
PFOS Inhibited Normal Functional Development of Placenta Cells via PPARγ Signaling


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cell Culture and Animal Treatment
2.3. si-PPARγ and pcDNA-PPARγ Transfection
2.4. Cell Viability Assay
2.5. Cell Migration Assay
2.6. Tube Formation Assay
2.7. Real-Time PCR (RT-PCR)
2.8. Statistical Analysis
3. Results
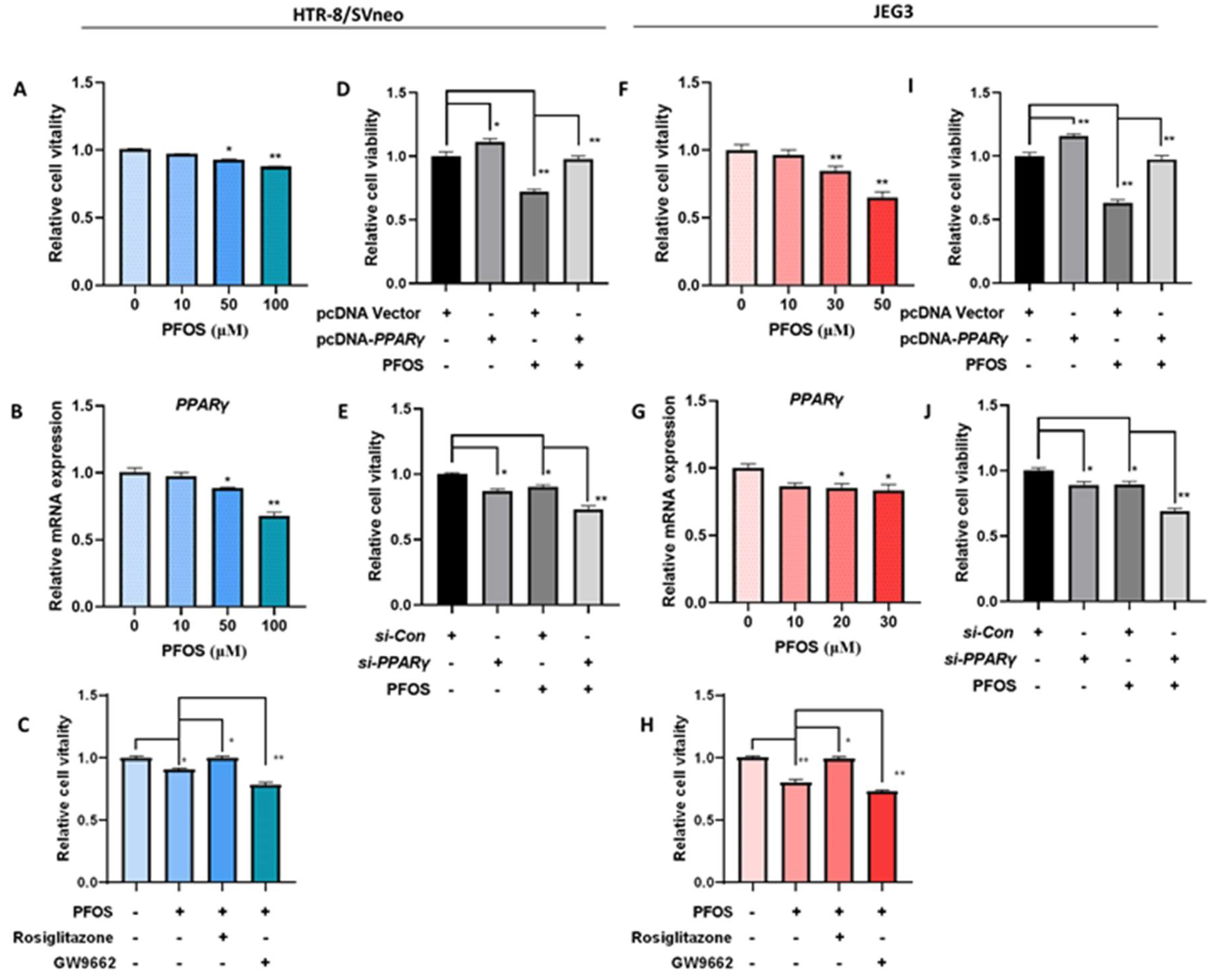
3.1. PPARγ Mediates PFOS-Induced Inhibition of Trophoblast Cells Survival and Proliferation In Vitro
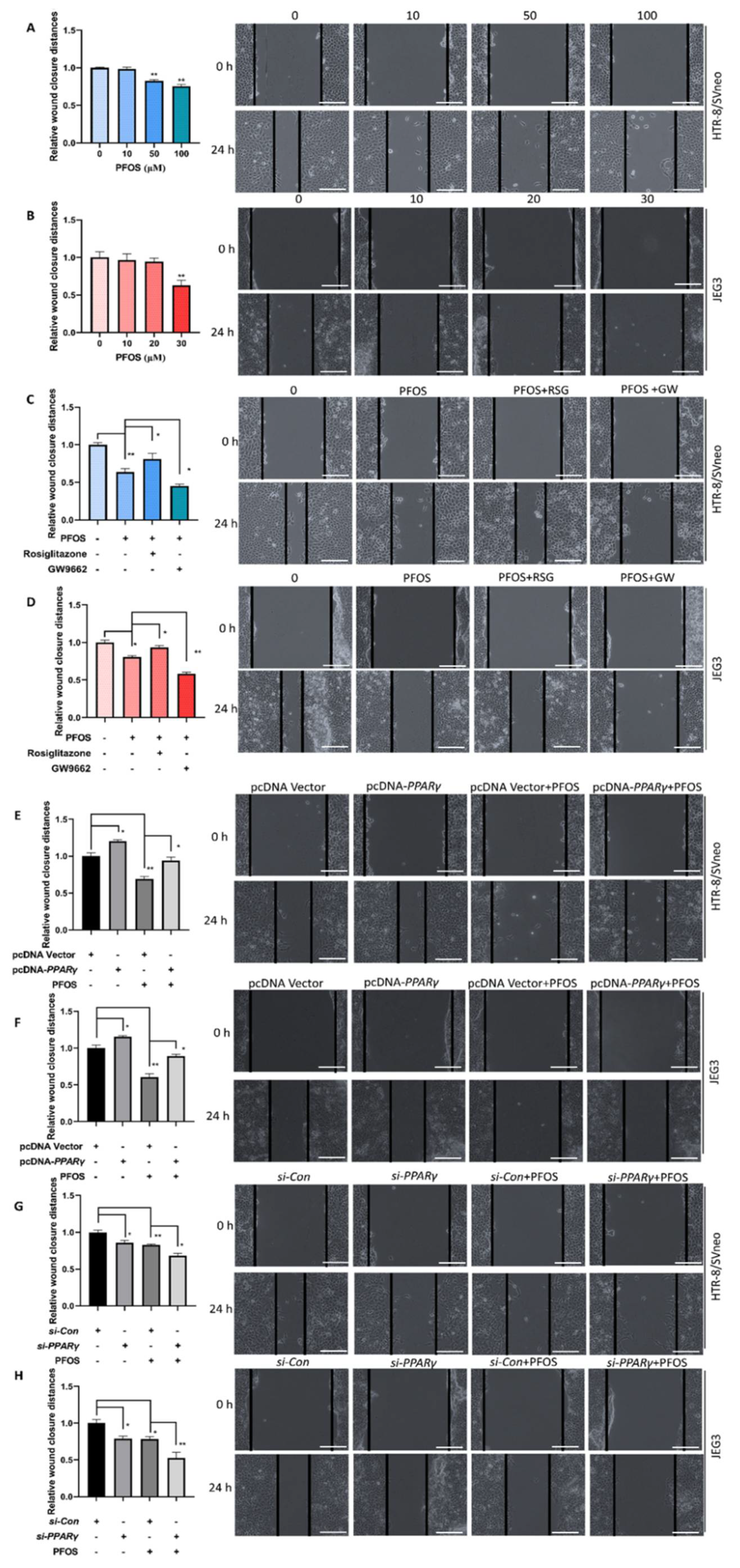
3.2. PPARγ Is Important for Inhibition Effect of PFOS on the Cell Migration
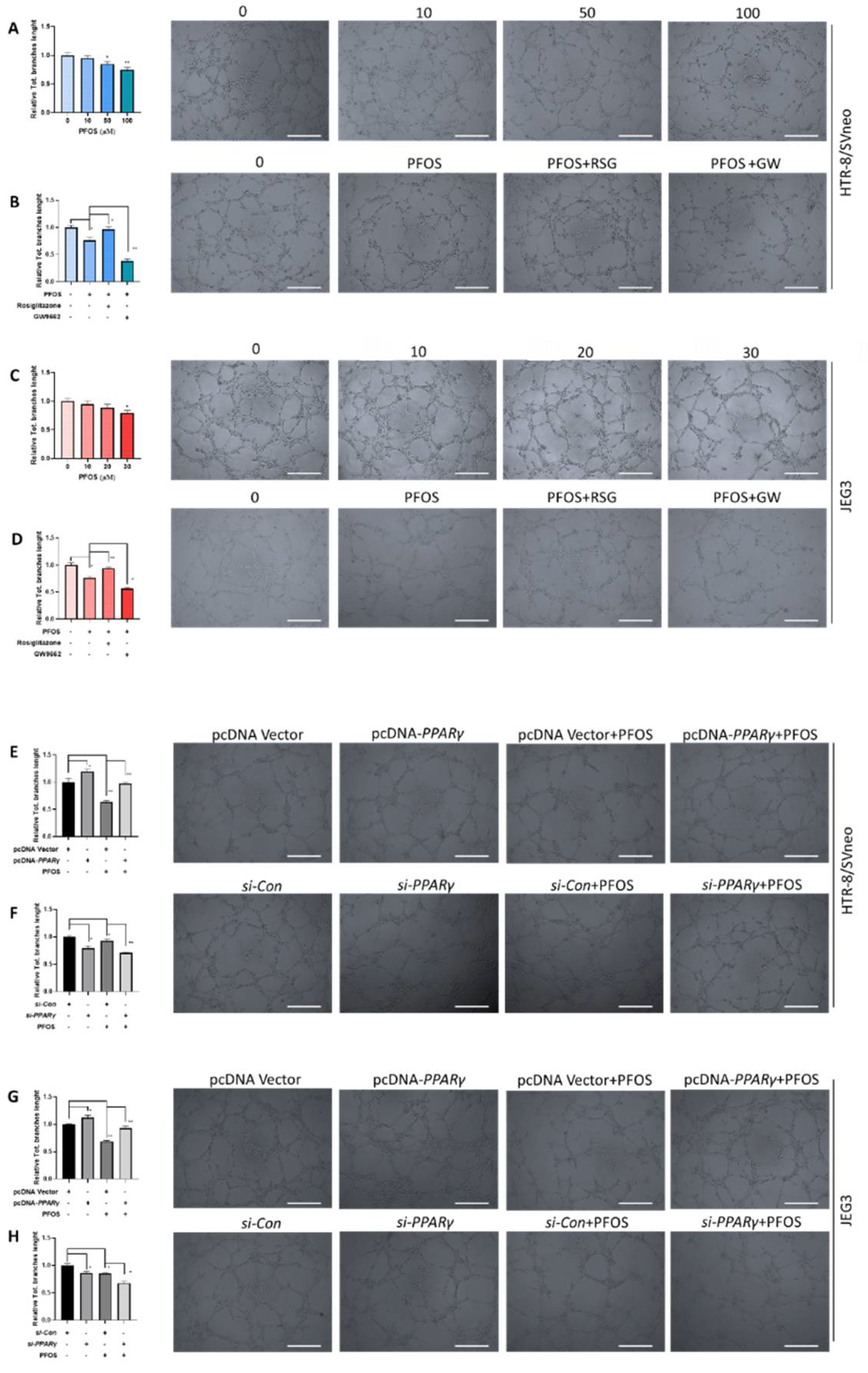
3.3. PPARγ Is Involved in Impaired PFOS–Induced Angiogenesis
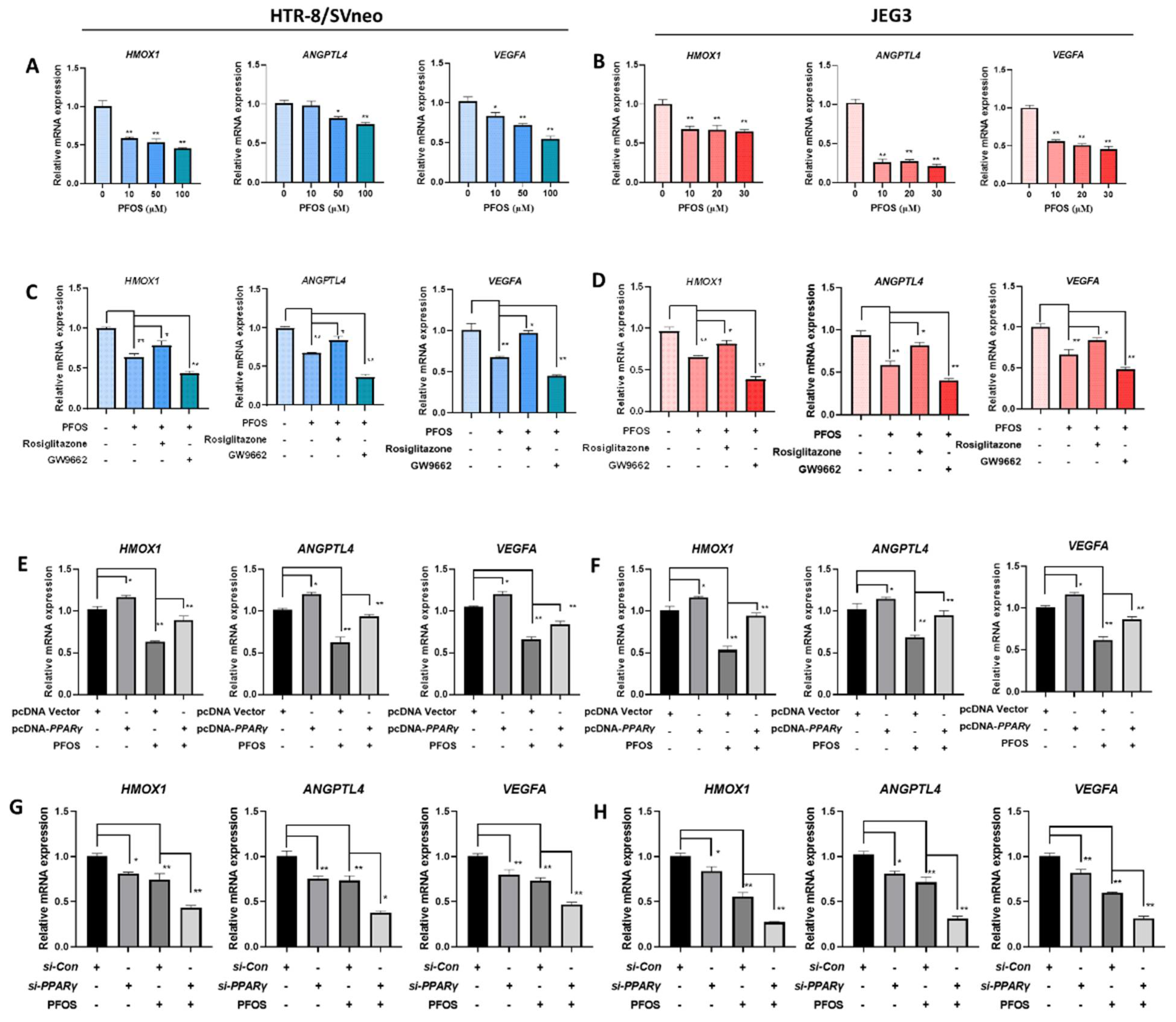
3.4. PFOS Alters mRNA Level of PPARγ Target Genes Associated with Proliferation and Angiogenesis
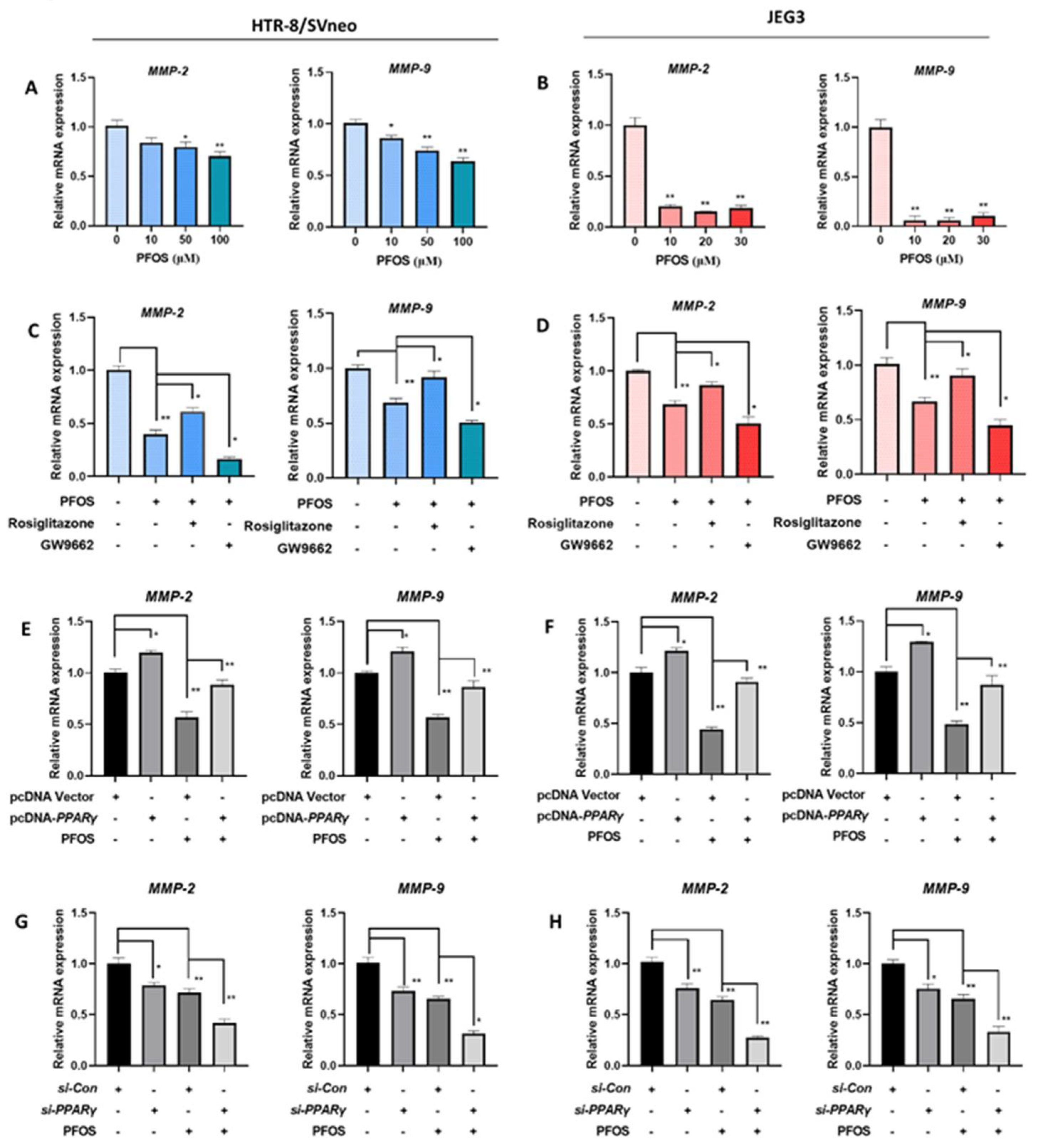
3.5. PFOS Alters mRNA Level of PPARγ Target Genes Associated with Migration
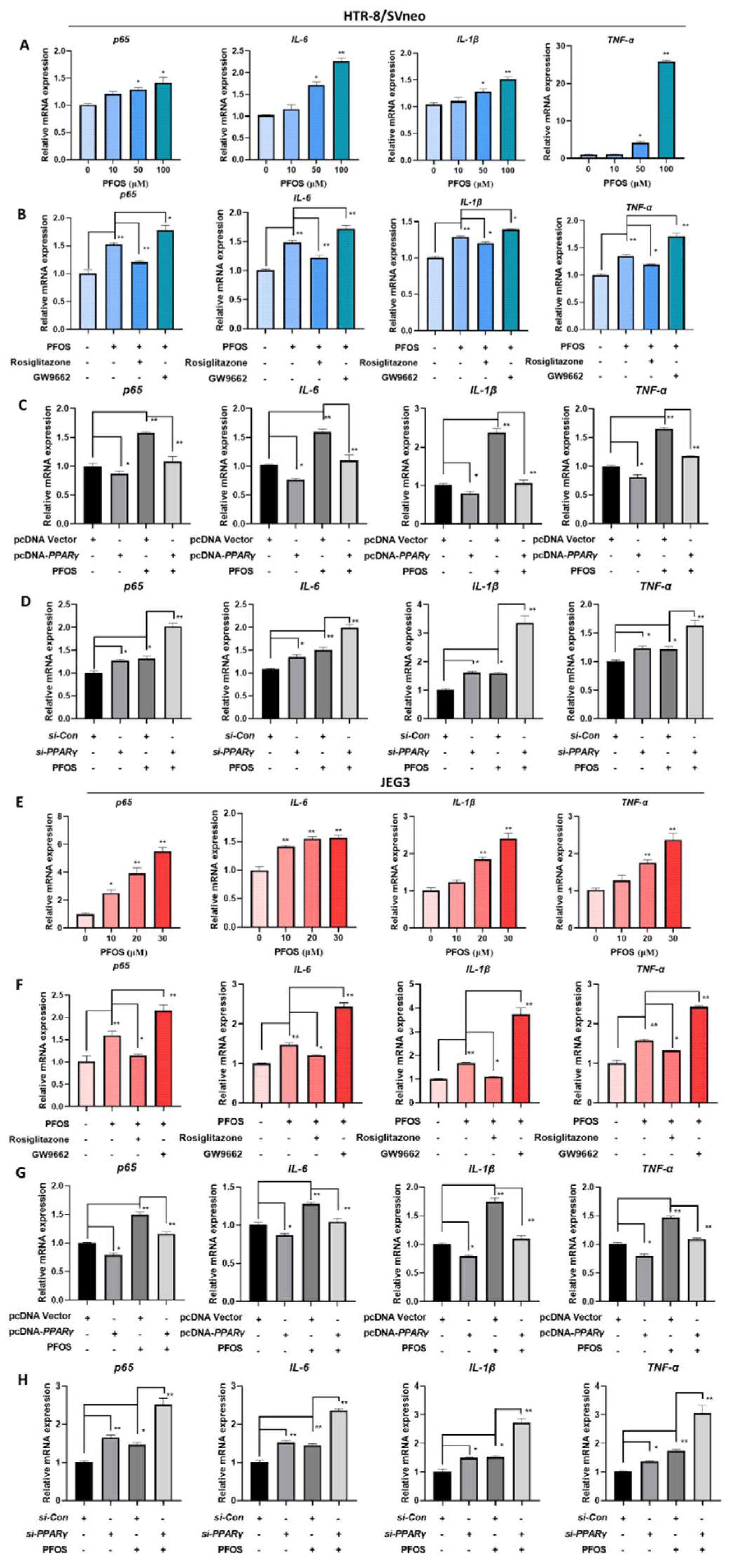
3.6. PFOS Alters mRNA Level of PPARγ Target Genes Associated with Inflammation
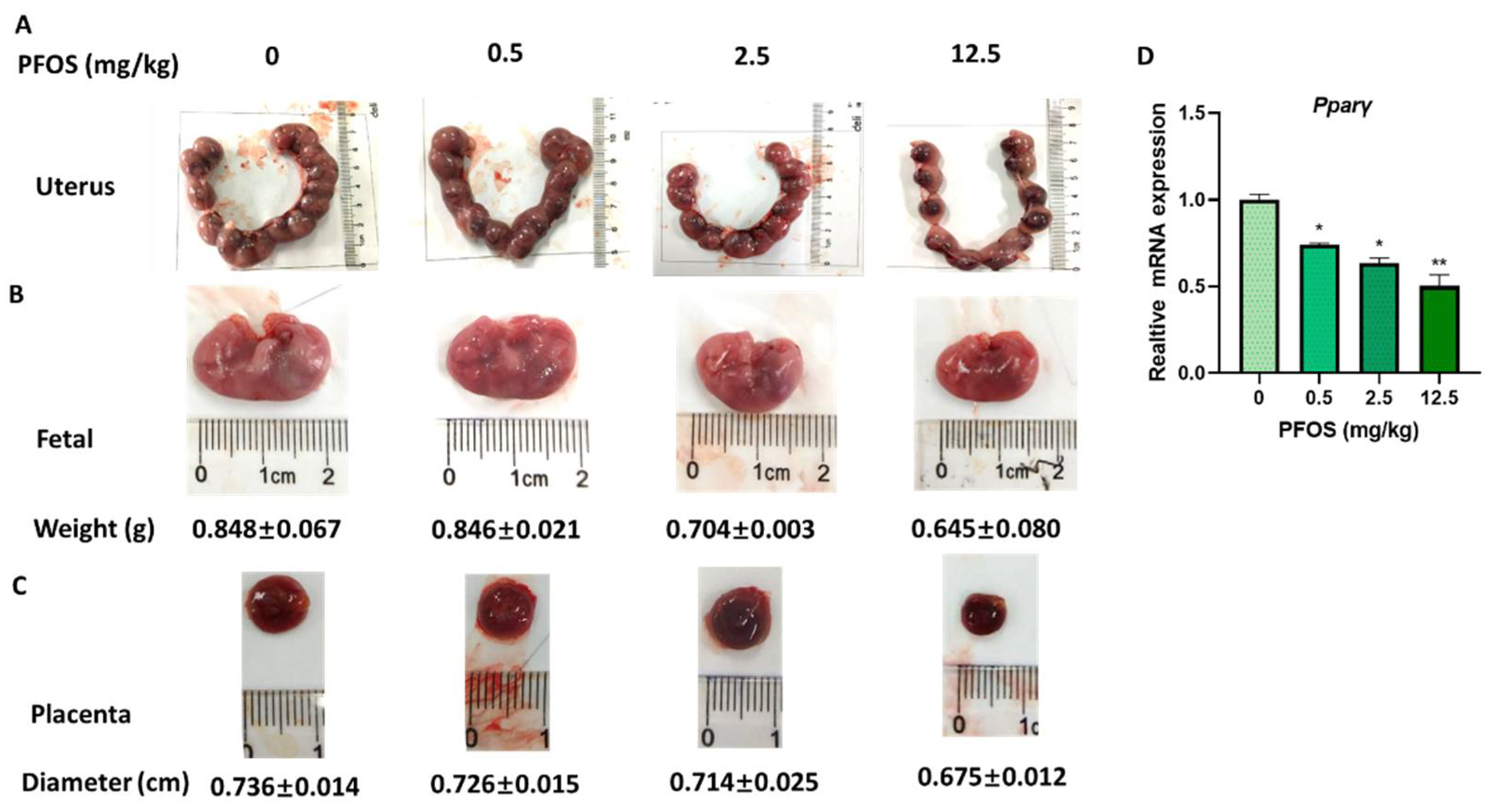
3.7. PFOS Induces Placenta Dysfunction in Mice
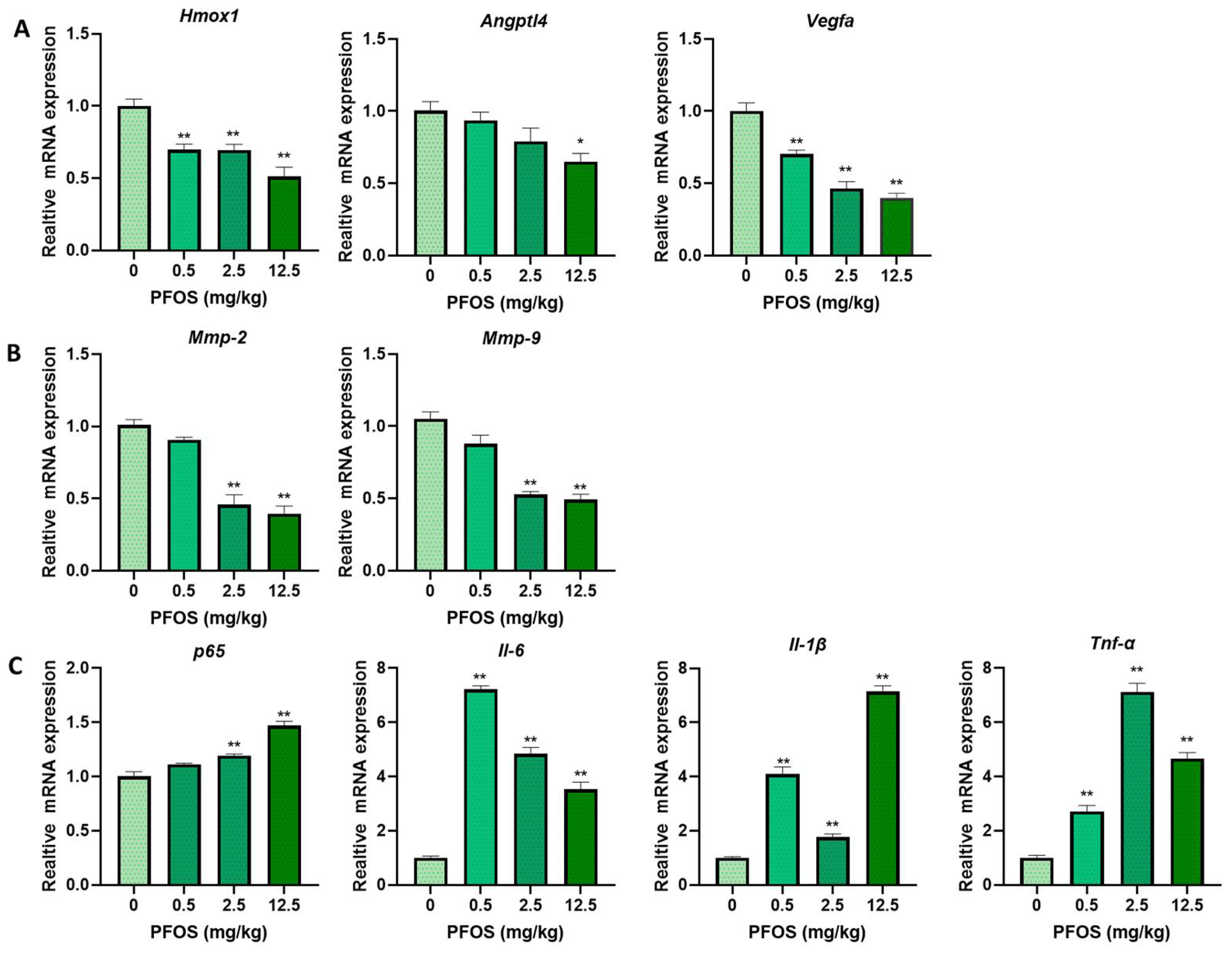
3.8. PFOS Alters PPARγ Target Genes mRNA Expression in Mice Placenta
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zoeller, R.T.; Brown, T.R.; Doan, L.L.; Gore, A.C.; Skakkebaek, N.E.; Soto, A.M.; Woodruff, T.J.; Vom Saal, F.S. Endocrine-disrupting chemicals and public health protection: A statement of principles from The Endocrine Society. Endocrinology 2012, 153, 4097–4110. [Google Scholar] [CrossRef]
- Saikat, S.; Kreis, I.; Davies, B.; Bridgman, S.; Kamanyire, R. The impact of PFOS on health in the general population: A review. Environ. Sci. Process. Impacts 2013, 15, 329–335. [Google Scholar] [CrossRef]
- Hansmeier, N.; Chao, T.-C.; Herbstman, J.B.; Goldman, L.R.; Witter, F.R.; Halden, R.U. Elucidating the molecular basis of adverse health effects from exposure to anthropogenic polyfluorinated compounds using toxicoproteomic approaches. J. Proteome Res. 2015, 14, 51–58. [Google Scholar] [CrossRef]
