
Mucus, Microbiomes and Pulmonary Disease


Abstract
1. Introduction
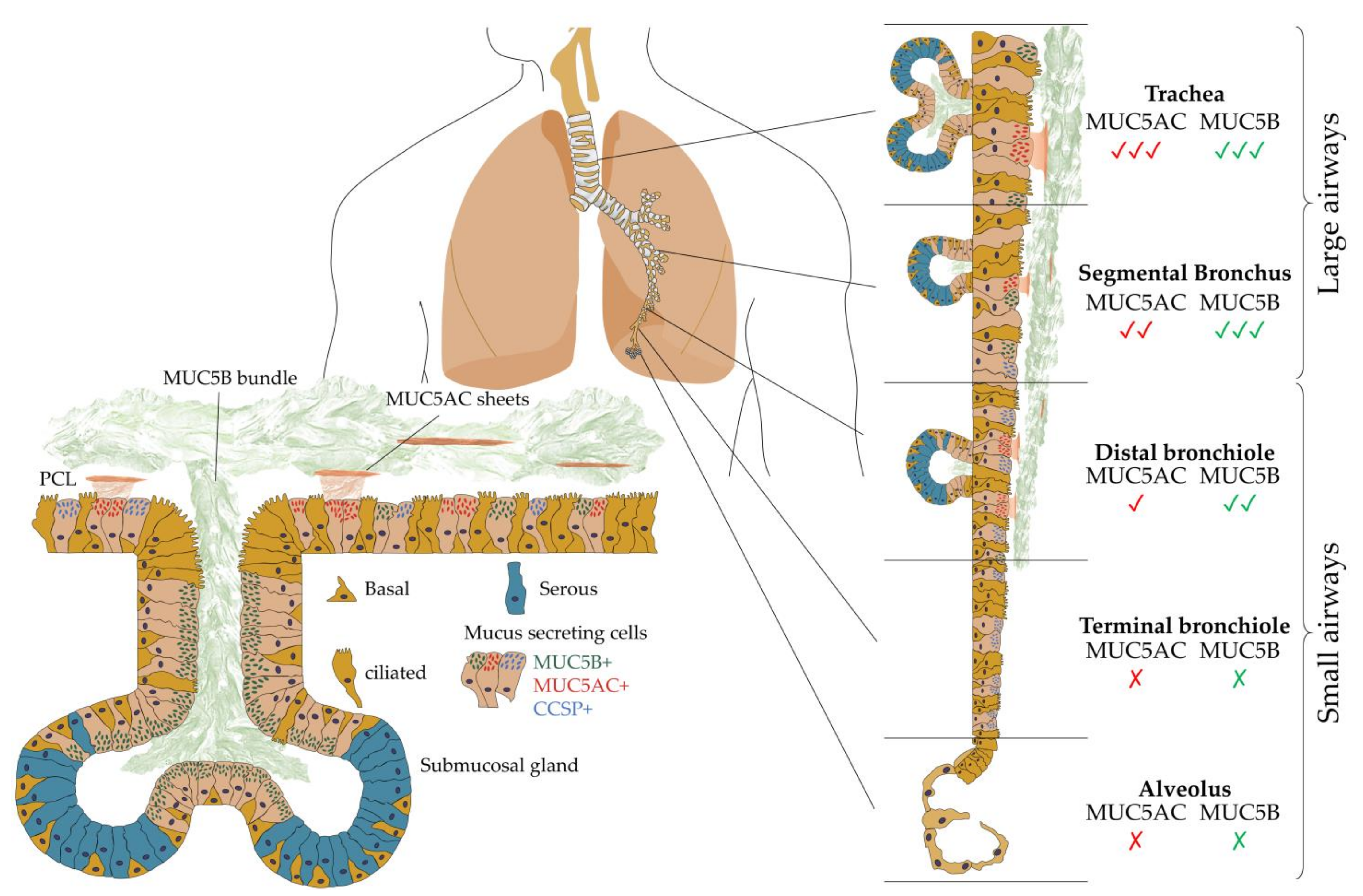
2. The Structure and Function of Pulmonary Mucus
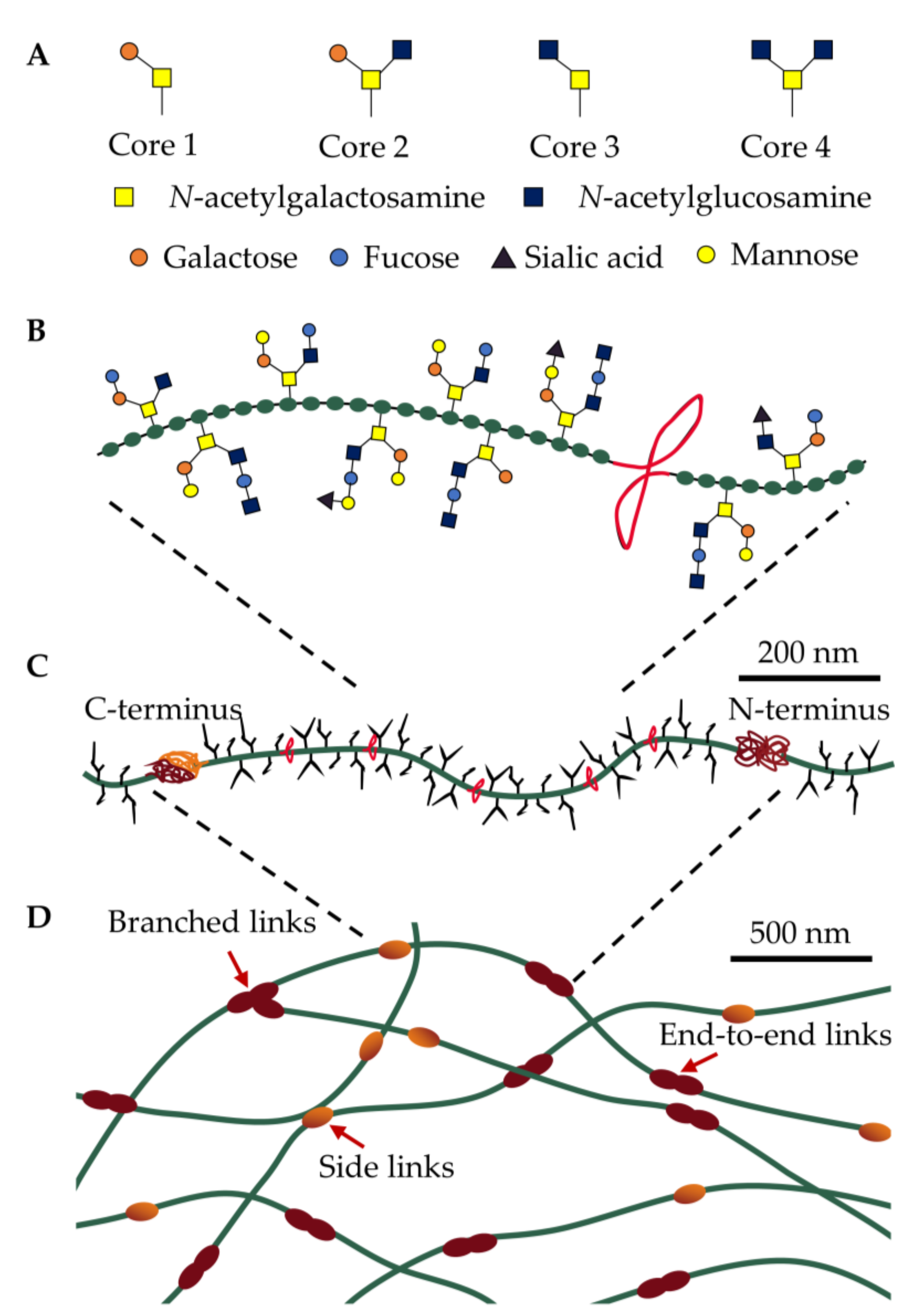
2.1. Polymerizing Mucins
2.2. Translating Mucin Properties into Understanding Pulmonary Disease
3. Going Beyond Mucus as a Barrier: The Role of Mucin in Bacterial Pathogenesis and Chronic Respiratory Disease
3.1. Mucin as a Nutritional Source
3.2. Mucin Permits the Spatial Organization of Lung Microbial Communities
3.3. Mucin Signaling and Microbial Behavior in the Lungs
3.4. Mucin in Chronic Pulmonary Disease
4. Targeting Mucin in Pulmonary Infection and Disease
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Palacios-Ceña, D.; Hernández-Barrera, V.; López-de-Andrés, A.; Fernández-de-las-Peñas, C.; Palacios-Ceña, M.; de Miguel-Díez, J.; Carrasco-Garrido, P.; Jiménez-García, R. Time trends in incidence and outcomes of hospitalizations for aspiration pneumonia among elderly people in Spain (2003–2013). Eur. J. Intern. Med. 2017, 38, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Ticinesi, A.; Nouvenne, A.; Folesani, G.; Prati, B.; Morelli, I.; Guida, L.; Lauretani, F.; Maggio, M.; Meschi, T. An investigation of multimorbidity measures as risk factors for pneumonia in elderly frail patients admitted to hospital. Eur. J. Intern. Med. 2016, 28, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Troeger, C.; Blacker, B.; Khalil, I.A.; Rao, P.C.; Cao, J.; Zimsen, S.R.M.; Albertson, S.B.; Deshpande, A.; Farag, T.; Abebe, Z.; et al. Estimates of the global, regional, and national morbidity, mortality, and aetiologies of lower respiratory infections in 195 countries, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Infect. Dis. 2018, 18, 1191–1210. [Google Scholar] [CrossRef]
