
The Role of Oxidative Stress in NAFLD–NASH–HCC Transition—Focus on NADPH Oxidases




Abstract
1. Introduction
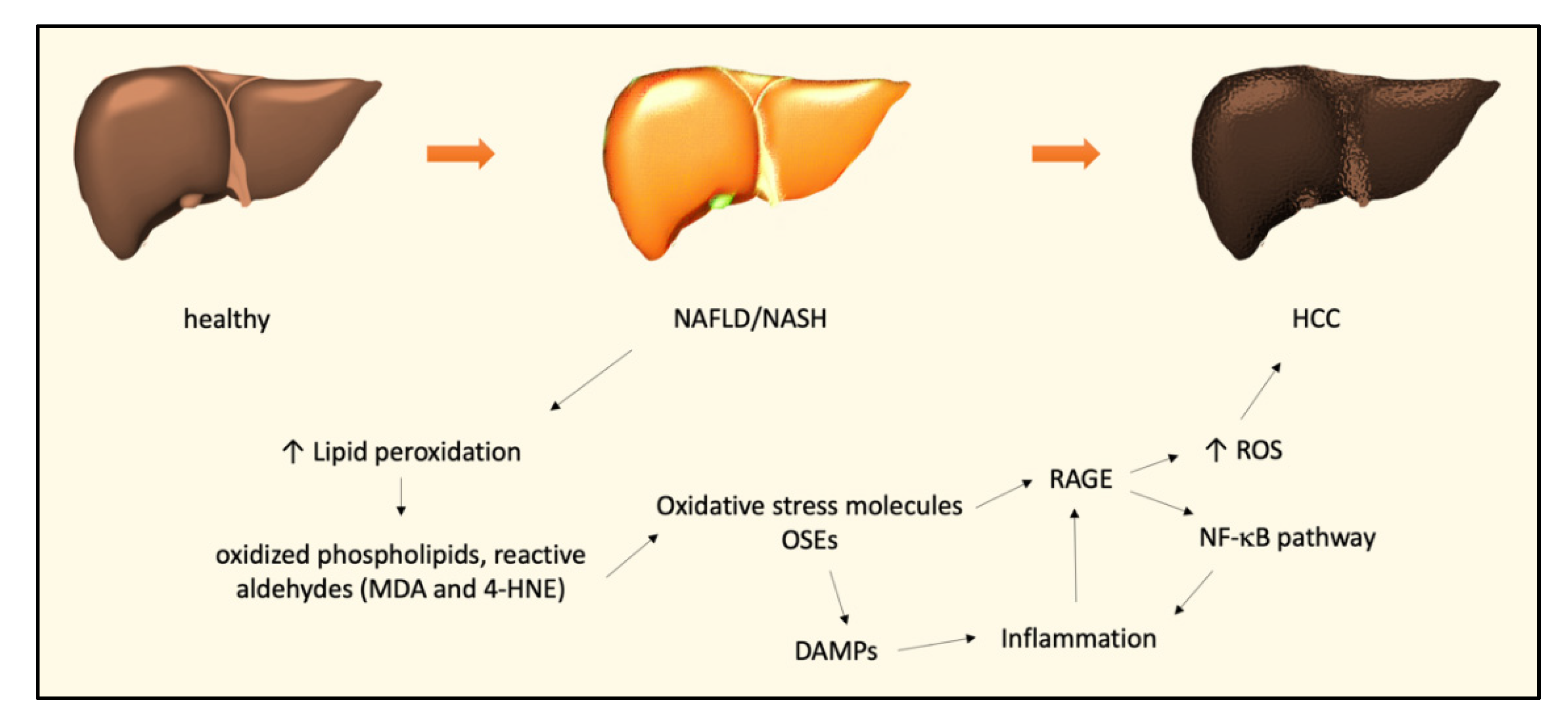
2. The Role of Oxidative Stress in NAFLD and NASH
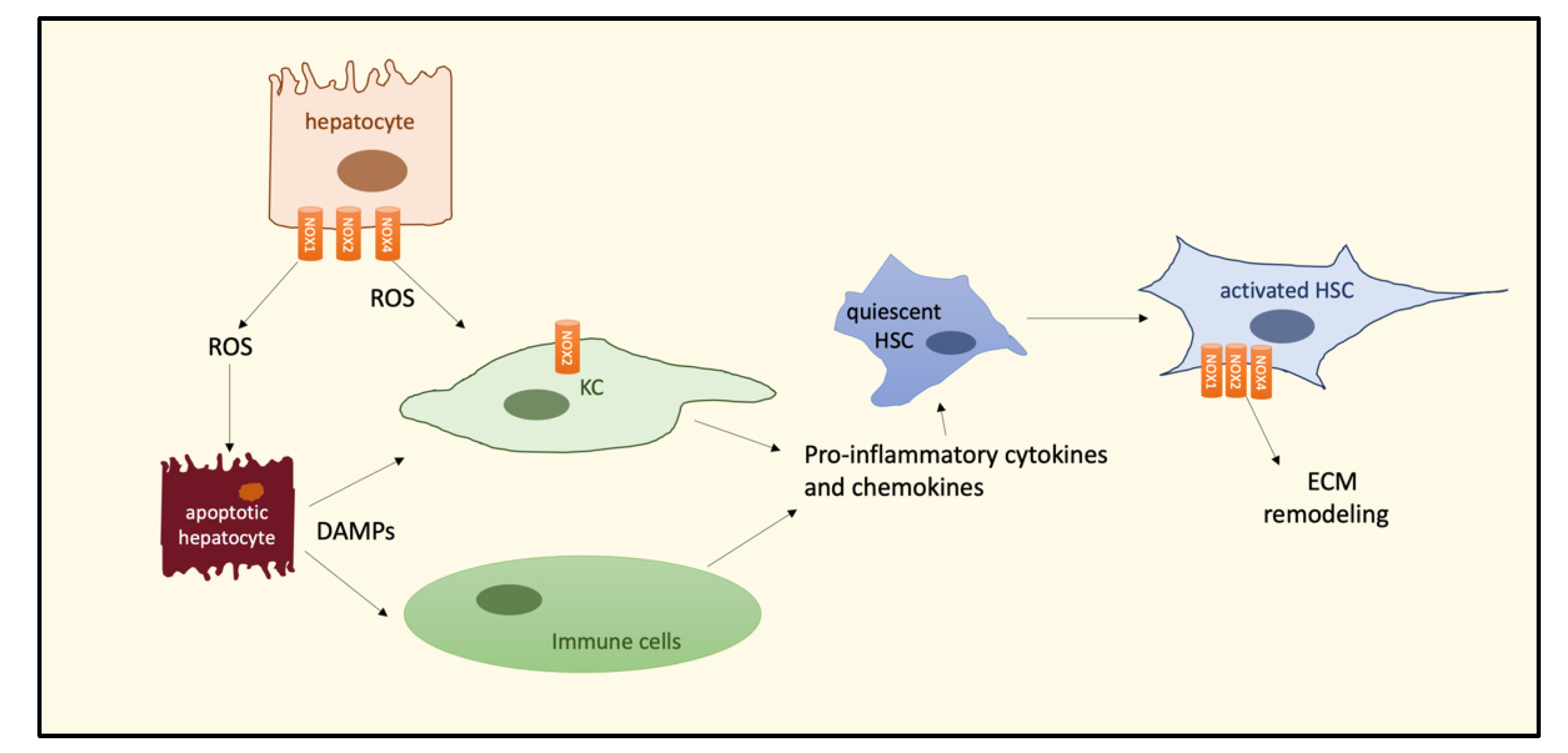
3. The Role of Oxidative Stress in the NASH–HCC Transition
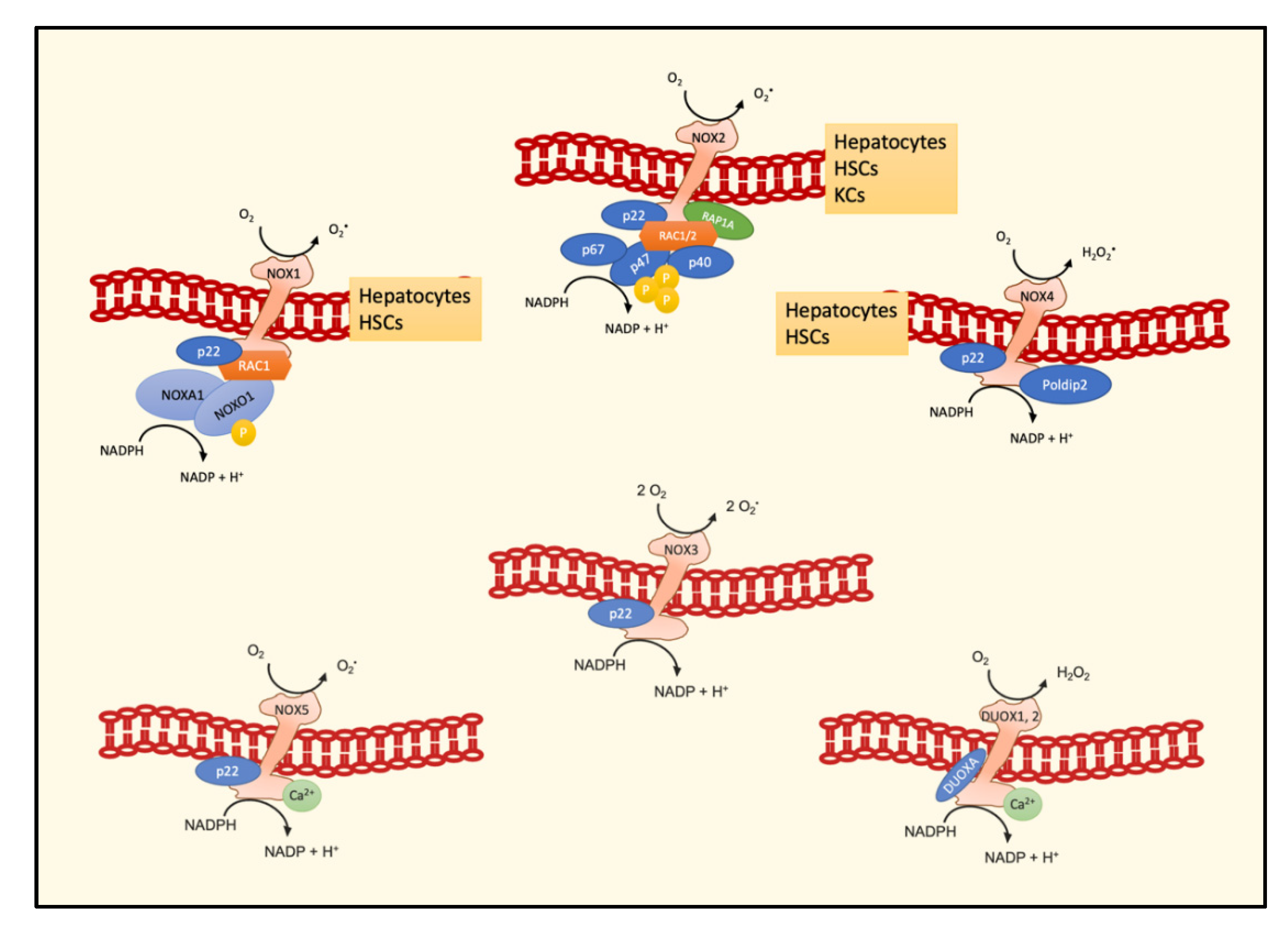
4. NOXs and the NASH–HCC Transition
4.1. NOX1
4.2. NOX2
4.3. NOX4
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Huang, T.; Behary, J.; Zekry, A. Non-alcoholic fatty liver disease: A review of epidemiology, risk factors, diagnosis and management. Intern. Med. J. 2020, 50, 1038–1047. [Google Scholar] [CrossRef]
- Straś, W.; Małkowski, P.; Tronina, O. Hepatocellular carcinoma in patients with non-alcoholic steatohepatitis–epimiology, risk factors, clinical implications and treatment. Clin. Exp. Hepatol. 2020, 6, 170–175. [Google Scholar] [CrossRef] [PubMed]
- Povsic, M.; Wong, O.Y.; Perry, R.; Bottomley, J. A structured literature review of the epidemiology and disease burden of non-alcoholic steatohepatitis (NASH). Adv. Ther. 2019, 36, 1574–1594. [Google Scholar] [CrossRef] [PubMed]
- Pierantonelli, I.; Svegliati-Baroni, G. Nonalcoholic Fatty Liver Disease: Basic Pathogenetic Mechanisms in the Progression From NAFLD to NASH. Transplantation 2019, 103, e1–e13. [Google Scholar] [CrossRef]
- Cholankeril, G.; Patel, R.; Khurana, S.; Satapathy, S.K. Hepatocellular carcinoma in non-alcoholic steatohepatitis: Current knowledge and implications for management. World J. Hepatol. 2017, 9, 533–543. [Google Scholar] [CrossRef]
- Llovet, J.M.; Kelley, R.K.; Villanueva, A.; Singal, A.G.; Pikarsky, E.; Roayaie, S.; Lencioni, R.; Koike, K.; Zucman-Rossi, J.; Finn, R.S. Hepatocellular carcinoma. Nat. Rev. Dis. Prim. 2021, 7, 1–28. [Google Scholar] [CrossRef]
- Global Burden of Disease Liver Cancer Collaboration; Akinyemiju, T.; Abera, S.; Ahmed, M.; Alam, N.; Alemayohu, M.A.; Allen, C.; Al-Raddadi, R.; Alvis-Guzman, N.; Amoako, Y.; et al. The Burden of Primary Liver Cancer and Underlying Etiologies From 1990 to 2015 at the Global, Regional, and National Level: Results from the Global Burden of Disease Study 2015. JAMA Oncol. 2017, 3, 1683–1691. [Google Scholar] [CrossRef]
- Giraud, J.; Chalopin, D.; Blanc, J.-F.; Saleh, M. Hepatocellular Carcinoma Immune Landscape and the Potential of Immunotherapies. Front. Immunol. 2021, 12, 655697. [Google Scholar] [CrossRef]
- Arroyave-Ospina, J.C.; Wu, Z.; Geng, Y.; Moshage, H. Role of Oxidative Stress in the Pathogenesis of Non-Alcoholic Fatty Liver Disease: Implications for Prevention and Therapy. Antioxidants 2021, 10, 174. [Google Scholar] [CrossRef]
- Sutti, S.; Albano, E. Adaptive immunity: An emerging player in the progression of NAFLD. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Bellanti, F.; Villani, R.; Facciorusso, A.; Vendemiale, G.; Serviddio, G. Lipid oxidation products in the pathogenesis of non-alcoholic steatohepatitis. Free Radic. Biol. Med. 2017, 111, 173–185. [Google Scholar] [CrossRef] [PubMed]
- Busch, C.J.-L.; Hendrikx, T.; Weismann, D.; Jäckel, S.; Walenbergh, S.M.A.; Rendeiro, A.F.; Weißer, J.; Puhm, F.; Hladik, A.; Göderle, L.; et al. Malondialdehyde epitopes are sterile mediators of hepatic inflammation in hypercholesterolemic mice: Steatohepatitis/Metabolic Liver Disease. Hepatology 2017, 65, 1181–1195. [Google Scholar] [CrossRef] [PubMed]
- Hendrikx, T.; Binder, C.J. Oxidation-Specific Epitopes in Non-Alcoholic Fatty Liver Disease. Front. Endocrinol. 2020, 11, 607011. [Google Scholar] [CrossRef] [PubMed]