- Lee, C.K.; Kang, S.G.; Lee, J.T.; Lee, S.-W.; Kim, J.H.; Kim, D.H.; Son, B.C.; Kim, K.H.; Suh, C.H.; Kim, S.Y.; et al. Effects of perfluorooctane sulfuric acid on placental PRL-family hormone production and fetal growth retardation in mice. Mol. Cell. Endocrinol. 2015, 401, 165–172. [Google Scholar] [CrossRef]
- Marks, K.J.; Cutler, A.J.; Jeddy, Z.; Northstone, K.; Kato, K.; Hartman, T.J. Maternal serum concentrations of perfluoroalkyl substances and birth size in British boys. Int. J. Hyg. Environ. Health 2019, 222, 889–895. [Google Scholar] [CrossRef] [PubMed]
- Fuentes, S.; Colomina, M.T.; Rodriguez, J.; Vicens, P.; Domingo, J.L. Interactions in developmental toxicology: Concurrent exposure to perfluorooctane sulfonate (PFOS) and stress in pregnant mice. Toxicol. Lett. 2006, 164, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Ye, L.; Ge, Y.; Yuan, K.; Zhang, Y.; Liang, Y.; Wei, J.; Zhao, C.; Lian, Q.-Q.; Zhu, X.; et al. In utero perfluorooctane sulfonate exposure causes low body weights of fetal rats: A mechanism study. Placenta 2016, 39, 125–133. [Google Scholar] [CrossRef]
- Li, J.; Quan, X.-J.; Chen, G.; Hong, J.-W.; Wang, Q.; Xu, L.-L.; Wang, B.-H.; Yu, Z.-H.; Yu, H.-M. PFOS-induced placental cell growth inhibition is partially mediated by lncRNA H19 through interacting with miR-19a and miR-19b. Chemosphere 2020, 261, 127640. [Google Scholar] [CrossRef]
- Hong, F.; Xu, P.; Zhai, Y. The opportunities and challenges of peroxisome proliferator-activated receptors ligands in clinical drug discovery and development. Int. J. Mol. Sci. 2018, 19, 2189. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Zhai, Y.; Wang, J. The role of PPAR and its cross-talk with CAR and LXR in obesity and atherosclerosis. Int. J. Mol. Sci. 2018, 19, 1260. [Google Scholar] [CrossRef]
