. Death Receptor-Mediated Cell Death and Proinflammatory Signaling in Nonalcoholic Steatohepatitis. Cell. Mol. Gastroenterol. Hepatol. 2015, 1, 17–27. [Google Scholar] [CrossRef] [Green Version]
- Moon, H.-S.; Dalamaga, M.; Kim, S.-Y.; Polyzos, S.A.; Hamnvik, O.-P.; Magkos, F.; Paruthi, J.; Mantzoros, C.S. Leptin’s role in lipodystrophic and nonlipodystrophic insulin-resistant and diabetic individuals. Endocr. Rev. 2013, 34, 377–412. [Google Scholar] [CrossRef] [Green Version]
- Asilmaz, E.; Cohen, P.; Miyazaki, M.; Dobrzyn, P.; Ueki, K.; Fayzikhodjaeva, G.; Soukas, A.A.; Kahn, C.R.; Ntambi, J.M.; Socci, N.D.; et al. Site and mechanism of leptin action in a rodent form of congenital lipodystrophy. J. Clin. Investig. 2004, 113, 414–424. [Google Scholar] [CrossRef]
- Wang, M.-Y.; Chen, L.; Clark, G.O.; Lee, Y.; Stevens, R.D.; Ilkayeva, O.R.; Wenner, B.R.; Bain, J.R.; Charron, M.J.; Newgard, C.B.; et al. Leptin therapy in insulin-deficient type I diabetes. Proc. Natl. Acad. Sci. USA 2010, 107, 4813–4819. [Google Scholar] [CrossRef] [Green Version]
- Denechaud, P.-D.; Dentin, R.; Girard, J.; Postic, C. Role of ChREBP in hepatic steatosis and insulin resistance. FEBS Lett. 2008, 582, 68–73. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.; Metlakunta, A.; Dedousis, N.; Ortmeyer, H.K.; Stefanovic-Racic, M.; O’Doherty, R.M. Leptin augments the acute suppressive effects of insulin on hepatic very low-density lipoprotein production in rats. Endocrinology 2009, 150, 2169–2174. [Google Scholar] [CrossRef] [Green Version]
- Cortés, V.A.; Cautivo, K.M.; Rong, S.; Garg, A.; Horton, J.D.; Agarwal, A.K. Leptin ameliorates insulin resistance and hepatic steatosis in Agpat2-/- lipodystrophic mice independent of hepatocyte leptin receptors. J. Lipid Res. 2014, 55, 276–288. [Google Scholar] [CrossRef] [Green Version]
- Hackl, M.T.; Fürnsinn, C.; Schuh, C.M.; Krssak, M.; Carli, F.; Guerra, S.; Freudenthaler, A.; Baumgartner-Parzer, S.; Helbich, T.H.; Luger, A.; et al. Brain leptin reduces liver lipids by increasing hepatic triglyceride secretion and lowering lipogenesis. Nat. Commun. 2019, 10, 2717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, X.; Li, J.; Xiang, W.; Cui, Y.; Xie, B.; Wang, X.; Xu, Z.; Gan, L. Metformin increases hepatic leptin receptor and decreases steatosis in mice. J. Endocrinol. 2016, 230, 227–237. [Google Scholar] [CrossRef] [Green Version]
- Lim, D.-W.; Bose, S.; Wang, J.-H.; Choi, H.S.; Kim, Y.-M.; Chin, Y.-M.; Jeon, S.-H.; Kim, J.-F.; Kim, H. Modified SJH alleviates FFAs-induced hepatic steatosis through leptin signaling pathways. Sci. Rep. 2017, 7, 45425. [Google Scholar] [CrossRef] [Green Version]