- Binder, C.J.; Papac-Milicevic, N.; Witztum, J.L. Innate sensing of oxidation-specific epitopes in health and disease. Nat. Rev. Immunol. 2016, 16, 485–497. [Google Scholar] [CrossRef] [PubMed]
- Mihm, S. Danger-Associated Molecular Patterns (DAMPs): Molecular Triggers for Sterile Inflammation in the Liver. Int. J. Mol. Sci. 2018, 19, 3104. [Google Scholar] [CrossRef] [PubMed]
- Guéraud, F.; Atalay, M.; Bresgen, N.; Cipak, A.; Eckl, P.M.; Huc, L.; Jouanin, I.; Siems, W.; Uchida, K. Chemistry and biochemistry of lipid peroxidation products. Free Radic. Res. 2010, 44, 1098–1124. [Google Scholar] [CrossRef]
- Zelber-Sagi, S.; Ivancovsky-Wajcman, D.; Fliss-Isakov, N.; Hahn, M.; Webb, M.; Shibolet, O.; Kariv, R.; Tirosh, O. Serum Malondialdehyde is Associated with Non-Alcoholic Fatty Liver and Related Liver Damage Differentially in Men and Women. Antioxidants 2020, 9, 578. [Google Scholar] [CrossRef] [PubMed]
- Bieghs, V.; Walenbergh, S.M.A.; Hendrikx, T.; van Gorp, P.J.; Verheyen, F.; Olde Damink, S.W.; Masclee, A.A.; Koek, G.H.; Hofker, M.H.; Binder, C.J.; et al. Trapping of oxidized LDL in lysosomes of Kupffer cells is a trigger for hepatic inflammation. Liver Int. 2013, 33, 1056–1061. [Google Scholar] [CrossRef]
- Sun, X.; Seidman, J.S.; Zhao, P.; Troutman, T.D.; Spann, N.J.; Que, X.; Zhou, F.; Liao, Z.; Pasillas, M.; Yang, X.; et al. Neutralization of Oxidized Phospholipids Ameliorates Non-Alcoholic Steatohepatitis. Cell Metab. 2020, 31, 189–206.e8. [Google Scholar] [CrossRef] [PubMed]
- Mol, M.; Degani, G.; Coppa, C.; Baron, G.; Popolo, L.; Carini, M.; Aldini, G.; Vistoli, G.; Altomare, A. Advanced lipoxidation end products (ALEs) as RAGE binders: Mass spectrometric and computational studies to explain the reasons why. Redox Biol. 2019, 23, 101083. [Google Scholar] [CrossRef]
- Palanissami, G.; Paul, S.F.D. RAGE and Its Ligands: Molecular Interplay Between Glycation, Inflammation, and Hallmarks of Cancer—A Review. Horm. Cancer 2018, 9, 295–325. [Google Scholar] [CrossRef] [PubMed]
- Zeng, S.; Feirt, N.; Goldstein, M.; Guarrera, J.; Ippagunta, N.; Ekong, U.; Dun, H.; Lu, Y.; Qu, W.; Schmidt, A.M.; et al. Blockade of receptor for advanced glycation end product (RAGE) attenuates ischemia and reperfusion injury to the liver in mice. Hepatology 2004, 39, 422–432. [Google Scholar] [CrossRef] [PubMed]
- Bijnen, M.; Beelen, N.; Wetzels, S.; van de Gaar, J.; Vroomen, M.; Wijnands, E.; Scheijen, J.L.; van de Waarenburg, M.P.H.; Gijbels, M.J.; Cleutjens, J.P.; et al. RAGE deficiency does not affect non-alcoholic steatohepatitis and atherosclerosis in Western type diet-fed Ldlr−/−mice. Sci. Rep. 2018, 8, 15256. [Google Scholar] [CrossRef] [PubMed]
- Pang, Q.; Sun, Z.; Shao, C.; Cai, H.; Bao, Z.; Wang, L.; Li, L.; Jing, L.; Zhang, L.; Wang, Z. CML/RAGE Signal Bridges a Common Pathogenesis Between Atherosclerosis and Non-Alcoholic Fatty Liver. Front. Med. 2020, 7, 583943. [Google Scholar] [CrossRef]
- Albano, E. Immune response towards lipid peroxidation products as a predictor of progression of non-alcoholic fatty liver disease to advanced fibrosis. Gut 2005, 54, 987–993. [Google Scholar] [CrossRef]
- Sutti, S.; Jindal, A.; Locatelli, I.; Vacchiano, M.; Gigliotti, L.; Bozzola, C.; Albano, E. Adaptive immune responses triggered by oxidative stress contribute to hepatic inflammation in NASH. Hepatology 2014, 59, 886–897. [Google Scholar] [CrossRef]
- Nobili, V.; Parola, M.; Alisi, A.; Marra, F.; Piemonte, F.; Mombello, C.; Sutti, S.; Povero, D.; Maina, V.; Novo, E.; et al. Oxidative stress parameters in paediatric non-alcoholic fatty liver disease. Int. J. Mol. Med. 2010, 26, 471–476. [Google Scholar] [CrossRef]
