
Sugar Fructose Triggers Gut Dysbiosis and Metabolic Inflammation with Cardiac Arrhythmogenesis

Abstract
:1. Introduction
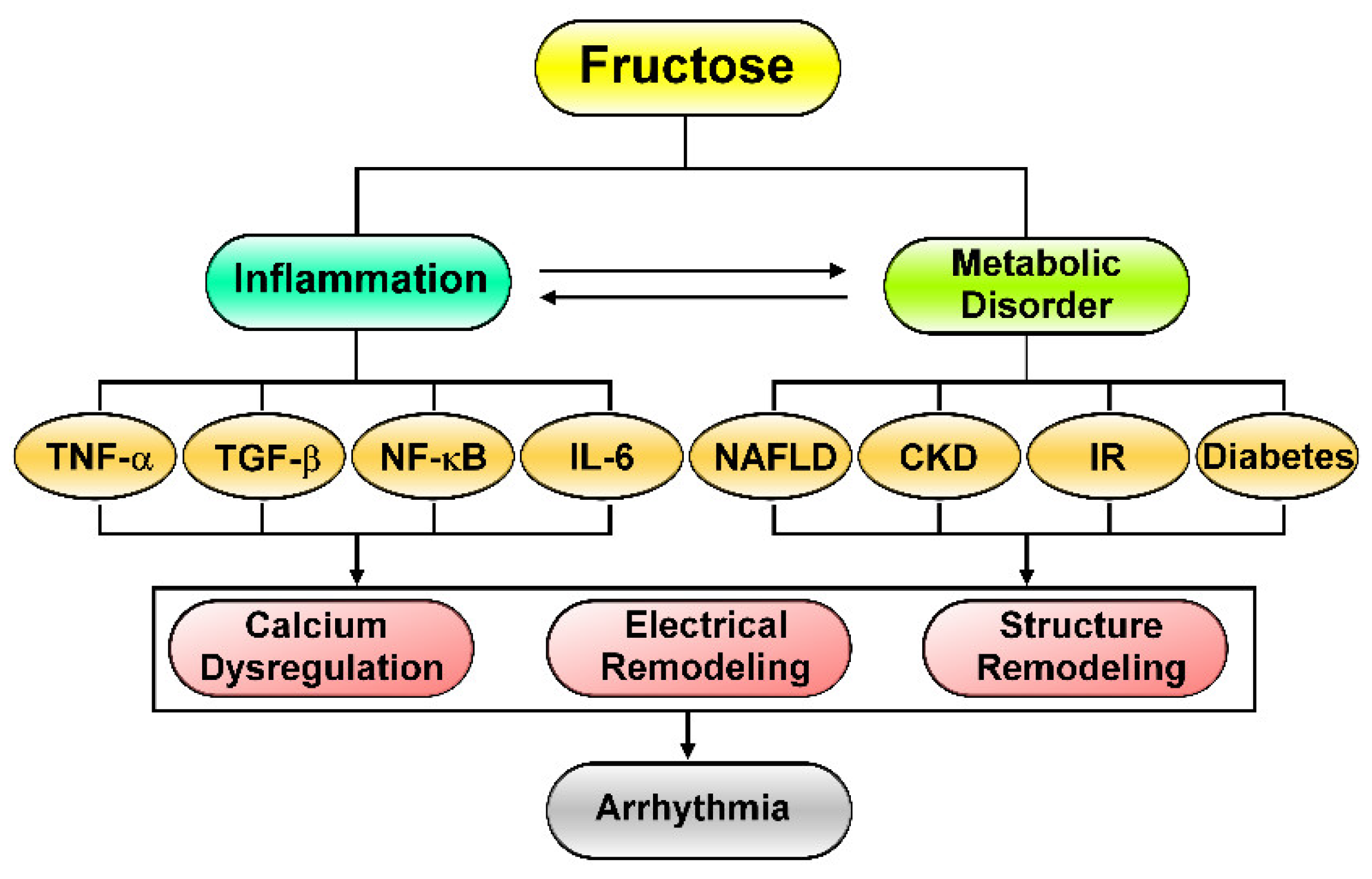
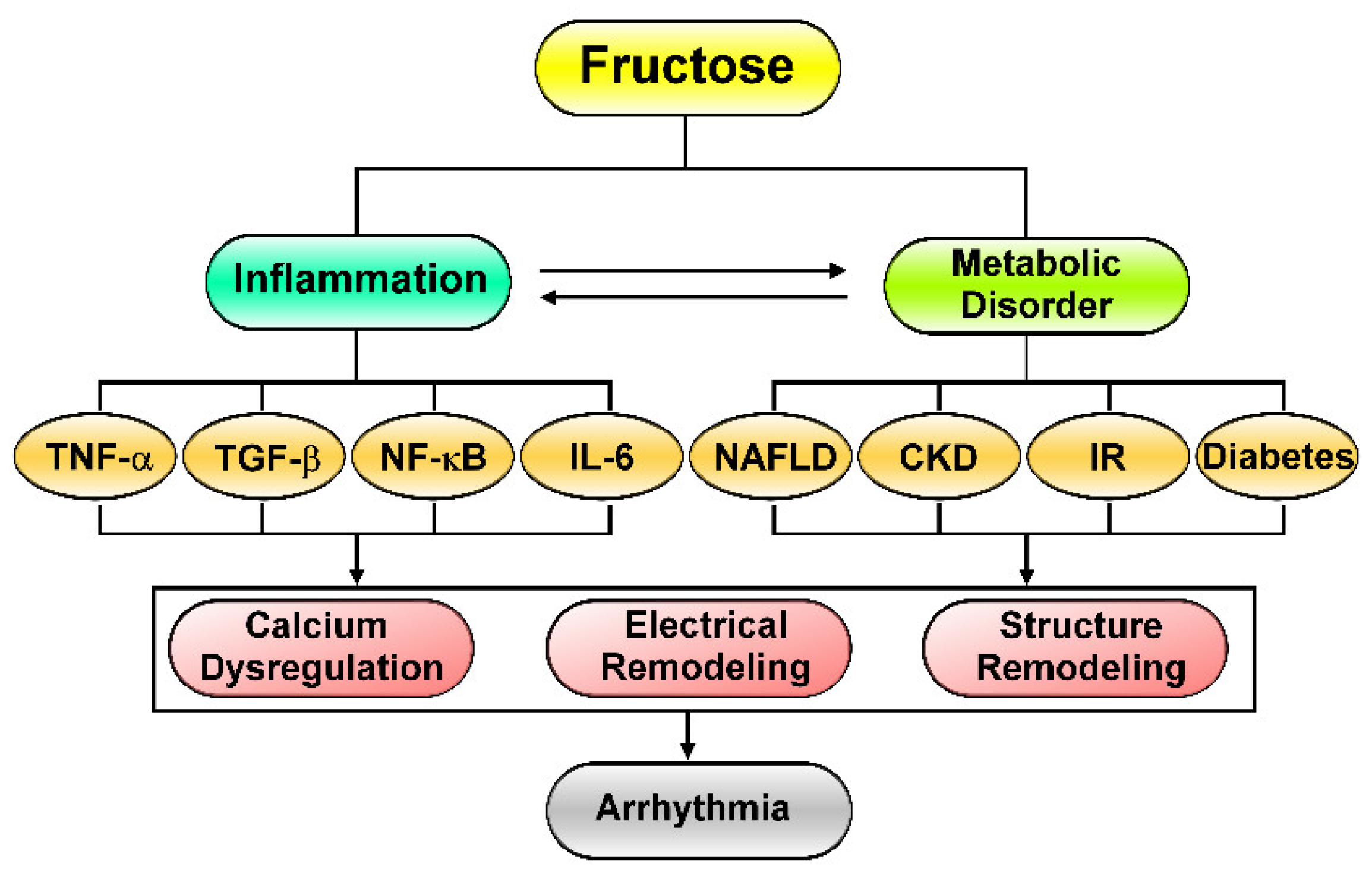
2. Fructose-Mediated Metabolic Disorders as a Driver of Cardiac Arrhythmia
3. Effects of Excess Fructose Intake on Cardiac Arrhythmogenesis
4. Fructose’s Effects on Cardiac Inflammation and Fibrosis
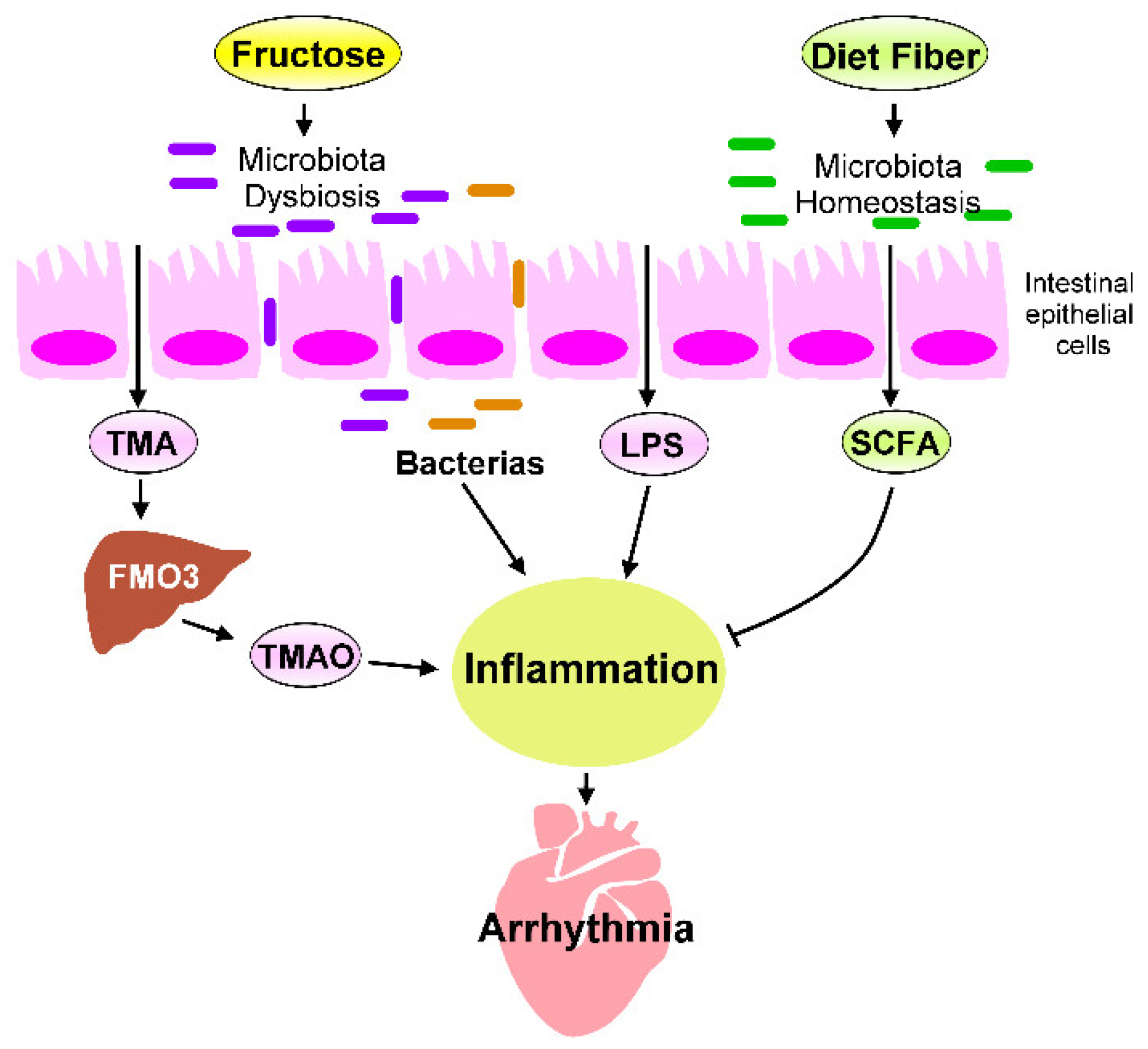
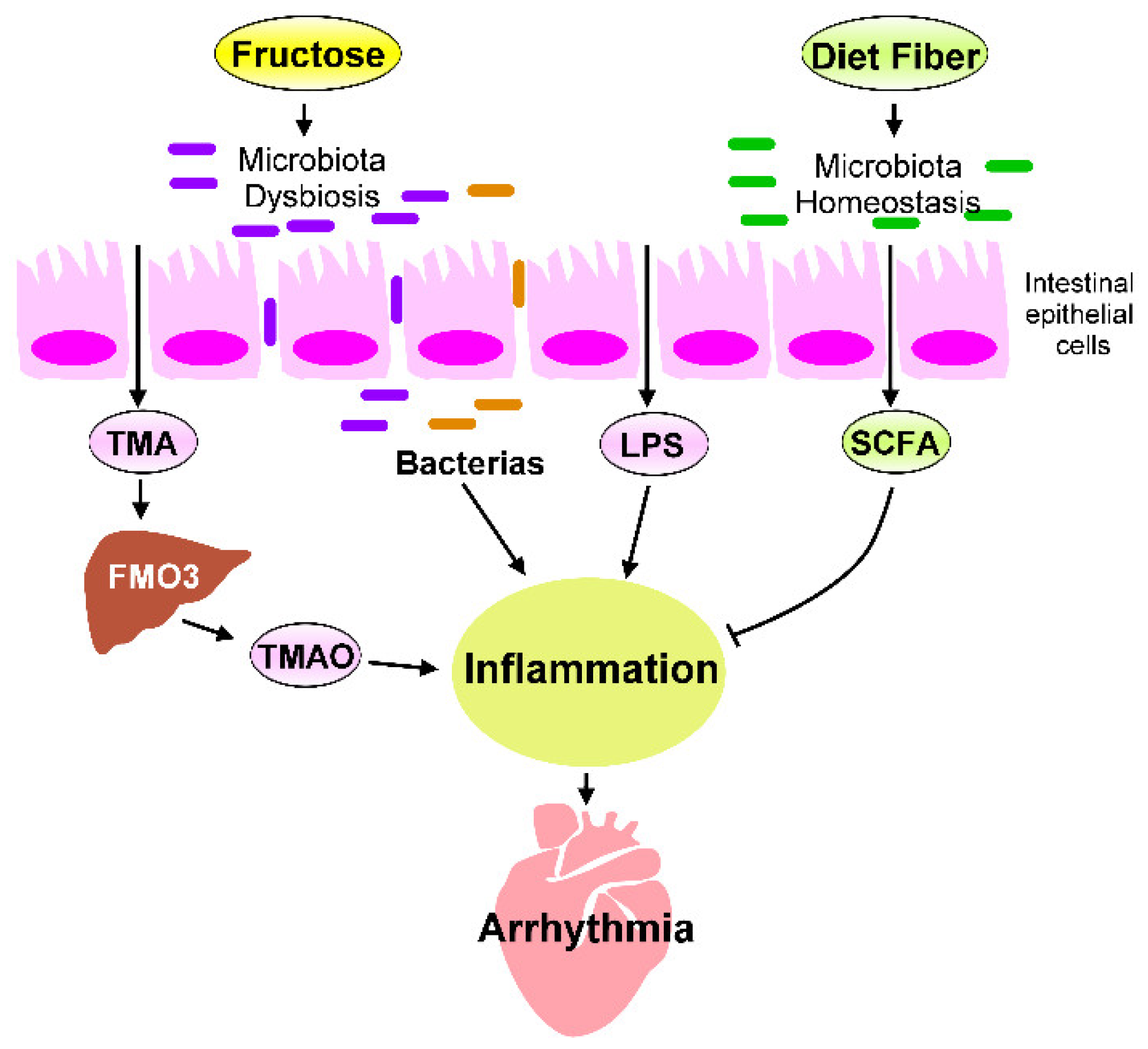
5. Heart–Gut Axis Regulates Fructose-Induced Cardiac Inflammation
5.1. Microbiota Dysbiosis in Fructose-Mediated Cardiac Arrhythmia
5.2. Effects of Microbial Metabolites on Fructose-Mediated Inflammation
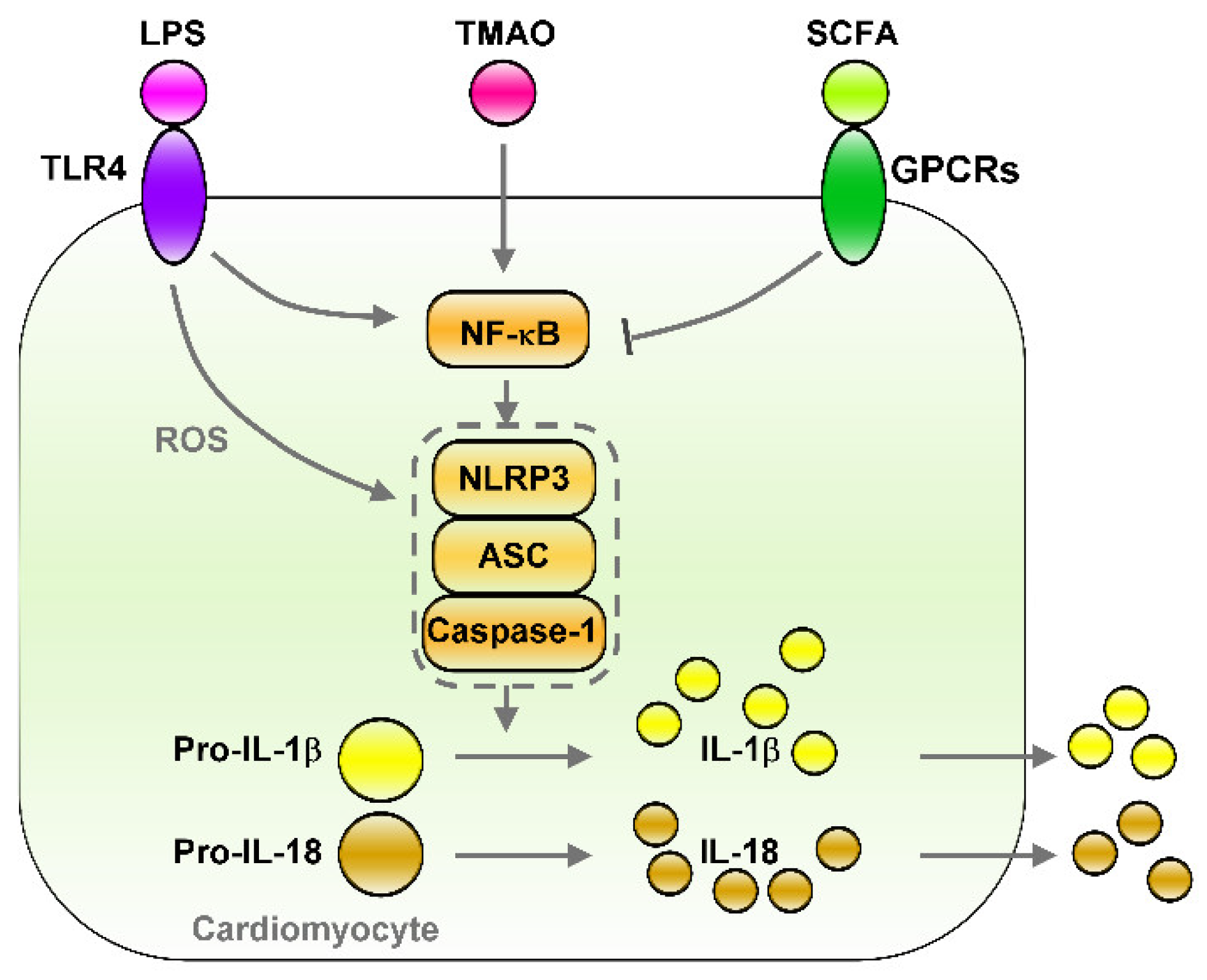
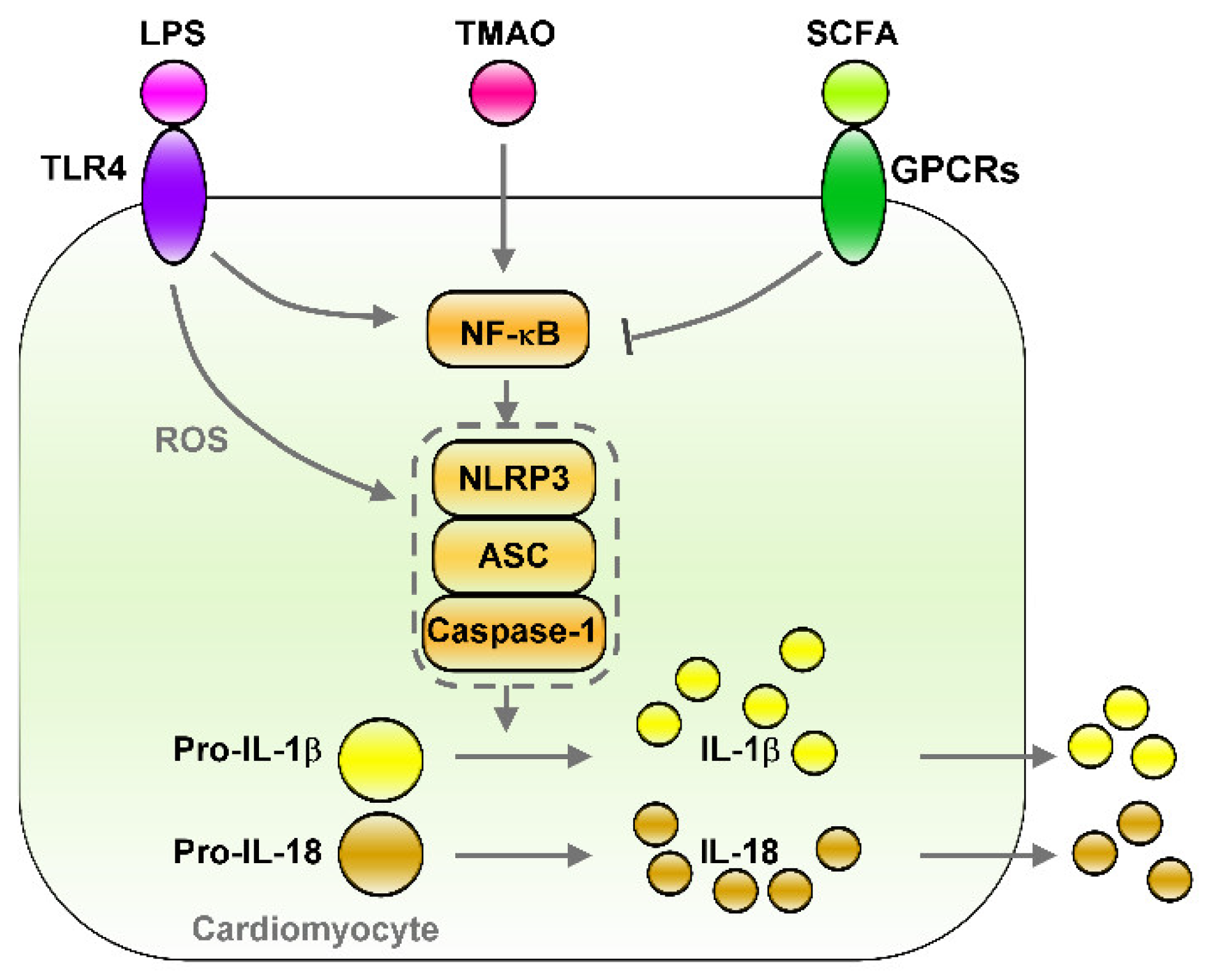
5.3. The LPS–TLRs Axis Mediates Inflammation
5.4. TMAO Induces Inflammation
6. Therapeutic Strategies for Fructose-Mediated Inflammation
6.1. Dietary Interventions
6.2. Probiotics for Controlling Cardiac Inflammation
6.3. Effects of SCFAs on Controlling Inflammation
6.4. HDACs’ Inhibition of Cardiac Inflammation
7. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Howard, B.V.; Wylie-Rosett, J. Sugar and cardiovascular disease: A statement for healthcare professionals from the Committee on Nutrition of the Council on Nutrition, Physical Activity, and Metabolism of the American Heart Association. Circulation 2002, 106, 523–527. [Google Scholar] [CrossRef] [Green Version]
- Mirtschink, P.; Jang, C.; Arany, Z.; Krek, W. Fructose metabolism, cardiometabolic risk, and the epidemic of coronary artery disease. Eur. Heart J. 2018, 39, 2497–2505. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.K.; Appel, L.J.; Brands, M.; Howard, B.V.; Lefevre, M.; Lustig, R.H.; Sacks, F.; Steffen, L.M.; Wylie-Rosett, J.; American Heart Association Nutrition Committee of the Council on Nutrition; et al. Dietary sugars intake and cardiovascular health: A scientific statement from the American Heart Association. Circulation 2009, 120, 1011–1020. [Google Scholar] [CrossRef] [PubMed]
- Bray, G.A. Fructose and risk of cardiometabolic disease. Curr. Atheroscler. Rep. 2012, 14, 570–578. [Google Scholar] [CrossRef] [PubMed]
- Vartanian, L.R.; Schwartz, M.B.; Brownell, K.D. Effects of soft drink consumption on nutrition and health: A systematic review and meta-analysis. Am. J. Public Health 2007, 97, 667–675. [Google Scholar] [CrossRef]
- Bray, G.A. Fructose: Pure, white, and deadly? Fructose, by any other name, is a health hazard. J. Diabetes Sci. Technol. 2010, 4, 1003–1007. [Google Scholar]
- Malik, V.S.; Hu, F.B. Fructose and cardiometabolic health: What the evidence from sugar-sweetened beverages tells us. J. Am. Coll. Cardiol. 2015, 66, 1615–1624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malik, V.S.; Hu, F.B. Sugar-sweetened beverages and cardiometabolic health: An update of the evidence. Nutrients 2019, 11, 8. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.M.; Jiao, R.Q.; Kong, L.D. High dietary fructose: Direct or indirect dangerous factors disturbing tissue and organ functions. Nutrients 2017, 9, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porto, M.L.; Lirio, L.M.; Dias, A.T.; Batista, A.T.; Campagnaro, B.P.; Mill, J.G.; Meyrelles, S.S.; Baldo, M.P. Increased oxidative stress and apoptosis in peripheral blood mononuclear cells of fructose-fed rats. Toxicol. In Vitro 2015, 29, 1977–1981. [Google Scholar] [CrossRef]
- Chan, W.; Smith, B.; Stegall, M.; Borrows, R. Obesity and metabolic syndrome in kidney transplantation: The role of dietary fructose and systemic endotoxemia. Transplantation 2019, 103, 191–201. [Google Scholar] [CrossRef]
- Miller, A.; Adeli, K. Dietary fructose and the metabolic syndrome. Curr. Opin. Gastroenterol. 2008, 24, 204–209. [Google Scholar] [CrossRef] [Green Version]
- Havel, P.J. Dietary fructose: Implications for dysregulation of energy homeostasis and lipid/carbohydrate metabolism. Nutr. Rev. 2005, 63, 133–157. [Google Scholar] [CrossRef]
- Gross, L.S.; Li, L.; Ford, E.S.; Liu, S. Increased consumption of refined carbohydrates and the epidemic of type 2 diabetes in the United States: An ecologic assessment. Am. J. Clin. Nutr. 2004, 79, 774–779. [Google Scholar] [CrossRef]
- Elliott, S.S.; Keim, N.L.; Stern, J.S.; Teff, K.; Havel, P.J. Fructose, weight gain, and the insulin resistance syndrome. Am. J. Clin. Nutr. 2002, 76, 911–922. [Google Scholar] [CrossRef] [PubMed]
- Dhingra, R.; Sullivan, L.; Jacques, P.F.; Wang, T.J.; Fox, C.S.; Meigs, J.B.; D’Agostino, R.B.; Gaziano, J.M.; Vasan, R.S. Soft drink consumption and risk of developing cardiometabolic risk factors and the metabolic syndrome in middle-aged adults in the community. Circulation 2007, 116, 480–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caliceti, C.; Calabria, D.; Roda, A.; Cicero, A.F.G. Fructose intake, serum uric acid, and cardiometabolic disorders: A critical review. Nutrients 2017, 9, 4. [Google Scholar] [CrossRef] [PubMed]
- Jegatheesan, P.; De Bandt, J.P. Fructose and NAFLD: The multifaceted aspects of fructose metabolism. Nutrients 2017, 9, 230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diamond, D.M.; O’Neill, B.J.; Volek, J.S. Low carbohydrate diet: Are concerns with saturated fat, lipids, and cardiovascular disease risk justified? Curr. Opin. Endocrinol. Diabetes Obes. 2020, 27, 291–300. [Google Scholar] [CrossRef]
- Tappy, L. Q&A: ‘toxic’ effects of sugar: Should we be afraid of fructose? BMC Biol. 2012, 10, 42. [Google Scholar]
- Lambertz, J.; Weiskirchen, S.; Landert, S.; Weiskirchen, R. Fructose: A dietary sugar in crosstalk with microbiota contributing to the development and progression of non-alcoholic liver disease. Front. Immunol. 2017, 8, 1159. [Google Scholar] [CrossRef] [Green Version]