- Vernon, G.; Baranova, A.; Younossi, Z.M. Systematic review: The epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment. Pharmacol. Ther. 2011, 34, 274–285. [Google Scholar] [CrossRef]
- Samuel, V.T.; Shulman, G.I. Mechanisms for insulin resistance: Common threads and missing links. Cell 2012, 148, 852–871. [Google Scholar] [CrossRef] [Green Version]
- Rotundo, L.; Persaud, A.; Feurdean, M.; Ahlawat, S.; Kim, H.S. The Association of leptin with severity of non-alcoholic fatty liver disease: A population-based study. Clin. Mol. Hepatol. 2018, 24, 392–401. [Google Scholar] [CrossRef] [Green Version]
- Pavlidis, C.; Panoutsopoulos, G.I.; Tiniakos, D.; Koutsounas, S.; Vlachogiannakos, J.; Zouboulis-Vafiadis, I. Serum leptin and ghrelin in chronic hepatitis C patients with steatosis. World J. Gastroenterol. 2011, 17, 5097–5104. [Google Scholar] [CrossRef] [PubMed]
- Eshraghian, A.; Nikeghbalian, S.; Shamsaeefar, A.; Kazemi, K.; Fattahi, M.R.; Malek-Hosseini, S.A. Hepatic steatosis and liver fat contents in liver transplant recipients are associated with serum adipokines and insulin resistance. Sci. Rep. 2020, 10, 12701. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Adolph, T.E.; Moschen, A.R. Multiple Parallel Hits Hypothesis in Nonalcoholic Fatty Liver Disease: Revisited After a Decade. Hepatology 2021, 73, 833–842. [Google Scholar] [CrossRef]
- Hirsova, P.; Ibrahim, S.H.; Gores, G.J.; Malhi, H. Lipotoxic lethal and sublethal stress signaling in hepatocytes: Relevance to NASH pathogenesis. J. Lipid. Res. 2016, 57, 1758–1770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuppan, D.; Surabattula, R.; Wang, X.Y. Determinants of fibrosis progression and regression in NASH. J. Hepatol. 2018, 68, 238–250. [Google Scholar] [CrossRef]
- Stal, P. Liver fibrosis in non-alcoholic fatty liver disease-diagnostic challenge with prognostic significance. World J. Gastroenterol. 2015, 21, 11077–11087. [Google Scholar] [CrossRef]
- Koyama, Y.; Brenner, D.A. Liver inflammation and fibrosis. J. Clin. Investig. 2017, 127, 55–64. [Google Scholar] [CrossRef]
- Chatterjee, S.; Ganini, D.; Tokar, E.J.; Kumar, A.; Das, S.; Corbett, J.; Kaddiska, M.B.; Waalkes, M.P.; Diehl, A.M.; Mason, R.P. Leptin is key to peroxynitrite-mediated oxidative stress and Kupffer cell activation in experimental non-alcoholic steatohepatitis. J. Hepatol. 2013, 58, 778–784. [Google Scholar] [CrossRef] [Green Version]