- Castellani, G.; Contarini, G.; Mereu, M.; Albanesi, E.; Devroye, C.; D’Amore, C.; Ferretti, V.; De Martin, S.; Papaleo, F. Dopamine-mediated immunomodulation affects choroid plexus function. Brain Behav. Immun. 2019, 81, 138–150. [Google Scholar] [CrossRef]
- Ma, X.; Hua, J.; Mohamood, A.R.; Hamad, A.R.A.; Ravi, R.; Li, Z. A high-fat diet and regulatory T cells influence susceptibility to endotoxin-induced liver injury. Hepatology 2007, 46, 1519–1529. [Google Scholar] [CrossRef]
- Cairoli, V.; De Matteo, E.; Rios, D.; Lezama, C.; Galoppo, M.; Casciato, P.; Mullen, E.; Giadans, C.; Bertot, G.; Preciado, M.V.; et al. Hepatic lymphocytes involved in the pathogenesis of pediatric and adult non-alcoholic fatty liver disease. Sci. Rep. 2021, 11, 1–10. [Google Scholar] [CrossRef]
- Anstee, Q.M.; Reeves, H.L.; Kotsiliti, E.; Govaere, O.; Heikenwalder, M. From NASH to HCC: Current concepts and future challenges. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 411–428. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.; Stepanova, M.; Ong, J.P.; Jacobson, I.M.; Bugianesi, E.; Duseja, A.; Eguchi, Y.; Wong, V.W.; Negro, F.; Yilmaz, Y.; et al. Nonalcoholic Steatohepatitis Is the Fastest Growing Cause of Hepatocellular Carcinoma in Liver Transplant Candidates. Clin. Gastroenterol. Hepatol. 2019, 17, 748–755.e3. [Google Scholar] [CrossRef]
- Huang, A.; Yang, X.-R.; Chung, W.-Y.; Dennison, A.R.; Zhou, J. Targeted therapy for hepatocellular carcinoma. Sig. Transduct. Target. Ther. 2020, 5, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Uchida, D.; Takaki, A.; Oyama, A.; Adachi, T.; Wada, N.; Onishi, H.; Okada, H. Oxidative Stress Management in Chronic Liver Diseases and Hepatocellular Carcinoma. Nutrients 2020, 12, 1576. [Google Scholar] [CrossRef] [PubMed]
- Poetsch, A.R. The genomics of oxidative DNA damage, repair, and resulting mutagenesis. Comput. Struct. Biotechnol. J. 2020, 18, 207–219. [Google Scholar] [CrossRef] [PubMed]
- McLoughlin, M.R.; Orlicky, D.J.; Prigge, J.R.; Krishna, P.; Talago, E.A.; Cavigli, I.R.; Eriksson, S.; Miller, C.G.; Kundert, J.A.; Sayin, V.I.; et al. TrxR1, Gsr, and oxidative stress determine hepatocellular carcinoma malignancy. Proc. Natl. Acad. Sci. USA 2019, 116, 11408–11417. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; Dinkova-Kostova, A.T.; Tew, K.D. Oxidative Stress in Cancer. Cancer Cell 2020, 38, 167–197. [Google Scholar] [CrossRef]
- Cho, E.S.; Cha, Y.H.; Kim, H.S.; Kim, N.H.; Yook, J.I. The Pentose Phosphate Pathway as a Potential Target for Cancer Therapy. Biomol. Ther. 2018, 26, 29–38. [Google Scholar] [CrossRef]
- Giacomini, I.; Ragazzi, E.; Pasut, G.; Montopoli, M. The Pentose Phosphate Pathway and Its Involvement in Cisplatin Resistance. Int. J. Mol. Sci. 2020, 21, 937. [Google Scholar] [CrossRef]
- Bedard, K.; Krause, K.-H. The NOX Family of ROS-Generating NADPH Oxidases: Physiology and Pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Chen, Z.; Tian, R.; She, Z.; Cai, J.; Li, H. Role of oxidative stress in the pathogenesis of nonalcoholic fatty liver disease. Free Radic. Biol. Med. 2020, 152, 116–141. [Google Scholar] [CrossRef]
- Yoshida, T.; Murayama, H.; Kawashima, M.; Nagahara, R.; Kangawa, Y.; Mizukami, S.; Kimura, M.; Abe, H.; Hayashi, S.; Shibutani, M. Apocynin and enzymatically modified isoquercitrin suppress the expression of a NADPH oxidase subunit p22phox in steatosis-related preneoplastic liver foci of rats. Exp. Toxicol. Pathol. 2017, 69, 9–16. [Google Scholar] [CrossRef]