- Kretowicz, M.; Johnson, R.J.; Ishimoto, T.; Nakagawa, T.; Manitius, J. The impact of fructose on renal function and blood pressure. Int. J. Nephrol. 2011, 2011, 315879. [Google Scholar] [CrossRef] [Green Version]
- DiNicolantonio, J.J.; O’Keefe, J.H.; Lucan, S.C. Added fructose: A principal driver of type 2 diabetes mellitus and its consequences. Mayo Clin. Proc. 2015, 90, 372–381. [Google Scholar] [CrossRef] [Green Version]
- Panchal, S.K.; Brown, L. Rodent models for metabolic syndrome research. J. Biomed. Biotechnol. 2011, 2011, 351982. [Google Scholar] [CrossRef] [Green Version]
- Sodhi, K.; Puri, N.; Favero, G.; Stevens, S.; Meadows, C.; Abraham, N.G.; Rezzani, R.; Ansinelli, H.; Lebovics, E.; Shapiro, J.I. Fructose mediated non-alcoholic fatty liver is attenuated by HO-1-SIRT1 module in murine hepatocytes and mice fed a high fructose diet. PLoS ONE 2015, 10, e0128648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarz, J.M.; Noworolski, S.M.; Erkin-Cakmak, A.; Korn, N.J.; Wen, M.J.; Tai, V.W.; Jones, G.M.; Palii, S.P.; Velasco-Alin, M.; Pan, K.; et al. Effects of dietary fructose restriction on liver fat, de novo lipogenesis, and insulin kinetics in children with obesity. Gastroenterology 2017, 153, 743–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albillos, A.; de Gottardi, A.; Rescigno, M. The gut-liver axis in liver disease: Pathophysiological basis for therapy. J. Hepatol. 2020, 72, 558–577. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, A. Nonalcoholic fatty liver disease (NAFLD) and risk of cardiac arrhythmias: A new aspect of the liver-heart axis. J. Clin. Transl. Hepatol. 2017, 5, 134–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ismaiel, A.; Colosi, H.A.; Rusu, F.; Dumitrascu, D.L. Cardiac arrhythmias and electrocardiogram modifications in non-alcoholic fatty liver disease. A systematic review. J. Gastrointestin. Liver Dis. 2019, 28, 483–493. [Google Scholar] [CrossRef]
- Roselli, C.; Rienstra, M.; Ellinor, P.T. Genetics of atrial fibrillation in 2020: GWAS, genome sequencing, polygenic risk, and beyond. Circ. Res. 2020, 127, 21–33. [Google Scholar] [CrossRef]
- Mantovani, A.; Dauriz, M.; Sandri, D.; Bonapace, S.; Zoppini, G.; Tilg, H.; Byrne, C.D.; Targher, G. Association between non-alcoholic fatty liver disease and risk of atrial fibrillation in adult individuals: An updated meta-analysis. Liver Int. 2019, 39, 758–769. [Google Scholar] [CrossRef] [Green Version]
- Johnson, R.J.; Sanchez-Lozada, L.G.; Nakagawa, T. The effect of fructose on renal biology and disease. J. Am. Soc. Nephrol. 2010, 21, 2036–2039. [Google Scholar] [CrossRef] [PubMed]
- Chan, Y.H.; Chang, G.J.; Lai, Y.J.; Chen, W.J.; Chang, S.H.; Hung, L.M.; Kuo, C.T.; Yeh, Y.H. Atrial fibrillation and its arrhythmogenesis associated with insulin resistance. Cardiovasc. Diabetol. 2019, 18, 125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bohne, L.J.; Johnson, D.; Rose, R.A.; Wilton, S.B.; Gillis, A.M. The association between diabetes mellitus and atrial fibrillation: Clinical and mechanistic insights. Front. Physiol. 2019, 10, 135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dublin, S.; Glazer, N.L.; Smith, N.L.; Psaty, B.M.; Lumley, T.; Wiggins, K.L.; Page, R.L.; Heckbert, S.R. Diabetes mellitus, glycemic control, and risk of atrial fibrillation. J. Gen. Intern. Med. 2010, 25, 853–858. [Google Scholar] [CrossRef] [Green Version]
- Nizami, H.L.; Katare, P.; Prabhakar, P.; Kumar, Y.; Arava, S.K.; Chakraborty, P.; Maulik, S.K.; Banerjee, S.K. Vitamin D deficiency in rats causes cardiac dysfunction by inducing myocardial insulin resistance. Mol. Nutr. Food Res. 2019, 63, e1900109. [Google Scholar] [CrossRef]
- Morel, S.; Berthonneche, C.; Tanguy, S.; Toufektsian, M.C.; Foulon, T.; de Lorgeril, M.; de Leiris, J.; Boucher, F. Insulin resistance modifies plasma fatty acid distribution and decreases cardiac tolerance to in vivo ischaemia/reperfusion in rats. Clin. Exp. Pharmacol. Physiol. 2003, 30, 446–451. [Google Scholar] [CrossRef]
- Diez, E.R.; Renna, N.F.; Prado, N.J.; Lembo, C.; Ponce Zumino, A.Z.; Vazquez-Prieto, M.; Miatello, R.M. Melatonin, given at the time of reperfusion, prevents ventricular arrhythmias in isolated hearts from fructose-fed rats and spontaneously hypertensive rats. J. Pineal. Res. 2013, 55, 166–173. [Google Scholar] [CrossRef]
- Axelsen, L.N.; Calloe, K.; Braunstein, T.H.; Riemann, M.; Hofgaard, J.P.; Liang, B.; Jensen, C.F.; Olsen, K.B.; Bartels, E.D.; Baandrup, U.; et al. Diet-induced pre-diabetes slows cardiac conductance and promotes arrhythmogenesis. Cardiovasc. Diabetol. 2015, 14, 87. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Huang, X.; Gao, J.; Guo, Y.; Di, Y.; Sun, S.; Deng, X.; Cao, J. Improved endogenous epoxyeicosatrienoic acid production mends heart function via increased PGC 1alpha-mitochondrial functions in metabolic syndrome. J. Pharmacol. Sci. 2018, 138, 138–145. [Google Scholar] [CrossRef]
- Federico, M.; Portiansky, E.L.; Sommese, L.; Alvarado, F.J.; Blanco, P.G.; Zanuzzi, C.N.; Dedman, J.; Kaetzel, M.; Wehrens, X.H.T.; Mattiazzi, A.; et al. Calcium-calmodulin-dependent protein kinase mediates the intracellular signalling pathways of cardiac apoptosis in mice with impaired glucose tolerance. J. Physiol. 2017, 595, 4089–4108. [Google Scholar] [CrossRef] [Green Version]
- Sommese, L.; Valverde, C.A.; Blanco, P.; Castro, M.C.; Rueda, O.V.; Kaetzel, M.; Dedman, J.; Anderson, M.E.; Mattiazzi, A.; Palomeque, J. Ryanodine receptor phosphorylation by CaMKII promotes spontaneous Ca(2+) release events in a rodent model of early stage diabetes: The arrhythmogenic substrate. Int. J. Cardiol. 2016, 202, 394–406. [Google Scholar] [CrossRef] [Green Version]