- Bensinger, S.J.; Tontonoz, P. Integration of metabolism and inflammation by lipid-activated nuclear receptors. Nature 2008, 454, 470–477. [Google Scholar] [CrossRef]
- Kotlinowski, J.; Jozkowicz, A. PPAR gamma and angiogenesis: Endothelial cells perspective. J. Diabetes Res. 2016, 2016, 8492353. [Google Scholar] [CrossRef]
- McCarthy, F.P.; Delany, A.C.; Kenny, L.C.; Walsh, S.K. PPAR-γ—A possible drug target for complicated pregnancies. Br. J. Pharmacol. 2013, 168, 1074–1085. [Google Scholar] [CrossRef]
- Barak, Y.; Nelson, M.C.; Ong, E.S.; Jones, Y.Z.; Ruiz-Lozano, P.; Chien, K.R.; Koder, A.; Evans, R.M. PPARγ is required for placental, cardiac, and adipose tissue development. Mol. Cell 1999, 4, 585–595. [Google Scholar] [CrossRef]
- Asami-Miyagishi, R.; Iseki, S.; Usui, M.; Uchida, K.; Kubo, H.; Morita, I. Expression and function of PPARγ in rat placental development. Biochem. Biophys. Res. Commun. 2004, 315, 497–501. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Yang, H.; Ye, Y.; Ma, Z.; Kuhn, C.; Rahmeh, M.; Mahner, S.; Makrigiannakis, A.; Jeschke, U.; von Schönfeldt, V. Role of peroxisome proliferator-activated receptors (PPARs) in trophoblast functions. Int. J. Mol. Sci. 2021, 22, 433. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhao, H.-J.; Xia, X.-R.; Diao, F.-Y.; Ma, X.; Wang, J.; Gao, L.; Liu, J.; Gao, C.; Cui, Y.-G.; et al. Hypoxia-induced and HIF1α-VEGF-mediated tight junction dysfunction in choriocarcinoma cells: Implications for preeclampsia. Clin. Chim. Acta 2019, 489, 203–211. [Google Scholar] [CrossRef]
- Li, S.-J.; Shang, T.; Li, S.-Y.; Li, Q.-L. [Effects of peroxisome proliferator-activated receptor gamma and its ligands on cytotrophoblast invasion in first trimester of pregnancy and mechanism thereof]. Zhonghua Yi Xue Za Zhi 2007, 87, 174–178. [Google Scholar]
- Barak, Y.; Liao, D.; He, W.; Ong, E.S.; Nelson, M.C.; Olefsky, J.M.; Boland, R.; Evans, R.M. Effects of peroxisome proliferator-activated receptor delta on placentation, adiposity, and colorectal cancer. Proc. Natl. Acad. Sci. USA 2002, 99, 303–308. [Google Scholar] [CrossRef] [PubMed]