- Prior, K.-K.; Leisegang, M.S.; Josipovic, I.; Löwe, O.; Shah, A.M.; Weissmann, N.; Schröder, K.; Brandes, R.P. CRISPR/Cas9-mediated knockout of p22phox leads to loss of Nox1 and Nox4, but not Nox5 activity. Redox Biol. 2016, 9, 287–295. [Google Scholar] [CrossRef]
- Jiang, J.X.; Török, N.J. NADPH Oxidases in Chronic Liver Diseases. Adv. Hepatol. 2014, 2014, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Gabbia, D.; Pozzo, L.; Zigiotto, G.; Roverso, M.; Sacchi, D.; Dalla Pozza, A.; Carrara, M.; Bogialli, S.; Floreani, A.; Guido, M.; et al. Dexamethasone counteracts hepatic inflammation and oxidative stress in cholestatic rats via CAR activation. PLoS ONE 2018, 13, e0204336. [Google Scholar] [CrossRef]
- Cremonini, E.; Oteiza, P.I. (-)-Epicatechin and its metabolites prevent palmitate-induced NADPH oxidase upregulation, oxidative stress and insulin resistance in HepG2 cells. Arch. Biochem. Biophys. 2018, 646, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Bettaieb, A.; Vazquez Prieto, M.A.; Rodriguez Lanzi, C.; Miatello, R.M.; Haj, F.G.; Fraga, C.G.; Oteiza, P.I. (−)-Epicatechin mitigates high-fructose-associated insulin resistance by modulating redox signaling and endoplasmic reticulum stress. Free Radic. Biol. Med. 2014, 72, 247–256. [Google Scholar] [CrossRef]
- Rabelo, F.; Stefano, J.T.; Cavaleiro, A.M.; Lima, R.V.C.; de Campos Mazo, D.F.; Carrilho, F.J.; Correa-Giannella, M.L.; Oliveira, C.P. Association between the CYBA and NOX4 genes of NADPH oxidase and its relationship with metabolic syndrome in non-alcoholic fatty liver disease in Brazilian population. Hepatobiliary Pancreat. Dis. Int. 2018, 17, 330–335. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi-Bardbori, A.; Vikström Bergander, L.; Rannug, U.; Rannug, A. NADPH Oxidase-Dependent Mechanism Explains How Arsenic and Other Oxidants Can Activate Aryl Hydrocarbon Receptor Signaling. Chem. Res. Toxicol. 2015, 28, 2278–2286. [Google Scholar] [CrossRef]
- Floreani, M.; Gabbia, D.; Barbierato, M.; De Martin, S.; Palatini, P. Differential Inducing Effect of Benzo [a] Pyrene on Gene Expression and Enzyme Activity of Cytochromes P450 1A1 and 1A2 in Sprague-Dawley and Wistar Rats. Drug Metab. Pharm. 2012, 27, 640–652. [Google Scholar] [CrossRef]
- Uno, S.; Nebert, D.W.; Makishima, M. Cytochrome P450 1A1 (CYP1A1) protects against nonalcoholic fatty liver disease caused by Western diet containing benzo [a] pyrene in mice. Food Chem. Toxicol. 2018, 113, 73–82. [Google Scholar] [CrossRef]
- Liang, S.; Kisseleva, T.; Brenner, D.A. The Role of NADPH Oxidases (NOXs) in Liver Fibrosis and the Activation of Myofibroblasts. Front. Physiol. 2016, 7, 17. [Google Scholar] [CrossRef]
- Valdivia, A.; Duran, C.; Martin, A. The role of Nox-mediated oxidation in the regulation of cytoskeletal dynamics. Curr. Pharm. Des. 2015, 21, 6009–6022. [Google Scholar] [CrossRef]
- Eun, H.S.; Cho, S.Y.; Joo, J.S.; Kang, S.H.; Moon, H.S.; Lee, E.S.; Kim, S.H.; Lee, B.S. Gene expression of NOX family members and their clinical significance in hepatocellular carcinoma. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef]
- Eun, H.S.; Chun, K.; Song, I.-S.; Oh, C.-H.; Seong, I.-O.; Yeo, M.-K.; Kim, K.-H. High nuclear NADPH oxidase 4 expression levels are correlated with cancer development and poor prognosis in hepatocellular carcinoma. Pathology 2019, 51, 579–585. [Google Scholar] [CrossRef]
- Ha, S.Y.; Paik, Y.-H.; Yang, J.W.; Lee, M.J.; Bae, H.; Park, C.-K. NADPH Oxidase 1 and NADPH Oxidase 4 Have Opposite Prognostic Effects for Patients with Hepatocellular Carcinoma after Hepatectomy. Gut Liver 2016, 10, 826–835. [Google Scholar] [CrossRef]