- Charalampous, T.; Kay, G.L.; Richardson, H.; Aydin, A.; Baldan, R.; Jeanes, C.; Rae, D.; Grundy, S.; Turner, D.J.; Wain, J.; et al. Nanopore metagenomics enables rapid clinical diagnosis of bacterial lower respiratory infection. Nat. Biotechnol. 2019, 37, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Hilty, M.; Burke, C.; Pedro, H.; Cardenas, P.; Bush, A.; Bossley, C.; Davies, J.; Ervine, A.; Poulter, L.; Pachter, L.; et al. Disordered microbial communities in asthmatic airways. PLoS ONE 2010, 5, e8578. [Google Scholar] [CrossRef]
- Paganin, P.; Fiscarelli, E.V.; Tuccio, V.; Chiancianesi, M.; Bacci, G.; Morelli, P.; Dolce, D.; Dalmastri, C.; De Alessandri, A.; Lucidi, V.; et al. Changes in Cystic Fibrosis Airway Microbial Community Associated with a Severe Decline in Lung Function. PLoS ONE 2015, 10, e0124348. [Google Scholar] [CrossRef]
- Tunney, M.M.; Einarsson, G.G.; Wei, L.; Drain, M.; Klem, E.R.; Cardwell, C.; Ennis, M.; Boucher, R.C.; Wolfgang, M.C.; Elborn, J.S. Lung microbiota and bacterial abundance in patients with bronchiectasis when clinically stable and during exacerbation. Am. J. Respir. Crit. Care Med. 2013, 187, 1118–1126. [Google Scholar] [CrossRef]
- Leung, J.M.; Tiew, P.Y.; Mac Aogáin, M.; Budden, K.F.; Yong, V.F.; Thomas, S.S.; Pethe, K.; Hansbro, P.M.; Chotirmall, S.H. The role of acute and chronic respiratory colonization and infections in the pathogenesis of COPD. Respirology 2017, 22, 634–650. [Google Scholar] [CrossRef]
- Dicker, A.; Lonergan, M.; Keir, H.; Fong, C.; Tan, B.; Cassidy, A.; Finch, S.; Mullerova, H.; Huang, J.; Miller, B.; et al. Lung microbiome dysbiosis is associated with mortality in COPD. Eur. Respir. J. 2019, 54, OA3581. [Google Scholar] [CrossRef]
- Poh, T.Y.; Ali, N.A.t.B.M.; Mac Aogáin, M.; Kathawala, M.H.; Setyawati, M.I.; Ng, K.W.; Chotirmall, S.H. Inhaled nanomaterials and the respiratory microbiome: Clinical, immunological and toxicological perspectives. Part. Fibre Toxicol. 2018, 15, 46. [Google Scholar] [CrossRef]
- Fahy, J.V.; Dickey, B.F. Airway mucus function and dysfunction. N. Engl. J. Med. 2010, 363, 2233–2247. [Google Scholar] [CrossRef]