- Seth, R.K.; Das, S.; Kumar, A.; Chanda, A.; Kadiiska, M.B.; Michelotti, G.; Manautou, J.; Diehl, A.M.; Chatterjee, S. CYP2E1-dependent and leptin-mediated hepatic CD57 expression on CD8+ T cells aid progression of environment-linked nonalcoholic steatohepatitis. Toxicol. Appl. Pharmacol. 2014, 274, 42–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mundi, M.S.; Velapati, S.; Patel, J.; Kellogg, T.A.; Abu-Dayyeh, B.K.; Hurt, R.T. Evolution of NAFLD and Its Management. Nutr. Clin. Pract. 2020, 35, 72–84. [Google Scholar] [CrossRef]
- Procaccini, C.; Galgani, M.; De Rosa, V.; Carbone, F.; La Rocca, C.; Ranucci, G.; Iorio, R.; Matarese, G. Leptin: The prototypic adipocytokine and its role in NAFLD. Curr. Pharm. Des. 2010, 16, 1902–1912. [Google Scholar] [CrossRef]
- Otte, C.; Otte, J.-M.; Strodthoff, D.; Bornstein, S.R.; Fölsch, U.R.; Mönig, H.; Kloehn, S. Expression of leptin and leptin receptor during the development of liver fibrosis and cirrhosis. Exp. Clin. Endocrinol. Diabetes 2004, 112, 10–17. [Google Scholar] [CrossRef]
- Sanyal, A.J. Past, present and future perspectives in nonalcoholic fatty liver disease. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 377–386. [Google Scholar] [CrossRef]
- Duan, X.F.; Tang, P.; Li, Q.; Yu, Z.T. Obesity, adipokines and hepatocellular carcinoma. Int. J. Cancer 2013, 133, 1776–1783. [Google Scholar] [CrossRef] [Green Version]
- Vanni, E.; Bugianesi, E. Obesity and liver cancer. Clin. Liver Dis. 2014, 18, 191–203. [Google Scholar] [CrossRef]
- Wang, S.-N.; Lee, K.-T.; Ker, C.-G. Leptin in hepatocellular carcinoma. World J. Gastroenterol. 2010, 16, 5801–5809. [Google Scholar] [CrossRef]
- Zhang, L.; Yuan, Q.; Li, M.; Chai, D.; Deng, W.; Wang, W. The association of leptin and adiponectin with hepatocellular carcinoma risk and prognosis: A combination of traditional, survival, and dose-response meta-analysis. BMC Cancer 2020, 20, 1167. [Google Scholar] [CrossRef]
- Ribatti, D.; Belloni, A.S.; Nico, B.; Di Comite, M.; Crivellato, E.; Vacca, A. Leptin-leptin receptor are involved in angiogenesis in human hepatocellular carcinoma. Peptides 2008, 29, 1596–1602. [Google Scholar] [CrossRef]
- Saxena, N.K.; Titus, M.A.; Ding, X.; Floyd, J.; Srinivasan, S.; Sitaraman, S.V.; Anania, F.A. Leptin as a novel profibrogenic cytokine in hepatic stellate cells: Mitogenesis and inhibition of apoptosis mediated by extracellular regulated kinase (Erk) and Akt phosphorylation. FASEB J. 2004, 18, 1612–1614. [Google Scholar] [CrossRef]
- Kitade, M.; Yoshiji, H.; Kojima, H.; Ikenaka, Y.; Noguchi, R.; Kaji, K.; Yoshii, J.; Yanase, K.; Namisaki, T.; Asada, K.; et al. Leptin-mediated neovascularization is a prerequisite for progression of nonalcoholic steatohepatitis in rats. Hepatology 2006, 44, 983–991. [Google Scholar] [CrossRef]