- Chen, S.; Ling, Q.; Yu, K.; Huang, C.; Li, N.; Zheng, J.; Bao, S.; Cheng, Q.; Zhu, M.; Chen, M. Dual oxidase 1: A predictive tool for the prognosis of hepatocellular carcinoma patients. Oncol. Rep. 2016, 35, 3198–3208. [Google Scholar] [CrossRef] [PubMed]
- Crosas-Molist, E.; Bertran, E.; Sancho, P.; López-Luque, J.; Fernando, J.; Sánchez, A.; Fernández, M.; Navarro, E.; Fabregat, I. The NADPH oxidase NOX4 inhibits hepatocyte proliferation and liver cancer progression. Free Radic. Biol. Med. 2014, 69, 338–347. [Google Scholar] [CrossRef]
- Matsumoto, M.; Zhang, J.; Zhang, X.; Liu, J.; Jiang, J.X.; Yamaguchi, K.; Taruno, A.; Katsuyama, M.; Iwata, K.; Ibi, M.; et al. The NOX1 isoform of NADPH oxidase is involved in dysfunction of liver sinusoids in nonalcoholic fatty liver disease. Free Radic. Biol. Med. 2018, 115, 412–420. [Google Scholar] [CrossRef]
- Pierantonelli, I.; Lioci, G.; Gurrado, F.; Giordano, D.M.; Rychlicki, C.; Bocca, C.; Trozzi, L.; Novo, E.; Panera, N.; De Stefanis, C.; et al. HDL cholesterol protects from liver injury in mice with intestinal specific LXRα activation. Liver Int. 2020, 40, 3127–3139. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.; Ma, H.-Y.; Zhong, Z.; Dhar, D.; Liu, X.; Xu, J.; Koyama, Y.; Nishio, T.; Karin, D.; Karin, G.; et al. NADPH Oxidase 1 in Liver Macrophages Promotes Inflammation and Tumor Development in Mice. Gastroenterology 2019, 156, 1156–1172.e6. [Google Scholar] [CrossRef] [PubMed]
- Lambeth, J.D.; Kawahara, T.; Diebold, B. Regulation of Nox and Duox enzymatic activity and expression. Free Radic. Biol. Med. 2007, 43, 319–331. [Google Scholar] [CrossRef]
- Stalin, J.; Garrido-Urbani, S.; Heitz, F.; Szyndralewiez, C.; Jemelin, S.; Coquoz, O.; Ruegg, C.; Imhof, B.A. Inhibition of host NOX1 blocks tumor growth and enhances checkpoint inhibitor–based immunotherapy. Life Sci. Alliance 2019, 2, e201800265. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Qiao, J.; Fu, Y.-X. Immunotherapy and tumor microenvironment. Cancer Lett. 2016, 370, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Vandierendonck, A.; Degroote, H.; Vanderborght, B.; Verhelst, X.; Geerts, A.; Devisscher, L.; Van Vlierberghe, H. NOX1 inhibition attenuates the development of a pro-tumorigenic environment in experimental hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2021, 40, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Dou, C.; Xu, Q.; Liu, J.; Wang, Y.; Zhou, Z.; Yao, W.; Jiang, K.; Cheng, J.; Zhang, C.; Tu, K. SHMT1 inhibits the metastasis of HCC by repressing NOX1-mediated ROS production. J. Exp. Clin. Cancer Res. 2019, 38, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Mardinoglu, A.; Agren, R.; Kampf, C.; Asplund, A.; Uhlen, M.; Nielsen, J. Genome-Scale metabolic modelling of hepatocytes reveals serine deficiency in patients with non-alcoholic fatty liver disease. Nat. Commun. 2014, 5, 1–11. [Google Scholar] [CrossRef]
- Kim, S.Y.; Jeong, J.-M.; Kim, S.J.; Seo, W.; Kim, M.-H.; Choi, W.-M.; Yoo, W.; Lee, J.-H.; Shim, Y.-R.; Yi, H.-S.; et al. Pro-inflammatory hepatic macrophages generate ROS through NADPH oxidase 2 via endocytosis of monomeric TLR4–MD2 complex. Nat. Commun. 2017, 8, 1–15. [Google Scholar] [CrossRef]
- Sarkar, S.; Saha, P.; Seth, R.K.; Mondal, A.; Bose, D.; Kimono, D.; Albadrani, M.; Mukherjee, A.; Porter, D.E.; Scott, G.I.; et al. Higher intestinal and circulatory lactate associated NOX2 activation leads to an ectopic fibrotic pathology following microcystin co-exposure in murine fatty liver disease. Comp. Biochem. Physiol. Part. C Toxicol. Pharmacol. 2020, 238, 108854. [Google Scholar] [CrossRef]