- Cheng, W.L.; Kao, Y.H.; Chen, Y.C.; Lin, Y.K.; Chen, S.A.; Chen, Y.J. Macrophage migration inhibitory factor increases atrial arrhythmogenesis through CD74 signaling. Transl. Res. 2020, 216, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.F.; Chen, Y.J.; Lin, Y.J.; Chen, S.A. Inflammation and the pathogenesis of atrial fibrillation. Nat. Rev. Cardiol. 2015, 12, 230–243. [Google Scholar] [CrossRef] [PubMed]
- Harada, M.; Van Wagoner, D.R.; Nattel, S. Role of inflammation in atrial fibrillation pathophysiology and management. Circ. J. 2015, 79, 495–502. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Zhang, L.; Zhang, Y.; Xu, J.J.; Sun, L.L.; Li, S.Z. The protective role of liquiritin in high fructose-induced myocardial fibrosis via inhibiting NF-kappaB and MAPK signaling pathway. Biomed. Pharmacother. 2016, 84, 1337–1349. [Google Scholar] [CrossRef] [PubMed]
- Kang, L.L.; Zhang, D.M.; Ma, C.H.; Zhang, J.H.; Jia, K.K.; Liu, J.H.; Wang, R.; Kong, L.D. Cinnamaldehyde and allopurinol reduce fructose-induced cardiac inflammation and fibrosis by attenuating CD36-mediated TLR4/6-IRAK4/1 signaling to suppress NLRP3 inflammasome activation. Sci. Rep. 2016, 6, 27460. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.W. Liquiritigenin attenuates cardiac injury induced by high fructose-feeding through fibrosis and inflammation suppression. Biomed. Pharmacother. 2017, 86, 694–704. [Google Scholar] [CrossRef] [PubMed]
- Yao, C.; Veleva, T.; Scott, L., Jr.; Cao, S.; Li, L.; Chen, G.; Jeyabal, P.; Pan, X.; Alsina, K.M.; Abu-Taha, I.D.; et al. Enhanced Cardiomyocyte NLRP3 Inflammasome Signaling Promotes Atrial Fibrillation. Circulation 2018, 138, 2227–2242. [Google Scholar] [CrossRef]
- Chiazza, F.; Couturier-Maillard, A.; Benetti, E.; Mastrocola, R.; Nigro, D.; Cutrin, J.C.; Serpe, L.; Aragno, M.; Fantozzi, R.; Ryffel, B.; et al. Targeting the NLRP3 inflammasome to reduce diet-induced metabolic abnormalities in mice. Mol. Med. 2016, 21, 1025–1037. [Google Scholar] [CrossRef]
- Lian, Y.G.; Zhao, H.Y.; Wang, S.J.; Xu, Q.L.; Xia, X.J. NLRP4 is an essential negative regulator of fructose-induced cardiac injury in vitro and in vivo. Biomed. Pharmacother. 2017, 91, 590–601. [Google Scholar] [CrossRef]
- Qin, B.; Polansky, M.M.; Harry, D.; Anderson, R.A. Green tea polyphenols improve cardiac muscle mRNA and protein levels of signal pathways related to insulin and lipid metabolism and inflammation in insulin-resistant rats. Mol. Nutr. Food Res. 2010, 54, S14–S23. [Google Scholar] [CrossRef]
- Lee, H.J.; Cha, J.Y. Recent insights into the role of ChREBP in intestinal fructose absorption and metabolism. BMB Rep. 2018, 51, 429–436. [Google Scholar] [CrossRef] [Green Version]
- Jang, C.; Hui, S.; Lu, W.; Cowan, A.J.; Morscher, R.J.; Lee, G.; Liu, W.; Tesz, G.J.; Birnbaum, M.J.; Rabinowitz, J.D. The Small intestine converts dietary fructose into glucose and organic acids. Cell Metab. 2018, 27, 351–361 e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Do, M.H.; Lee, E.; Oh, M.J.; Kim, Y.; Park, H.Y. High-glucose or-fructose diet cause changes of the gut microbiota and metabolic disorders in mice without body weight change. Nutrients 2018, 10, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tappy, L.; Rosset, R. Health outcomes of a high fructose intake: The importance of physical activity. J. Physiol. 2019, 597, 3561–3571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, K.A.; Faeh, D.; Stettler, R.; Debard, C.; Loizon, E.; Vidal, H.; Boesch, C.; Ravussin, E.; Tappy, L. Effects of four-week high-fructose diet on gene expression in skeletal muscle of healthy men. Diabetes Metab. 2008, 34, 82–85. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.W.; Kao, Y.H.; Chen, Y.J.; Chao, T.F.; Lee, T.I. Therapeutic potential of vitamin D in AGE/RAGE-related cardiovascular diseases. Cell Mol. Life Sci. 2019, 76, 4103–4115. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S.; Ahn, H.; Park, Y.K. High dietary fructose intake on cardiovascular disease related parameters in growing rats. Nutrients 2016, 9, 1. [Google Scholar] [CrossRef] [Green Version]
- Ussher, J.R.; Lopaschuk, G.D.; Arduini, A. Gut microbiota metabolism of L-carnitine and cardiovascular risk. Atherosclerosis 2013, 231, 456–461. [Google Scholar] [CrossRef]
- Wang, Y.; Qi, W.; Song, G.; Pang, S.; Peng, Z.; Li, Y.; Wang, P. High-fructose diet increases inflammatory cytokines and alters gut microbiota composition in rats. Mediat. Inflamm. 2020, 2020, 6672636. [Google Scholar] [CrossRef]
- Forkosh, E.; Ilan, Y. The heart-gut axis: New target for atherosclerosis and congestive heart failure therapy. Open Heart 2019, 6, e000993. [Google Scholar] [CrossRef]
- Jin, M.; Qian, Z.; Yin, J.; Xu, W.; Zhou, X. The role of intestinal microbiota in cardiovascular disease. J. Cell Mol. Med. 2019, 23, 2343–2350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamo, T.; Akazawa, H.; Suzuki, J.I.; Komuro, I. Novel concept of a heart-gut axis in the pathophysiology of heart failure. Korean Circ. J. 2017, 47, 663–669. [Google Scholar] [CrossRef] [Green Version]
- Lezutekong, J.N.; Nikhanj, A.; Oudit, G.Y. Imbalance of gut microbiome and intestinal epithelial barrier dysfunction in cardiovascular disease. Clin. Sci. (Lond.) 2018, 132, 901–904. [Google Scholar] [CrossRef] [PubMed]
- Sandek, A.; Bauditz, J.; Swidsinski, A.; Buhner, S.; Weber-Eibel, J.; von Haehling, S.; Schroedl, W.; Karhausen, T.; Doehner, W.; Rauchhaus, M.; et al. Altered intestinal function in patients with chronic heart failure. J. Am. Coll Cardiol. 2007, 50, 1561–1569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, X.; Ye, L.; Li, J.; Jin, L.; Wang, W.; Li, S.; Bao, M.; Wu, S.; Li, L.; Geng, B.; et al. Metagenomic and metabolomic analyses unveil dysbiosis of gut microbiota in chronic heart failure patients. Sci. Rep. 2018, 8, 635. [Google Scholar] [CrossRef] [PubMed]