- O’Dwyer, D.N.; Dickson, R.P.; Moore, B.B. The Lung Microbiome, Immunity, and the Pathogenesis of Chronic Lung Disease. J. Immunol. 2016, 196, 4839–4847. [Google Scholar] [CrossRef]
- Bassis, C.M.; Erb-Downward, J.R.; Dickson, R.P.; Freeman, C.M.; Schmidt, T.M.; Young, V.B.; Beck, J.M.; Curtis, J.L.; Huffnagle, G.B. Analysis of the Upper Respiratory Tract Microbiotas as the Source of the Lung and Gastric Microbiotas in Healthy Individuals. mBio 2015, 6, e00037-15. [Google Scholar] [CrossRef]
- Hanada, S.; Pirzadeh, M.; Carver, K.Y.; Deng, J.C. Respiratory Viral Infection-Induced Microbiome Alterations and Secondary Bacterial Pneumonia. Front. Immunol. 2018, 9, 2640. [Google Scholar] [CrossRef]
- Soret, P.; Vandenborght, L.-E.; Francis, F.; Coron, N.; Enaud, R.; Avalos, M.; Schaeverbeke, T.; Berger, P.; Fayon, M.; Thiebaut, R.; et al. Respiratory mycobiome and suggestion of inter-kingdom network during acute pulmonary exacerbation in cystic fibrosis. Sci. Rep. 2020, 10, 3589. [Google Scholar] [CrossRef]
- Ali, N.; Ivan, F.X.; Mac Aogáin, M.; Narayana, J.K.; Lee, S.Y.; Lim, C.L.; Chotirmall, S.H. The Healthy Airway Mycobiome in Individuals of Asian Descent. Chest 2021, 159, 544–548. [Google Scholar] [CrossRef]
- Mac Aogáin, M.; Narayana, J.K.; Tiew, P.Y.; Ali, N.A.t.B.M.; Yong, V.F.L.; Jaggi, T.K.; Lim, A.Y.H.; Keir, H.R.; Dicker, A.J.; Thng, K.X.; et al. Integrative microbiomics in bronchiectasis exacerbations. Nat. Med. 2021, 27, 688–699. [Google Scholar] [CrossRef]
- Siegel, S.J.; Weiser, J.N. Mechanisms of Bacterial Colonization of the Respiratory Tract. Annu. Rev. Microbiol. 2015, 69, 425–444. [Google Scholar] [CrossRef]
- Mendez, R.; Banerjee, S.; Bhattacharya, S.K.; Banerjee, S. Lung inflammation and disease: A perspective on microbial homeostasis and metabolism. IUBMB Life 2019, 71, 152–165. [Google Scholar] [CrossRef]