- Luo, X.; Bai, Y.; He, S.; Sun, S.; Jiang, X.; Yang, Z.; Lu, D.; Wei, P.; Liang, Y.; Peng, C.; et al. Sirtuin 1 ameliorates defenestration in hepatic sinusoidal endothelial cells during liver fibrosis via inhibiting stress-induced premature senescence. Cell Prolif. 2021, 54, e12991. [Google Scholar] [CrossRef]
- Albadrani, M.; Seth, R.K.; Sarkar, S.; Kimono, D.; Mondal, A.; Bose, D.; Porter, D.E.; Scott, G.I.; Brooks, B.; Raychoudhury, S.; et al. Exogenous PP2A inhibitor exacerbates the progression of nonalcoholic fatty liver disease via NOX2-dependent activation of miR21. Am. J. Physiol. Gastrointest Liver Physiol. 2019, 317, G408–G428. [Google Scholar] [CrossRef]
- Shiau, D.-J.; Kuo, W.-T.; Davuluri, G.V.N.; Shieh, C.-C.; Tsai, P.-J.; Chen, C.-C.; Lin, Y.-S.; Wu, Y.-Z.; Hsiao, Y.-P.; Chang, C.-P. Hepatocellular carcinoma-derived high mobility group box 1 triggers M2 macrophage polarization via a TLR2/NOX2/autophagy axis. Sci. Rep. 2020, 10, 1–14. [Google Scholar] [CrossRef]
- Murayama, H.; Eguchi, A.; Nakamura, M.; Kawashima, M.; Nagahara, R.; Mizukami, S.; Kimura, M.; Makino, E.; Takahashi, N.; Ohtsuka, R.; et al. Spironolactone in Combination with α-glycosyl Isoquercitrin Prevents Steatosis-related Early Hepatocarcinogenesis in Rats through the Observed NADPH Oxidase Modulation. Toxicol. Pathol. 2018, 46, 530–539. [Google Scholar] [CrossRef]
- Bettaieb, A.; Jiang, J.X.; Sasaki, Y.; Chao, T.-I.; Kiss, Z.; Chen, X.; Tian, J.; Katsuyama, M.; Yabe-Nishimura, C.; Xi, Y.; et al. Hepatocyte Nicotinamide Adenine Dinucleotide Phosphate Reduced Oxidase 4 Regulates Stress Signaling, Fibrosis, and Insulin Sensitivity During Development of Steatohepatitis in Mice. Gastroenterology 2015, 149, 468–480.e10. [Google Scholar] [CrossRef]
- Carmona-Cuenca, I.; Roncero, C.; Sancho, P.; Caja, L.; Fausto, N.; Fernández, M.; Fabregat, I. Upregulation of the NADPH oxidase NOX4 by TGF-beta in hepatocytes is required for its pro-apoptotic activity. J. Hepatol. 2008, 49, 965–976. [Google Scholar] [CrossRef] [PubMed]
- Herranz-Itúrbide, M.; López-Luque, J.; Gonzalez-Sanchez, E.; Caballero-Díaz, D.; Crosas-Molist, E.; Martín-Mur, B.; Gut, M.; Esteve-Codina, A.; Jaquet, V.; Jiang, J.X.; et al. NADPH oxidase 4 (Nox4) deletion accelerates liver regeneration in mice. Redox Biol. 2021, 40, 101841. [Google Scholar] [CrossRef] [PubMed]
- Gu, Q.; He, Y.; Ji, J.; Yao, Y.; Shen, W.; Luo, J.; Zhu, W.; Cao, H.; Geng, Y.; Xu, J.; et al. Hypoxia-inducible factor 1α (HIF-1α) and reactive oxygen species (ROS) mediates radiation-induced invasiveness through the SDF-1α/CXCR4 pathway in non-small cell lung carcinoma cells. Oncotarget 2015, 6, 10893–10907. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Tu, K.; Wang, Y.; Yao, B.; Li, Q.; Wang, L.; Dou, C.; Liu, Q.; Zheng, X. Hypoxia Accelerates Aggressiveness of Hepatocellular Carcinoma Cells Involving Oxidative Stress, Epithelial-Mesenchymal Transition and Non-Canonical Hedgehog Signaling. Cell Physiol. Biochem. 2017, 44, 1856–1868. [Google Scholar] [CrossRef] [PubMed]
- Owada, S.; Endo, H.; Okada, C.; Yoshida, K.; Shida, Y.; Tatemichi, M. Setanaxib as a Potent Hypoxia-Specific Therapeutic Agent Against Liver Cancer. Anticancer Res. 2020, 40, 5071–5079. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Study | Model Used | Effect on NOXs | Outcome |
|---|---|---|---|
| Preclinical studies | |||
| Yoshida et al. [42] | Rat model of HCC (IP injection of N-diethylnitrosamine (DEN) and high-fat diet (HFD)) | The NOX inhibitor Apocynin downregulates p22phox (not p47phox and NOX4) in the hepatocytes of precancerous foci | NOX inhibition suppresses hyperlipidemia and steatosis-induced preneoplastic hepatic lesions |