- Kubota, N.; Terauchi, Y.; Miki, H.; Tamemoto, H.; Yamauchi, T.; Komeda, K.; Satoh, S.; Nakano, R.; Ishii, C.; Sugiyama, T.; et al. PPARγ mediates high-fat diet-induced adipocyte hypertrophy and insulin resistance. Mol. Cell 1999, 4, 597–609. [Google Scholar] [CrossRef]
- Vanden Heuvel, J.P.; Thompson, J.T.; Frame, S.R.; Gillies, P.J. Differential activation of nuclear receptors by perfluorinated fatty acid analogs and natural fatty acids: A comparison of human, mouse, and rat peroxisome proliferator-activated receptor-alpha, -beta, and -gamma, liver X receptor-beta, and retinoid X receptor-alpha. Toxicol. Sci. 2006, 92, 476–489. [Google Scholar] [PubMed]
- Wen, L.-L.; Lin, C.-Y.; Chou, H.-C.; Chang, C.-C.; Lo, H.-Y.; Juan, S.-H. Perfluorooctanesulfonate mediates renal tubular cell apoptosis through PPARgamma inactivation. PLoS ONE 2016, 11, e0155190. [Google Scholar] [CrossRef] [PubMed]
- Wan Ibrahim, W.N.; Tofighi, R.; Onishchenko, N.; Rebellato, P.; Bose, R.; Uhlén, P.; Ceccatelli, S. Perfluorooctane sulfonate induces neuronal and oligodendrocytic differentiation in neural stem cells and alters the expression of PPARγ in vitro and in vivo. Toxicol. Appl. Pharmacol. 2013, 269, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Lei, D.; Deng, N.; Wang, S.; Huang, J.; Fan, C. Upregulated ARRDC3 limits trophoblast cell invasion and tube formation and is associated with preeclampsia. Placenta 2020, 89, 10–19. [Google Scholar] [CrossRef]
- Chen, G.; Xu, L.L.; Huang, Y.F.; Wang, Q.; Wang, B.H.; Yu, Z.H.; Shi, Q.M.; Hong, J.W.; Li, J.; Xu, L.C. Prenatal exposure to perfluorooctane sulfonate impairs placental angiogenesis and induces aberrant expression of LncRNA Xist. Biomed. Environ. Sci. 2018, 31, 843–847. [Google Scholar] [CrossRef] [PubMed]
- Eaton, M.; Davies, A.H.; Devine, J.; Zhao, X.; Simmons, D.G.; Maríusdóttir, E.; Natale, D.R.C.; Matyas, J.R.; Bering, E.A.; Workentine, M.L.; et al. Complex patterns of cell growth in the placenta in normal pregnancy and as adaptations to maternal diet restriction. PLoS ONE 2020, 15, e0226735. [Google Scholar] [CrossRef]