- Zuo, K.; Li, J.; Li, K.; Hu, C.; Gao, Y.; Chen, M.; Hu, R.; Liu, Y.; Chi, H.; Wang, H.; et al. Disordered gut microbiota and alterations in metabolic patterns are associated with atrial fibrillation. Gigascience 2019, 8, 6. [Google Scholar] [CrossRef] [Green Version]
- Zuo, K.; Li, J.; Wang, P.; Liu, Y.; Liu, Z.; Yin, X.; Liu, X.; Yang, X. Duration of persistent atrial fibrillation is associated with alterations in human gut microbiota and metabolic phenotypes. mSystems 2019, 4, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, N.; Yamashita, T.; Hirata, K.I. Gut microbiome and cardiovascular diseases. Diseases 2018, 6, 3. [Google Scholar] [CrossRef] [Green Version]
- Pastori, D.; Carnevale, R.; Nocella, C.; Novo, M.; Santulli, M.; Cammisotto, V.; Menichelli, D.; Pignatelli, P.; Violi, F. Gut-derived serum lipopolysaccharide is associated with enhanced risk of major adverse cardiovascular events in atrial fibrillation: Effect of adherence to Mediterranean diet. J. Am. Heart Assoc. 2017, 6, 6. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.H.W.; Li, D.Y.; Hazen, S.L. Dietary metabolism, the gut microbiome, and heart failure. Nat. Rev. Cardiol. 2019, 16, 137–154. [Google Scholar] [CrossRef]
- Svingen, G.F.T.; Zuo, H.; Ueland, P.M.; Seifert, R.; Loland, K.H.; Pedersen, E.R.; Schuster, P.M.; Karlsson, T.; Tell, G.S.; Schartum-Hansen, H.; et al. Increased plasma trimethylamine-N-oxide is associated with incident atrial fibrillation. Int. J. Cardiol. 2018, 267, 100–106. [Google Scholar] [CrossRef]
- Leustean, A.M.; Ciocoiu, M.; Sava, A.; Costea, C.F.; Floria, M.; Tarniceriu, C.C.; Tanase, D.M. Implications of the intestinal microbiota in diagnosing the progression of diabetes and the presence of cardiovascular complications. J. Diabetes Res. 2018, 2018, 5205126. [Google Scholar] [CrossRef] [Green Version]
- Tomlinson, J.A.P.; Wheeler, D.C. The role of trimethylamine N-oxide as a mediator of cardiovascular complications in chronic kidney disease. Kidney Int. 2017, 92, 809–815. [Google Scholar] [CrossRef] [Green Version]
- Janeiro, M.H.; Ramirez, M.J.; Milagro, F.I.; Martinez, J.A.; Solas, M. Implication of trimethylamine N-oxide (TMAO) in disease: Potential biomarker or new therapeutic target. Nutrients 2018, 10, 10. [Google Scholar] [CrossRef] [Green Version]
- Pan, Y.; Kong, L.D. High fructose diet-induced metabolic syndrome: Pathophysiological mechanism and treatment by traditional Chinese medicine. Pharmacol. Res. 2018, 130, 438–450. [Google Scholar] [CrossRef] [PubMed]
- Manco, M.; Putignani, L.; Bottazzo, G.F. Gut microbiota, lipopolysaccharides, and innate immunity in the pathogenesis of obesity and cardiovascular risk. Endocr. Rev. 2010, 31, 817–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neves, A.L.; Coelho, J.; Couto, L.; Leite-Moreira, A.; Roncon-Albuquerque, R., Jr. Metabolic endotoxemia: A molecular link between obesity and cardiovascular risk. J. Mol. Endocrinol. 2013, 51, R51–R64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barton, G.M.; Kagan, J.C. A cell biological view of Toll-like receptor function: Regulation through compartmentalization. Nat. Rev. Immunol. 2009, 9, 535–542. [Google Scholar] [CrossRef] [PubMed]
- Guzzo, C.; Ayer, A.; Basta, S.; Banfield, B.W.; Gee, K. IL-27 enhances LPS-induced proinflammatory cytokine production via upregulation of TLR4 expression and signaling in human monocytes. J. Immunol. 2012, 188, 864–873. [Google Scholar] [CrossRef] [Green Version]
- Spruss, A.; Kanuri, G.; Wagnerberger, S.; Haub, S.; Bischoff, S.C.; Bergheim, I. Toll-like receptor 4 is involved in the development of fructose-induced hepatic steatosis in mice. Hepatology 2009, 50, 1094–1104. [Google Scholar] [CrossRef] [PubMed]
- Bergheim, I.; Weber, S.; Vos, M.; Kramer, S.; Volynets, V.; Kaserouni, S.; McClain, C.J.; Bischoff, S.C. Antibiotics protect against fructose-induced hepatic lipid accumulation in mice: Role of endotoxin. J. Hepatol. 2008, 48, 983–992. [Google Scholar] [CrossRef] [PubMed]
- Soares, J.B.; Pimentel-Nunes, P.; Roncon-Albuquerque, R.; Leite-Moreira, A. The role of lipopolysaccharide/toll-like receptor 4 signaling in chronic liver diseases. Hepatol. Int. 2010, 4, 659–672. [Google Scholar] [CrossRef] [Green Version]
- Dieterlen, M.T.; John, K.; Reichenspurner, H.; Mohr, F.W.; Barten, M.J. Dendritic cells and their role in cardiovascular diseases: A view on human studies. J. Immunol. Res. 2016, 2016, 5946807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavine, K.J.; Pinto, A.R.; Epelman, S.; Kopecky, B.J.; Clemente-Casares, X.; Godwin, J.; Rosenthal, N.; Kovacic, J.C. The macrophage in cardiac homeostasis and disease: JACC macrophage in CVD series (Part 4). J. Am. Coll. Cardiol. 2018, 72, 2213–2230. [Google Scholar] [CrossRef] [PubMed]
- Spruss, A.; Bergheim, I. Dietary fructose and intestinal barrier: Potential risk factor in the pathogenesis of nonalcoholic fatty liver disease. J. Nutr. Biochem. 2009, 20, 657–662. [Google Scholar] [CrossRef]
- Spruss, A.; Kanuri, G.; Stahl, C.; Bischoff, S.C.; Bergheim, I. Metformin protects against the development of fructose-induced steatosis in mice: Role of the intestinal barrier function. Lab. Investig. 2012, 92, 1020–1032. [Google Scholar] [CrossRef] [Green Version]
- Sellmann, C.; Priebs, J.; Landmann, M.; Degen, C.; Engstler, A.J.; Jin, C.J.; Garttner, S.; Spruss, A.; Huber, O.; Bergheim, I. Diets rich in fructose, fat or fructose and fat alter intestinal barrier function and lead to the development of nonalcoholic fatty liver disease over time. J. Nutr. Biochem. 2015, 26, 1183–1192. [Google Scholar] [CrossRef]