| Cremonini et al. [46] | High-fat-diet-fed mice and palmitate-treated HepG2 cells | NOX3/4 increased in palmitate-treated HepG2 cells, as well as redox-sensitive kinases and oxidative stress | (-)-Epicatechin decreases the hepatic expression of NOX3 and NOX4, improving oxidative stress in high-fat-diet fed mice |
| Bettaieb et al. [47] | High fructose-fed rats | A high-fructose diet upregulates NOX2, p47phox, and NOX4 in the adipose tissue and NOX4 in the liver | (-)-Epicatechin prevents hepatic NOX activation and decreases the upregulation of NOX2 and NOX4, modulating superoxide production in the liver and adipose tissue |
| Liang et al. [61] | Mouse model of HCC (IP DEN injection in wild-type (WT) C57BL/6 mice and in Nox1−/−, Nox4−/−, and double knockout mice) | NOX1 expression in macrophages promotes hepatic tumorigenesis through the release of pro-inflammatory cytokines. | Blocking NOX1- and/or NOX1-mediated cytokine release might slow HCC progression. NOX1 ablation in macrophages inhibits cancer growth |
| Kim et al. [68] | Mouse models of HFD-induced NAFLD: WT C57BL/6 mice, NOX2 (gp91phox)-, TLR4-, MyD88- and Trif-KO mice | NAFLD is reduced in NOX2 (gp91phox)- and TLR4-KO mice | NOX2 deficiency attenuates HFD-induced steatosis and insulin resistance |
| Sarkar et al. [69] | WT C57BL/6 mice and p47phox KO mice fed with methionine choline-deficient and high-fat diet (MCD-HFD) | Lactate activates NOX2, which mediates cell differentiation and fibrosis | The NOX inhibitor Apocynin inhibits ectopic steatosis |
| Albadrani et al. [71] | WT C57BL/6 mice and p47phox and miR21 KO mice fed with MCD-HFD | PP2A inhibition exacerbates NAFLD by activating p47phox. | Microcystin-induced NAFLD exacerbation is reduced in p47phox- and miR21-KO mice |
| Murayama et al. [73] | HCC model in HFD-fed rats | The NOX subunit p22phox is expressed in HFD-induced preneoplastic hepatic foci | α -glycosyl isoquercitrin and spironolactone prevent precancerous lesions, suppress HFD-induced hyperlipidemia and early hepatocarcinogenesis by reducing p67phox and p22phox expression in precancerous lesions |
| Liu et al. [78] | Healthy immortalized human hepatocytes and HCC cell lines (MHCC-97H, Hep3B, Huh7, MHCC-97L, and HCCLM3) | Hypoxia upregulates NOX4 and prompts ROS-mediated progression and invasion of HCC cells | siRNA-mediated knockdown of NOX4 results in deletion of ROS generation and GLI1-dependent activation and invasion of hypoxic HCC cells |
| Owada et al. [79] | Hepatic cancer cell lines (HepG2, HLE, and Alexander, PLC/PRF/5) | NOX1/4 inhibition induces selective cytotoxicity and apoptosis of hypoxic cancer cells. | The NOX1/4 inhibitor setanaxib (GKT137831) is able to induce apoptosis in cancer cell, thus representing a promising drug candidate for HCC |
| Vandierendonck et al. [65] | Hepatic cancer (Huh7, Hep3B Hepa1-6) and monocytic human cell lines, murine macrophages. HCC mouse model (DEN injection) | NOX1 inhibition modulates the polarization of macrophages and affects pro-inflammatory, angiogenic, and fibrotic markers | The NOX1 inhibitor GKT771 reduces inflammation, angiogenesis, and fibrosis |
| Shiau et al. [72] | Murine HCC model (hepatoma sh-luciferase (Luc) or shHMGB1-ML-14a cells) | NOX2 affects M2 macrophage polarization, sustaining the development of HCC | The HMGB1 and ROS inhibitors ethyl pyruvate and N-acetylcysteine amide decrease M2 macrophage accumulation and liver nodule formation in HCC-bearing mice |