- Aleffi, S.; Petrai, I.; Bertolani, C.; Parola, M.; Colombatto, S.; Novo, E.; Vizzutti, F.; Anania, F.A.; Milani, S.; Rombouts, K.; et al. Upregulation of proinflammatory and proangiogenic cytokines by leptin in human hepatic stellate cells. Hepatology 2005, 42, 1339–1348. [Google Scholar] [CrossRef] [PubMed]
- Sadik, N.A.; Ahmed, A.; Ahmed, S. The significance of serum levels of adiponectin, leptin, and hyaluronic acid in hepatocellular carcinoma of cirrhotic and noncirrhotic patients. Hum. Exp. Toxicol. 2012, 31, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Mittenbühler, M.J.; Sprenger, H.-G.; Gruber, S.; Wunderlich, C.M.; Kern, L.; Brüning, J.C.; Wunderich, F.T. Hepatic leptin receptor expression can partially compensate for IL-6Rα deficiency in DEN-induced hepatocellular carcinoma. Mol. Metab. 2018, 17, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Zhan, J.; Ling, F.; Huang, Y.; Yang, M.; Zhang, Y.; Wei, Y.; Zhang, Q.; Wang, H.; Song, L.; et al. Leptin Receptor (LEPR) promotes proliferation, migration, and invasion and inhibits apoptosis in hepatocellular carcinoma by regulating ANXA7. Cancer Cell Int. 2021, 21, 4. [Google Scholar] [CrossRef] [PubMed]
- Elinav, E.; Abd-Elnabi, A.; Pappo, O.; Bernstein, I.; Klein, A.; Engelhardt, D.; Rabbani, E.; Ilan, Y. Supression of hepatocellular carcinoma growth in mice via leptin is associated with inhibition of tumor cell growth and natural killer cell activation. J. Hepatol. 2006, 44, 529–536. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.N.; Yeh, Y.T.; Yang, S.F.; Chai, C.Y.; Lee, K.T. Potential role of leptin expression in hepatocellular carcinoma. J. Clin. Pathol. 2006, 59, 930–934. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.N.; Chuang, S.C.; Yeh, Y.T.; Yang, S.F.; Chai, C.Y.; Chen, W.T.; Kuo, K.K.; Chen, J.S.; Lee, K.T. Potential prognostic value of leptin receptor in hepatocellular carcinoma. J. Clin. Pathol. 2006, 59, 1267–1271. [Google Scholar] [CrossRef] [PubMed]

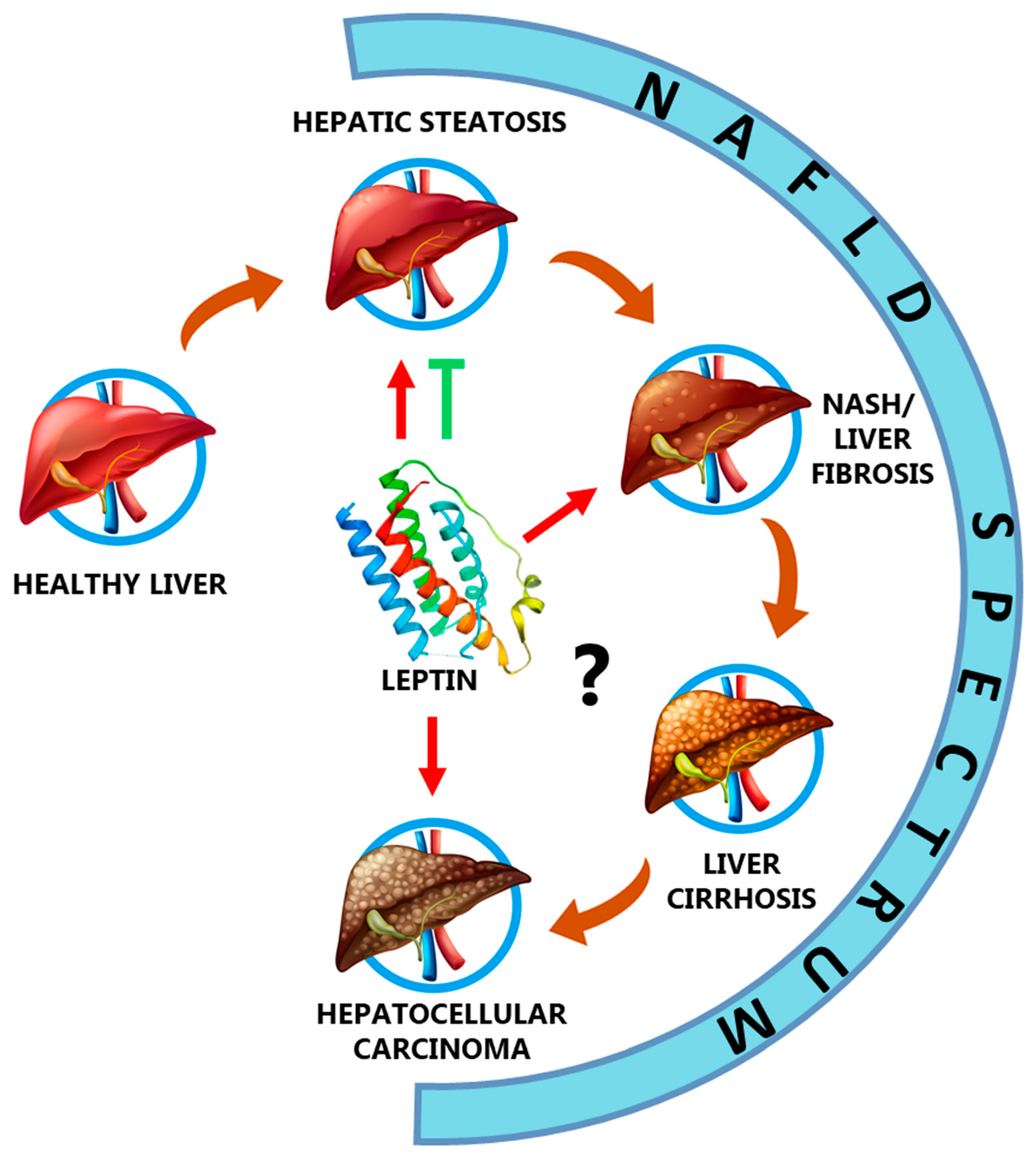

{kind=link}
{kind=link}
| Author (Year) | Country | No Patients | Conclusions |
|---|---|---|---|
| Jacobs et al. (2011) [29] | Netherlands | 434 | Insulin resistance (IR) mediated between 75–80% of the association of the metabolic syndrome with alanine aminotransferase, as well as suggesting that IR, adipose tissue inflammation and endothelial dysfunction may contribute to NAFLD progression. |
| Hossain et al. (2015) [36] | Bangladesh | 110 | IR was independently associated with serum leptin levels irrespective of adiposity and glycemic status in male prediabetic subjects. In addition, serum leptin was increased in the female patients, accompanied by pancreatic beta cell dysfunction and IR. However, their relationship with NAFLD was not affected by the degree of adiposity. |