- Burton, G.J.; Jauniaux, E. Pathophysiology of placental-derived fetal growth restriction. Am. J. Obstet. Gynecol. 2018, 218, S745–S761. [Google Scholar] [CrossRef]
- Pham, A.; Zhang, J.; Feng, L. Exposure to perfluorobutane sulfonate and perfluorooctanesulfonic acid disrupts the production of angiogenesis factors and stress responses in human placental syncytiotrophoblast. Reprod. Toxicol. 2020, 98, 269–277. [Google Scholar] [CrossRef]
- Ferretti, C.; Bruni, L.; Dangles-Marie, V.; Pecking, A.P.; Bellet, D. Molecular circuits shared by placental and cancer cells, and their implications in the proliferative, invasive and migratory capacities of trophoblasts. Hum. Reprod. Update 2007, 13, 121–141. [Google Scholar] [CrossRef]
- Szilagyi, J.T.; Freedman, A.N.; Kepper, S.L.; Keshava, A.M.; Bangma, J.T.; Fry, R.C. Per- and polyfluoroalkyl substances differentially inhibit placental trophoblast migration and invasion in vitro. Toxicol. Sci. 2020, 175, 210–219. [Google Scholar] [CrossRef]
- Folkman, J.; Klagsbrun, M. Angiogenic factors. Science 1987, 235, 442–447. [Google Scholar] [CrossRef]
- Reynolds, L.P.; Redmer, D.A. Angiogenesis in the placenta. Biol. Reprod. 2001, 64, 1033–1040. [Google Scholar] [CrossRef]
- Meegdes, B.H.; Ingenhoes, R.; Peeters, L.L.; Exalto, N. Early pregnancy wastage: Relationship between chorionic vascularization and embryonic development. Fertil. Steril. 1988, 49, 216–220. [Google Scholar] [CrossRef]
- Bassil, S.; Magritte, J.P.; Roth, J.; Nisolle, M.; Donnez, J.; Gordts, S. Uterine vascularity during stimulation and its correlation with implantation in in-vitro fertilization. Hum. Reprod. 1995, 10, 1497–1501. [Google Scholar] [CrossRef]
- Reynolds, L.P.; Grazul-Bilska, A.T.; Redmer, D.A. Angiogenesis in the corpus luteum. Endocrine 2000, 12, 1–9. [Google Scholar] [CrossRef]