| Carmona-Cuenca et al. [75] | Primary cultures of rat and human hepatocytes, HepG2, and Hep3B cell lines | TGF-β upregulates NOX4. NOX4 deletion attenuates caspase activation and death of rat hepatocytes | TGF-β displays its proapoptotic activity by the upregulation of NOX4, and its impairment causes HCC cell resistance toward apoptosis |
| Clinical studies | |||
| Rabelo et al. [48] | NAFLD and NASH patients | The rs3017887 SNP of NOX4 is associated with higher ALT concentration in NAFLD patients, the AA genotype in the CYBA-675 T/A CYBA polymorphism with higher triglyceride and lower HDL levels in NASH patients | Genetic NOX polymorphisms are correlated with specific phenotypes in NAFLD/NASH patients |
| Eun et al. [54] | Liver tissues of HCC patients and healthy subjects | NOX1, NOX2, and NOX5 correlate with metastasis-associated genes, NOX4 and DUOX1 are linked to tumor progression | Higher mRNA levels of NOX4 and DUOX1 correlate with prolonged overall survival, whereas higher levels of NOX1, NOX2, and NOX5 are associated with a poor overall survival |
| Ha et al. [56] | Liver tissues of HCC patients | In HCC patients, high levels of NOX1 and low levels of NOX4 are observed | NOX1 and NOX4 expression displays an opposite prognostic effect in HCC: high NOX1 and low NOX4 levels correlate with shorter recurrence-free survival and overall survival |
| Matsumoto et al. [59] | NASH patients | In NASH patients, NOX1 is upregulated in liver sinusoidal endothelial cells (LSECs) | NOX1 upregulation prompts peroxy-nitrite-mediated cellular injury and impairs hepatic microcirculation, helping NAFLD progression |
| Dou et al. [66] | HCC tissues and cell lines | NOX1 is a downstream target of the tumor suppressor SHMT1 in HCC | NOX1 expression is negatively correlated with SHMT1 expression in HCC |
| Eun et al. [55] | Liver tissues of HCC patients and healthy subjects | Cytoplasmic NOX2 and nuclear NOX4 expression are increased in HCC cells. NOX4 translocation into the nucleus affects HCC development and progression | NOX2 and NOX4 increased expression in HCC cells correlates with liver cirrhosis. NOX2 and NOX4 could represent diagnostic markers of HCC prognosis |
| Bettaieb et al. [74] | NAFLD and NASH patients | NASH increases hepatic NOX4 expression, and its deletion in hepatocytes reduces oxidative stress, lipid peroxidation, and liver fibrosis in NASH mice | Hepatic NOX4 deletion is reduces diet-induced hepatic injury and fibrosis. NOX inhibition reduces liver inflammation and fibrosis and increases insulin sensitivity |
| Chen et al. [57] | HCC patients | High DUOX1 levels in HCC patients correlate with a better prognosis in terms of disease-free survival and overall survival after resection | DUOX1 represents a valuable predictor of HCC patients’ survival |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gabbia, D.; Cannella, L.; De Martin, S. The Role of Oxidative Stress in NAFLD–NASH–HCC Transition—Focus on NADPH Oxidases. Biomedicines 2021, 9, 687. https://doi.org/10.3390/biomedicines9060687
Gabbia D, Cannella L, De Martin S. The Role of Oxidative Stress in NAFLD–NASH–HCC Transition—Focus on NADPH Oxidases. Biomedicines. 2021; 9(6):687. https://doi.org/10.3390/biomedicines9060687
Chicago/Turabian StyleGabbia, Daniela, Luana Cannella, and Sara De Martin. 2021. "The Role of Oxidative Stress in NAFLD–NASH–HCC Transition—Focus on NADPH Oxidases" Biomedicines 9, no. 6: 687. https://doi.org/10.3390/biomedicines9060687
APA StyleGabbia, D., Cannella, L., & De Martin, S. (2021). The Role of Oxidative Stress in NAFLD–NASH–HCC Transition—Focus on NADPH Oxidases. Biomedicines, 9(6), 687. https://doi.org/10.3390/biomedicines9060687