- Chen, Y.Y.; Sun, Z.W.; Jiang, J.P.; Kang, X.D.; Wang, L.L.; Shen, Y.L.; Xie, X.D.; Zheng, L.R. alpha-adrenoceptor-mediated enhanced inducibility of atrial fibrillation in a canine system inflammation model. Mol. Med. Rep. 2017, 15, 3767–3774. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, X.; Shi, H.; Yu, Y.; Yu, Y.; Li, M.; Chen, R. NLRP3 inflammasome, an immune-inflammatory target in pathogenesis and treatment of cardiovascular diseases. Clin. Transl. Med. 2020, 10, 91–106. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, S.; Li, B.; Luo, Y.; Gong, Y.; Jin, X.; Zhang, J.; Zhou, Y.; Zhuo, X.; Wang, Z.; et al. Gut microbiota dysbiosis promotes age-related atrial fibrillation by lipopolysaccharide and glucose-induced activation of NLRP3-inflammasome. Cardiovasc. Res. 2021. online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.N.; Chang-Chien, G.P.; Lin, S.; Hou, C.Y.; Tain, Y.L. Targeting on gut microbial metabolite trimethylamine-N-oxide and short-chain fatty acid to prevent maternal high-fructose-diet-induced developmental programming of hypertension in adult male offspring. Mol. Nutr. Food Res. 2019, 63, e1900073. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.N.; Chan, J.Y.H.; Yu, H.R.; Lee, W.C.; Wu, K.L.H.; Chang-Chien, G.P.; Lin, S.; Hou, C.Y.; Tain, Y.L. Targeting on gut microbiota-derived metabolite trimethylamine to protect adult male rat offspring against hypertension programmed by combined maternal high-fructose intake and dioxin exposure. Int. J. Mol. Sci. 2020, 21, 15. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Yu, H.; Lei, P.; Huang, S.; Ren, J.; Fan, W.; Han, L.; Yu, H.; Wang, Y.; Ren, M.; et al. Determination of trimethylamine N-oxide and betaine in serum and food by targeted metabonomics. Molecules 2021, 26, 5. [Google Scholar] [CrossRef]
- Ji, Y.; Yin, Y.; Sun, L.; Zhang, W. The molecular and mechanistic insights based on gut-liver axis: Nutritional target for non-alcoholic fatty liver disease (NAFLD) improvement. Int. J. Mol. Sci. 2020, 21, 9. [Google Scholar] [CrossRef] [PubMed]
- Schugar, R.C.; Shih, D.M.; Warrier, M.; Helsley, R.N.; Burrows, A.; Ferguson, D.; Brown, A.L.; Gromovsky, A.D.; Heine, M.; Chatterjee, A.; et al. The TMAO-producing enzyme flavin-containing monooxygenase 3 regulates obesity and the beiging of white adipose tissue. Cell Rep. 2017, 19, 2451–2461. [Google Scholar] [CrossRef] [Green Version]
- Savi, M.; Bocchi, L.; Bresciani, L.; Falco, A.; Quaini, F.; Mena, P.; Brighenti, F.; Crozier, A.; Stilli, D.; Del Rio, D. Trimethylamine-N-oxide (TMAO)-induced impairment of cardiomyocyte function and the protective role of urolithin B-glucuronide. Molecules 2018, 23, 3. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Wu, Z.; Yan, J.; Liu, H.; Liu, Q.; Deng, Y.; Ou, C.; Chen, M. Gut microbe-derived metabolite trimethylamine N-oxide induces cardiac hypertrophy and fibrosis. Lab. Investig. 2019, 99, 346–357. [Google Scholar] [CrossRef]
- Yang, W.; Zhang, S.; Zhu, J.; Jiang, H.; Jia, D.; Ou, T.; Qi, Z.; Zou, Y.; Qian, J.; Sun, A.; et al. Gut microbe-derived metabolite trimethylamine N-oxide accelerates fibroblast-myofibroblast differentiation and induces cardiac fibrosis. J. Mol. Cell Cardiol. 2019, 134, 119–130. [Google Scholar] [CrossRef]
- Seldin, M.M.; Meng, Y.; Qi, H.; Zhu, W.; Wang, Z.; Hazen, S.L.; Lusis, A.J.; Shih, D.M. Trimethylamine N-oxide promotes vascular inflammation through signaling of mitogen-activated protein kinase and nuclear factor-κB. J. Am. Heart Assoc. 2016, 5, 2. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Li, Y.; Yang, P.; Liu, X.; Lu, L.; Chen, Y.; Zhong, X.; Li, Z.; Liu, H.; Ou, C.; et al. Trimethylamine-N-oxide promotes vascular calcification through activation of NLRP3 (nucleotide-binding domain, leucine-rich-containing family, pyrin domain-containing-3) inflammasome and NF-κB (Nuclear Factor κB) signals. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 751–765. [Google Scholar] [CrossRef]
- Li, X.; Geng, J.; Zhao, J.; Ni, Q.; Zhao, C.; Zheng, Y.; Chen, X.; Wang, L. Trimethylamine N-oxide exacerbates cardiac fibrosis via activating the NLRP3 inflammasome. Front. Physiol. 2019, 10, 866. [Google Scholar] [CrossRef]
- Chen, M.L.; Zhu, X.H.; Ran, L.; Lang, H.D.; Yi, L.; Mi, M.T. Trimethylamine-N-oxide induces vascular inflammation by activating the NLRP3 inflammasome through the SIRT3-SOD2-mtROS signaling pathway. J. Am. Heart Assoc. 2017, 6, 9. [Google Scholar] [CrossRef] [PubMed]
- Roncal, C.; Martinez-Aguilar, E.; Orbe, J.; Ravassa, S.; Fernandez-Montero, A.; Saenz-Pipaon, G.; Ugarte, A.; Estella-Hermoso de Mendoza, A.; Rodriguez, J.A.; Fernandez-Alonso, S.; et al. Trimethylamine-N-oxide (TMAO) predicts cardiovascular mortality in peripheral artery disease. Sci. Rep. 2019, 9, 15580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, G.; Pan, B.; Chen, Y.; Guo, C.; Zhao, M.; Zheng, L.; Chen, B. Trimethylamine N-oxide in atherogenesis: Impairing endothelial self-repair capacity and enhancing monocyte adhesion. Biosci. Rep. 2017, 37, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haghikia, A.; Li, X.S.; Liman, T.G.; Bledau, N.; Schmidt, D.; Zimmermann, F.; Krankel, N.; Widera, C.; Sonnenschein, K.; Haghikia, A.; et al. Gut microbiota-dependent trimethylamine N-oxide predicts risk of cardiovascular events in patients with stroke and is related to proinflammatory monocytes. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 2225–2235. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.; Meng, G.; Huang, B.; Zhou, X.; Stavrakis, S.; Wang, M.; Li, X.; Zhou, L.; Wang, Y.; Wang, M.; et al. A potential relationship between gut microbes and atrial fibrillation: Trimethylamine N-oxide, a gut microbe-derived metabolite, facilitates the progression of atrial fibrillation. Int. J. Cardiol. 2018, 255, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Henderson, A.; Petriello, M.C.; Romano, K.A.; Gearing, M.; Miao, J.; Schell, M.; Sandoval-Espinola, W.J.; Tao, J.; Sha, B.; et al. Trimethylamine N-oxide binds and activates PERK to promote metabolic dysfunction. Cell Metab. 2019, 30, 1141–1151 e5. [Google Scholar] [CrossRef] [PubMed]