- Giaginis, C.; Spanopoulou, E.; Theocharis, S. PPARγ signaling pathway in placental development and function: A potential therapeutic target in the treatment of gestational diseases. Expert Opin. Ther. Targets 2008, 12, 1049–1063. [Google Scholar] [CrossRef] [PubMed]
- Schaiff, W.T.; Barak, Y.; Sadovsky, Y. The pleiotropic function of PPARγ in the placenta. Mol. Cell. Endocrinol. 2006, 249, 10–15. [Google Scholar] [CrossRef]
- Seargent, J.M.; Yates, E.A.; Gill, J.H. GW9662, a potent antagonist of PPARγ, inhibits growth of breast tumour cells and promotes the anticancer effects of the PPARγ agonist rosiglitazone, independently of PPARγ activation. Br. J. Pharmacol. 2004, 143, 933–937. [Google Scholar] [CrossRef] [PubMed]
- Kohan-Ghadr, H.-R.; Kilburn, B.A.; Kadam, L.; Johnson, E.; Kolb, B.L.; Rodriguez-Kovacs, J.; Hertz, M.; Armant, D.R.; Drewlo, S. Rosiglitazone augments antioxidant response in the human trophoblast and prevents apoptosis†. Biol. Reprod. 2019, 100, 479–494. [Google Scholar] [CrossRef]
- Zhou, G.; Han, X.; Wu, Z.; Shi, Q.; Bao, X. Rosiglitazone accelerates wound healing by improving endothelial precursor cell function and angiogenesis in mice. PeerJ 2019, 7, e7815. [Google Scholar] [CrossRef]
- Kadam, L.; Gomez-Lopez, N.; Mial, T.N.; Kohan-Ghadr, H.-R.; Drewlo, S. Rosiglitazone regulates TLR4 and rescues HO-1 and NRF2 expression in myometrial and decidual macrophages in inflammation-induced preterm birth. Reprod. Sci. 2017, 24, 1590–1599. [Google Scholar] [CrossRef]
- Li, C.-C.; Hou, Y.-C.; Yeh, C.-L.; Yeh, S.-L. Effects of eicosapentaenoic acid and docosahexaenoic acid on prostate cancer cell migration and invasion induced by tumor-associated macrophages. PLoS ONE 2014, 9, e99630. [Google Scholar] [CrossRef] [PubMed]
- Xi, Y.; Zhang, Y.; Zhu, S.; Luo, Y.; Xu, P.; Huang, Z. PPAR-mediated toxicology and applied pharmacology. Cells 2020, 9, 352. [Google Scholar] [CrossRef]