| Cernea et al. (2018) [37] | Romania | 159 | Hepatic steatosis was positively correlated with serum leptin and leptin resistance, and negatively with serum Ob-R. Leptin/Ob-R, and leptin resistance did not made a significant contribution to hepatic fibrosis. |
| Angulo et al. (2004) [38] | U.S.A. | 88 | There was no association between serum leptin and hepatic fibrosis. However, there was a correlation between leptin with more advanced NAFLD-related liver fibrosis. |
| Chitturi et al. (2002) [39] | Australia | 36 patients and 47 controls | Hyperleptinemia in NASH was correlated with some factors (e.g., age and extent of hepatic steatosis), but not with inflammation or fibrotic severity. |
| Ataseven et al. (2006) [40] | Turkey | 45 patients (23 cirrhosis + 22 HCC) and 25 controls | In cirrhosis and HCC patients there was a decrease of serum leptin levels due to, at least partly, the presence of nutritional and metabolic abnormalities, including malnutrition, and high ghrelin levels. |
| Naveau et al. (2006) [41] | France | 209 | Serum leptin was independently correlated with steatosis and may play an important role in severity of fibrosis. |
| Ockenga et al. (2007) [42] | Germany | 40 liver cirrhosis + 31 controls | Patients had bound leptin and soluble leptin receptor levels significantly increased compared with controls, without changes in free leptin. |
| Ertle et al. (2011) [43] | Germany | 162 | NAFLD/NASH posed a risk factor for HCC, even in the absence of cirrhosis. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiménez-Cortegana, C.; García-Galey, A.; Tami, M.; del Pino, P.; Carmona, I.; López, S.; Alba, G.; Sánchez-Margalet, V. Role of Leptin in Non-Alcoholic Fatty Liver Disease. Biomedicines 2021, 9, 762. https://doi.org/10.3390/biomedicines9070762
Jiménez-Cortegana C, García-Galey A, Tami M, del Pino P, Carmona I, López S, Alba G, Sánchez-Margalet V. Role of Leptin in Non-Alcoholic Fatty Liver Disease. Biomedicines. 2021; 9(7):762. https://doi.org/10.3390/biomedicines9070762
Chicago/Turabian StyleJiménez-Cortegana, Carlos, Alba García-Galey, Malika Tami, Pilar del Pino, Isabel Carmona, Soledad López, Gonzalo Alba, and Víctor Sánchez-Margalet. 2021. "Role of Leptin in Non-Alcoholic Fatty Liver Disease" Biomedicines 9, no. 7: 762. https://doi.org/10.3390/biomedicines9070762
APA StyleJiménez-Cortegana, C., García-Galey, A., Tami, M., del Pino, P., Carmona, I., López, S., Alba, G., & Sánchez-Margalet, V. (2021). Role of Leptin in Non-Alcoholic Fatty Liver Disease. Biomedicines, 9(7), 762. https://doi.org/10.3390/biomedicines9070762