- Meng, G.; Zhou, X.; Wang, M.; Zhou, L.; Wang, Z.; Wang, M.; Deng, J.; Wang, Y.; Zhou, Z.; Zhang, Y.; et al. Gut microbe-derived metabolite trimethylamine N-oxide activates the cardiac autonomic nervous system and facilitates ischemia-induced ventricular arrhythmia via two different pathways. EBioMedicine 2019, 44, 656–664. [Google Scholar] [CrossRef] [Green Version]
- Liang, Z.; Dong, Z.; Guo, M.; Shen, Z.; Yin, D.; Hu, S.; Hai, X. Trimethylamine N-oxide as a risk marker for ischemic stroke in patients with atrial fibrillation. J. Biochem. Mol. Toxicol. 2019, 33, e22246. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Jin, M.; Liu, L.; Yu, Z.; Lu, X.; Zhang, H. Trimethylamine N-oxide and cardiovascular outcomes in patients with chronic heart failure after myocardial infarction. ESC Heart Fail 2020, 7, 188–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, T.; Unwin, D.; Finucane, F. Low-carbohydrate diets in the management of obesity and type 2 diabetes: A review from clinicians using the approach in practice. Int. J. Environ. Res. Public Health 2020, 17, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Unwin, D.J.; Tobin, S.D.; Murray, S.W.; Delon, C.; Brady, A.J. Substantial and sustained improvements in blood pressure, weight and lipid profiles from a carbohydrate restricted diet: An observational study of insulin resistant patients in primary care. Int. J. Environ. Res. Public Health 2019, 16, 15. [Google Scholar] [CrossRef] [Green Version]
- McKenzie, A.L.; Hallberg, S.J.; Creighton, B.C.; Volk, B.M.; Link, T.M.; Abner, M.K.; Glon, R.M.; McCarter, J.P.; Volek, J.S.; Phinney, S.D. A novel intervention including individualized nutritional recommendations reduces hemoglobin A1c level, medication use, and weight in type 2 diabetes. JMIR Diabetes 2017, 2, e5. [Google Scholar] [CrossRef]
- De Filippis, F.; Pellegrini, N.; Vannini, L.; Jeffery, I.B.; La Storia, A.; Laghi, L.; Serrazanetti, D.I.; Di Cagno, R.; Ferrocino, I.; Lazzi, C.; et al. High-level adherence to a Mediterranean diet beneficially impacts the gut microbiota and associated metabolome. Gut 2016, 65, 1812–1821. [Google Scholar] [CrossRef]
- Pastori, D.; Carnevale, R.; Bartimoccia, S.; Nocella, C.; Tanzilli, G.; Cangemi, R.; Vicario, T.; Catena, M.; Violi, F.; Pignatelli, P. Does Mediterranean diet reduce cardiovascular events and oxidative stress in atrial fibrillation? Antioxid. Redox Signal. 2015, 23, 682–687. [Google Scholar] [CrossRef]
- Sofi, F.; Cesari, F.; Abbate, R.; Gensini, G.F.; Casini, A. Adherence to Mediterranean diet and health status: Meta-analysis. BMJ 2008, 337, a1344. [Google Scholar] [CrossRef] [Green Version]
- Wang, A.Y.; Sea, M.M.; Ng, K.; Wang, M.; Chan, I.H.; Lam, C.W.-K.; Sanderson, J.; Woo, J. Dietary fiber intake, myocardial injury, and major adverse cardiovascular events among end-stage kidney disease patients: A prospective cohort study. Kidney Int. Rep. 2019, 4, 814–823. [Google Scholar] [CrossRef] [Green Version]
- Hendijani, F.; Akbari, V. Probiotic supplementation for management of cardiovascular risk factors in adults with type II diabetes: A systematic review and meta-analysis. Clin. Nutr. 2018, 37, 532–541. [Google Scholar] [CrossRef]
- Gan, X.T.; Ettinger, G.; Huang, C.X.; Burton, J.P.; Haist, J.V.; Rajapurohitam, V.; Sidaway, J.E.; Martin, G.; Gloor, G.B.; Swann, J.R.; et al. Probiotic administration attenuates myocardial hypertrophy and heart failure after myocardial infarction in the rat. Circ. Heart Fail. 2014, 7, 491–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raygan, F.; Ostadmohammadi, V.; Bahmani, F.; Asemi, Z. The effects of vitamin D and probiotic co-supplementation on mental health parameters and metabolic status in type 2 diabetic patients with coronary heart disease: A randomized, double-blind, placebo-controlled trial. Prog. Neuropsychopharmacol. Biol. Psychiatry 2018, 84, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Sadeghzadeh, J.; Vakili, A.; Sameni, H.R.; Shadnoush, M.; Bandegi, A.R.; Zahedi Khorasani, M. The effect of oral consumption of probiotics in prevention of heart injury in a rat myocardial infarction model: A histopathological, hemodynamic and biochemical evaluation. Iran. Biomed. J. 2017, 21, 174–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, D.Y.; Ahn, Y.T.; Huh, C.S.; McGregor, R.A.; Choi, M.S. Dual probiotic strains suppress high fructose-induced metabolic syndrome. World J. Gastroenterol. 2013, 19, 274–283. [Google Scholar] [CrossRef]
- Xu, H.; Wang, J.; Cai, J.; Feng, W.; Wang, Y.; Liu, Q.; Cai, L. Protective effect of Lactobacillus rhamnosus GG and its supernatant against myocardial dysfunction in obese mice exposed to intermittent hypoxia is associated with the activation of Nrf2 pathway. Int. J. Biol. Sci. 2019, 15, 2471–2483. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Yan, H.; Lu, Y.; Li, X.; Wang, X.; Shan, Y.; Yi, Y.; Liu, B.; Zhou, Y.; Lu, X. Anti-obesity effect of Lactobacillus rhamnosus LS-8 and Lactobacillus crustorum MN047 on high-fat and high-fructose diet mice base on inflammatory response alleviation and gut microbiota regulation. Eur. J. Nutr. 2020, 59, 2709–2728. [Google Scholar] [CrossRef] [PubMed]
- Zubiria, M.G.; Gambaro, S.E.; Rey, M.A.; Carasi, P.; Serradell, M.L.A.; Giovambattista, A. Deleterious metabolic effects of high fructose intake: The preventive effect of Lactobacillus kefiri administration. Nutrients 2017, 9, 5. [Google Scholar] [CrossRef]