- Hong, F.; Pan, S.; Guo, Y.; Xu, P.; Zhai, Y. PPARs as nuclear receptors for nutrient and energy metabolism. Molecules 2019, 24, 2545. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zhuang, X.; Jiang, M.; Guan, F.; Fu, Q.; Lin, J. ANGPTL4 mediates the protective role of PPARγ activators in the pathogenesis of preeclampsia. Cell Death Dis. 2017, 8, e3054. [Google Scholar] [CrossRef] [PubMed]
- Cudmore, M.; Ahmad, S.; Al-Ani, B.; Fujisawa, T.; Coxall, H.; Chudasama, K.; Devey, L.R.; Wigmore, S.J.; Abbas, A.; Hewett, P.W.; et al. Negative regulation of soluble Flt-1 and soluble endoglin release by heme oxygenase-1. Circulation 2007, 115, 1789–1797. [Google Scholar] [CrossRef] [PubMed]
- Krönke, G.; Kadl, A.; Ikonomu, E.; Blüml, S.; Fürnkranz, A.; Sarembock, I.J.; Bochkov, V.N.; Exner, M.; Binder, B.R.; Leitinger, N. Expression of heme oxygenase-1 in human vascular cells is regulated by peroxisome proliferator-activated receptors. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1276–1282. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, F.P.; Drewlo, S.; Kingdom, J.; Johns, E.J.; Walsh, S.K.; Kenny, L.C. Peroxisome proliferator-activated receptor-γ as a potential therapeutic target in the treatment of preeclampsia. Hypertension 2011, 58, 280–286. [Google Scholar] [CrossRef]
- Nadeau-Vallée, M.; Obari, D.; Palacios, J.; Brien, M.-È.; Duval, C.; Chemtob, S.; Girard, S. Sterile inflammation and pregnancy complications: A review. Reproduction 2016, 152, R277–R292. [Google Scholar] [CrossRef]
- Svinarich, D.M.; Bitonti, O.M.; Romero, R.; Gonik, B. Induction and posttranslational expression of cytokines in a first-trimester trophoblast cell line by lipopolysaccharide. Am. J. Obstet. Gynecol. 1996, 175, 970–973. [Google Scholar] [CrossRef]
- Zaga-Clavellina, V.; Garcia-Lopez, G.; Flores-Herrera, H.; Espejel-Nuñez, A.; Flores-Pliego, A.; Soriano-Becerril, D.; Maida-Claros, R.; Merchant-Larios, H.; Vadillo-Ortega, F. In vitro secretion profiles of interleukin (IL)-1beta, IL-6, IL-8, IL-10, and TNF alpha after selective infection with Escherichia coli in human fetal membranes. Reprod. Biol. Endocrinol. 2007, 5, 46. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.R.; Cao, J.; Hong, J.W.; Li, J. [Effects of PFOS on inflammatory factors in human placental trophoblast cells]. Zhonghua Lao Dong Wei Sheng Zhi Ye Bing Za Zhi 2020, 38, 481–484. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.-X.; Ji, X.-J.; Li, Q.-Q.; Lu, X.-X.; Luo, L. Rosiglitazone reduces apoptosis and inflammation in lipopolysaccharide-induced human umbilical vein endothelial cells. Med. Sci. Monit. 2018, 24, 6200–6207. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Romero, R.; Miller, D.; Kadam, L.; Mial, T.N.; Plazyo, O.; Garcia-Flores, V.; Hassan, S.S.; Xu, Z.; Tarca, A.L.; et al. An M1-like macrophage polarization in decidual tissue during spontaneous preterm labor that is attenuated by rosiglitazone treatment. J. Immunol. 2016, 196, 2476–2491. [Google Scholar] [CrossRef]
- Zhang, Y.; Hu, L.; Cui, Y.; Qi, Z.; Huang, X.; Cai, L.; Zhang, T.; Yin, Y.; Lu, Z.; Xiang, J. Roles of PPARγ/NF-κB signaling pathway in the pathogenesis of intrahepatic cholestasis of pregnancy. PLoS ONE 2014, 9, e87343. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, J.; Quan, X.; Lei, S.; Huang, Z.; Wang, Q.; Xu, P. PFOS Inhibited Normal Functional Development of Placenta Cells via PPARγ Signaling. Biomedicines 2021, 9, 677. https://doi.org/10.3390/biomedicines9060677
Li J, Quan X, Lei S, Huang Z, Wang Q, Xu P. PFOS Inhibited Normal Functional Development of Placenta Cells via PPARγ Signaling. Biomedicines. 2021; 9(6):677. https://doi.org/10.3390/biomedicines9060677
Chicago/Turabian StyleLi, Jing, Xiaojie Quan, Saifei Lei, Zhenyao Huang, Qi Wang, and Pengfei Xu. 2021. "PFOS Inhibited Normal Functional Development of Placenta Cells via PPARγ Signaling" Biomedicines 9, no. 6: 677. https://doi.org/10.3390/biomedicines9060677
APA StyleLi, J., Quan, X., Lei, S., Huang, Z., Wang, Q., & Xu, P. (2021). PFOS Inhibited Normal Functional Development of Placenta Cells via PPARγ Signaling. Biomedicines, 9(6), 677. https://doi.org/10.3390/biomedicines9060677