- Priyadarshini, M.; Kotlo, K.U.; Dudeja, P.K.; Layden, B.T. Role of short chain fatty acid receptors in intestinal physiology and pathophysiology. Compr. Physiol. 2018, 8, 1091–1115. [Google Scholar]
- Koh, A.; De Vadder, F.; Kovatcheva-Datchary, P.; Backhed, F. From dietary fiber to host physiology: Short-chain fatty acids as key bacterial metabolites. Cell 2016, 165, 1332–1345. [Google Scholar] [CrossRef] [Green Version]
- Krautkramer, K.A.; Kreznar, J.H.; Romano, K.A.; Vivas, E.I.; Barrett-Wilt, G.A.; Rabaglia, M.E.; Keller, M.P.; Attie, A.D.; Rey, F.E.; Denu, J.M. Diet-microbiota interactions mediate global epigenetic programming in multiple host tissues. Mol. Cell 2016, 64, 982–992. [Google Scholar] [CrossRef] [Green Version]
- den Besten, G.; van Eunen, K.; Groen, A.K.; Venema, K.; Reijngoud, D.J.; Bakker, B.M. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J. Lipid Res. 2013, 54, 2325–2340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakraborti, C.K. New-found link between microbiota and obesity. World J. Gastrointest. Pathophysiol. 2015, 6, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Fachi, J.L.; Felipe, J.S.; Pral, L.P.; da Silva, B.K.; Correa, R.O.; de Andrade, M.C.P.; da Fonseca, D.M.; Basso, P.J.; Camara, N.O.S.; de Sales, E.S.E.L.; et al. Butyrate protects mice from Clostridium difficile-induced colitis through an HIF-1-dependent mechanism. Cell Rep. 2019, 27, 750–761 e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, G.; Wang, B.; Shafiei Jahani, P.; Hurrell, B.P.; Banie, H.; Aleman Muench, G.R.; Maazi, H.; Helou, D.G.; Howard, E.; Galle-Treger, L.; et al. Dietary fiber-induced microbial short chain fatty acids suppress ILC2-dependent airway inflammation. Front. Immunol. 2019, 10, 2051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.M.; Yu, R.; Zhang, L.P.; Wen, S.Y.; Wang, S.J.; Zhang, X.Y.; Xu, Q.; Kong, L.D. Dietary fructose-induced gut dysbiosis promotes mouse hippocampal neuroinflammation: A benefit of short-chain fatty acids. Microbiome 2019, 7, 98. [Google Scholar] [CrossRef] [PubMed]
- Depommier, C.; Everard, A.; Druart, C.; Plovier, H.; Van Hul, M.; Vieira-Silva, S.; Falony, G.; Raes, J.; Maiter, D.; Delzenne, N.M.; et al. Supplementation with Akkermansia muciniphila in overweight and obese human volunteers: A proof-of-concept exploratory study. Nat. Med. 2019, 25, 1096–1103. [Google Scholar] [CrossRef]
- Thangaraju, M.; Cresci, G.A.; Liu, K.; Ananth, S.; Gnanaprakasam, J.P.; Browning, D.D.; Mellinger, J.D.; Smith, S.B.; Digby, G.J.; Lambert, N.A.; et al. GPR109A is a G-protein-coupled receptor for the bacterial fermentation product butyrate and functions as a tumor suppressor in colon. Cancer Res. 2009, 69, 2826–2832. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; Wang, Y.; Wang, P.; Huang, Y.; Wang, F. Short-chain fatty acids manifest stimulative and protective effects on intestinal barrier function through the inhibition of NLRP3 inflammasome and autophagy. Cell Physiol. Biochem. 2018, 49, 190–205. [Google Scholar] [CrossRef]
- Xu, M.; Jiang, Z.; Wang, C.; Li, N.; Bo, L.; Zha, Y.; Bian, J.; Zhang, Y.; Deng, X. Acetate attenuates inflammasome activation through GPR43-mediated Ca(2+)-dependent NLRP3 ubiquitination. Exp. Mol. Med. 2019, 51, 83. [Google Scholar]
- Bartolomaeus, H.; Balogh, A.; Yakoub, M.; Homann, S.; Marko, L.; Hoges, S.; Tsvetkov, D.; Krannich, A.; Wundersitz, S.; Avery, E.G.; et al. Short-chain fatty acid propionate protects from hypertensive cardiovascular damage. Circulation 2019, 139, 1407–1421. [Google Scholar] [CrossRef]
- Subramanian, U.; Kumar, P.; Mani, I.; Chen, D.; Kessler, I.; Periyasamy, R.; Raghavaraju, G.; Pandey, K.N. Retinoic acid and sodium butyrate suppress the cardiac expression of hypertrophic markers and proinflammatory mediators in Npr1 gene-disrupted haplotype mice. Physiol. Genom. 2016, 48, 477–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, X.; Huang, X.; Tong, Y.; Gao, H. Butyrate improves cardiac function and sympathetic neural remodeling following myocardial infarction in rats. Can. J. Physiol. Pharmacol. 2020, 98, 391–399. [Google Scholar] [CrossRef] [PubMed]
- Pop, C.; Berce, C.; Ghibu, S.; Pop, A.; Kiss, B.; Irimie, A.; Popa, Ş.; Cismaru, G.; Loghin, F.; Mogosan, C. Validation and characterization of a heart failure animal model. Farmacia 2016, 64, 435–443. [Google Scholar]
- Patel, B.M. Sodium Butyrate Controls Cardiac Hypertrophy in Experimental Models of Rats. Cardiovasc. Toxicol. 2018, 18, 1–8. [Google Scholar] [CrossRef]
- Marques, F.Z.; Nelson, E.; Chu, P.Y.; Horlock, D.; Fiedler, A.; Ziemann, M.; Tan, J.K.; Kuruppu, S.; Rajapakse, N.W.; El-Osta, A.; et al. High-fiber diet and acetate supplementation change the gut microbiota and prevent the development of hypertension and heart failure in hypertensive mice. Circulation 2017, 135, 964–977. [Google Scholar] [CrossRef]
- Li, M.; van Esch, B.; Henricks, P.A.J.; Folkerts, G.; Garssen, J. The Anti-inflammatory effects of short chain fatty acids on lipopolysaccharide- or tumor necrosis factor alpha-stimulated endothelial cells via activation of GPR41/43 and inhibition of HDACs. Front. Pharmacol. 2018, 9, 533. [Google Scholar] [CrossRef] [Green Version]
- Lkhagva, B.; Kao, Y.H.; Chen, Y.C.; Chao, T.F.; Chen, S.A.; Chen, Y.J. Targeting histone deacetylases: A novel therapeutic strategy for atrial fibrillation. Eur. J. Pharmacol. 2016, 781, 250–257. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Levin, M.D.; Petrenko, N.B.; Lu, M.M.; Wang, T.; Yuan, L.J.; Stout, A.L.; Epstein, J.A.; Patel, V.V. Histone-deacetylase inhibition reverses atrial arrhythmia inducibility and fibrosis in cardiac hypertrophy independent of angiotensin. J. Mol. Cell Cardiol. 2008, 45, 715–723. [Google Scholar] [CrossRef] [Green Version]
- Fan, X.D.; Wan, L.L.; Duan, M.; Lu, S. HDAC11 deletion reduces fructose-induced cardiac dyslipidemia, apoptosis and inflammation by attenuating oxidative stress injury. Biochem. Biophys. Res. Commun. 2018, 503, 444–451. [Google Scholar] [CrossRef]
- McKinsey, T.A. Targeting inflammation in heart failure with histone deacetylase inhibitors. Mol. Med. 2011, 17, 434–441. [Google Scholar] [CrossRef]
- Lkhagva, B.; Kao, Y.H.; Lee, T.I.; Lee, T.W.; Cheng, W.L.; Chen, Y.J. Activation of Class I histone deacetylases contributes to mitochondrial dysfunction in cardiomyocytes with altered complex activities. Epigenetics 2018, 13, 376–385. [Google Scholar] [CrossRef] [PubMed]
- Lkhagva, B.; Chang, S.L.; Chen, Y.C.; Kao, Y.H.; Lin, Y.K.; Chiu, C.T.; Chen, S.A.; Chen, Y.J. Histone deacetylase inhibition reduces pulmonary vein arrhythmogenesis through calcium regulation. Int. J. Cardiol. 2014, 177, 982–989. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Choi, S.E.; Lee, H.B.; Song, M.W.; Kim, Y.H.; Jeong, J.Y.; Kang, Y.; Kim, H.J.; Kim, T.H.; Jeon, J.Y.; et al. A Class I histone deacetylase inhibitor attenuates insulin resistance and inflammation in palmitate-treated C2C12 myotubes and muscle of HF/HFr diet mice. Front. Pharmacol. 2020, 11, 601448. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Probiotics | Protocol | Outcomes | References |
|---|---|---|---|
| L. rhamnosus GR-1 | Coronary artery ligation rats fed rGR-1 (109 CFU/g, daily) in drinking water for 6 weeks. | Reduced cardiac hypertrophy and LV dysfunction. | [121] |
| L. acidophilus, Bifidobacterium bifidum, L. reuteri, L. fermentum | Patients with diabetic and coronary heart disease received vitamin D (50,000 IU) plus probiotics (8 × 109 CFU, every 2 weeks) for 12 weeks. | Reduced inflammation and increased antioxidant capacity, nitric oxide, glycemic control, and high-density lipoprotein. | [122] |
| B. breve, L. casei, L. bulgaricus L. acidophilus | Rats fed probiotics (2 × 106 CFU/mL, daily) for 2 weeks in response to isoproterenol-induced myocardial injury. | Reduced oxidative stress and inflammation and increased cardiac function. | [123] |
| L. curvatus HY7601, L. plantarum KY1032 | Rats fed a high-fructose diet (70% w/w) for 3 weeks followed by a probiotic (109–1010 CFU, daily) for 3 weeks. | Reduced oxidative stress, insulin resistance, and levels of plasma glucose and triglycerides. | [124] |
| L. rhamnosus LS-8, L. crustorum MN047 | Mice fed a high-fructose high fact diet (45% kcal fat, 10% w/v fructose) and a probiotic (109 CFU, daily) for 10 weeks. | Reduced insulin resistance and inflammation. | [126] |
| L. kefiri | Mice fed fructose (20% w/v) and a probiotic (108 CFU, every 2 days) for 6 weeks. | Reduced adipose tissue expansion, plasma triglyceride and leptin levels, and inflammation. | [127] |
| SCFAs | Study Design | Outcomes | Proposed Mechanisms | References |
|---|---|---|---|---|
| Propionate/Propionic Acid | Angiotensin II-treated wild-type or ApoE-KO mice fed sodium propionate (200 mmol/L, daily) in drinking water for 28–33 days. | Cardiac hypertrophy↓ Cardiac fibrosis↓ Ventricular tachyarrhythmias↓ Atherosclerotic lesion burden↓ | Protects cardiac functions though regulating T helper cell homeostasis. | [140] |
| Butyrate/Butyric Acid | Npr1 gene-disrupted heterozygous (Npr1+/−, 1 copy) mice received butyric acid (0.5 mg/kg/day, daily) intraperitoneally for 14 days. | Hypertrophic markers↓ Inflammatory mediators↓ HDAC activity↓ Cardiac dysfunction↓ | Suppress the cardiac expression of hypertrophic markers and proinflammatory mediators in Npr1 gene–disrupted haplotype mice. | [141] |
| Butyrate/Butyric Acid | Myocardial infarction rats received butyric acid (7.5 mmol/kg, daily) intraperitoneally for 3–7 days. | Cardiac dysfunction↓ Ventricular arrhythmias↓ Inflammation↓ Sympathetic neural remodeling↓ | Prevents ventricular arrhythmias by inhibiting inflammation and reducing sympathetic neural remodeling. | [142] |
| Butyrate/Butyric Acid | PAAC-induced cardiac hypertrophy in rats fed sodium butyrate (5 mg/kg, daily) for 56 days. | LV dysfunction↓ Cardiac fibrosis↓ | Prevents PAAC-induced cardiac hypertrophy through downregulation of class I HDACs. | [144] |
| Acetate/Acetic Acid | Mineralocorticoid-excess-treated mice fed a high-fiber diet (72.7% fiber) or acetate (200 mmol/L) in drinking water for 21 days. | Blood pressure↓ Cardiorenal fibrosis↓ Left ventricular hypertrophy↓ | Prevents hypertension and cardiac fibrosis though downregulating Egr-1. | [145] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, W.-L.; Li, S.-J.; Lee, T.-I.; Lee, T.-W.; Chung, C.-C.; Kao, Y.-H.; Chen, Y.-J. Sugar Fructose Triggers Gut Dysbiosis and Metabolic Inflammation with Cardiac Arrhythmogenesis. Biomedicines 2021, 9, 728. https://doi.org/10.3390/biomedicines9070728
Cheng W-L, Li S-J, Lee T-I, Lee T-W, Chung C-C, Kao Y-H, Chen Y-J. Sugar Fructose Triggers Gut Dysbiosis and Metabolic Inflammation with Cardiac Arrhythmogenesis. Biomedicines. 2021; 9(7):728. https://doi.org/10.3390/biomedicines9070728
Chicago/Turabian StyleCheng, Wan-Li, Shao-Jung Li, Ting-I Lee, Ting-Wei Lee, Cheng-Chih Chung, Yu-Hsun Kao, and Yi-Jen Chen. 2021. "Sugar Fructose Triggers Gut Dysbiosis and Metabolic Inflammation with Cardiac Arrhythmogenesis" Biomedicines 9, no. 7: 728. https://doi.org/10.3390/biomedicines9070728
APA StyleCheng, W.-L., Li, S.-J., Lee, T.-I., Lee, T.-W., Chung, C.-C., Kao, Y.-H., & Chen, Y.-J. (2021). Sugar Fructose Triggers Gut Dysbiosis and Metabolic Inflammation with Cardiac Arrhythmogenesis. Biomedicines, 9(7), 728. https://doi.org/10.3390/biomedicines9070728