- Rogers, G.B.; Bruce, K.D.; Martin, M.L.; Burr, L.D.; Serisier, D.J. The effect of long-term macrolide treatment on respiratory microbiota composition in non-cystic fibrosis bronchiectasis: An analysis from the randomised, double-blind, placebo-controlled BLESS trial. Lancet Respir. Med. 2014, 2, 988–996. [Google Scholar] [CrossRef]
- Cuthbertson, L.; Walker, A.W.; Oliver, A.E.; Rogers, G.B.; Rivett, D.W.; Hampton, T.H.; Ashare, A.; Elborn, J.S.; de Soyza, A.; Carroll, M.P.; et al. Lung function and microbiota diversity in cystic fibrosis. Microbiome 2020, 8, 45. [Google Scholar] [CrossRef]
- Vandeplassche, E.; Tavernier, S.; Coenye, T.; Crabbé, A. Influence of the lung microbiome on antibiotic susceptibility of cystic fibrosis pathogens. Eur. Respir. Rev. 2019, 28. [Google Scholar] [CrossRef]
- Montoro, D.T.; Haber, A.L.; Biton, M.; Vinarsky, V.; Lin, B.; Birket, S.E.; Yuan, F.; Chen, S.; Leung, H.M.; Villoria, J.; et al. A revised airway epithelial hierarchy includes CFTR-expressing ionocytes. Nature 2018, 560, 319–324. [Google Scholar] [CrossRef]
- Whitsett, J.A. Airway Epithelial Differentiation and Mucociliary Clearance. Ann. Am. Thorac. Soc. 2018, 15, S143–S148. [Google Scholar] [CrossRef] [PubMed]
- Thornton, D.J.; Rousseau, K.; McGuckin, M.A. Structure and Function of the Polymeric Mucins in Airways Mucus. Annu. Rev. Physiol. 2008, 70, 459–486. [Google Scholar] [CrossRef]
- Okuda, K.; Chen, G.; Subramani, D.B.; Wolf, M.; Gilmore, R.C.; Kato, T.; Radicioni, G.; Kesimer, M.; Chua, M.; Dang, H.; et al. Localization of Secretory Mucins MUC5AC and MUC5B in Normal/Healthy Human Airways. Am. J. Respir. Crit. Care Med. 2019, 199, 715–727. [Google Scholar] [CrossRef]
- Tran, D.T.; ten Hagen, K.G. Mucin-type O-glycosylation during development. J. Biol. Chem. 2013, 288, 6921–6929. [Google Scholar] [CrossRef]
- Kesimer, M.; Makhov, A.M.; Griffith, J.D.; Verdugo, P.; Sheehan, J.K. Unpacking a gel-forming mucin: A view of MUC5B organization after granular release. Am. J. Physiol. Lung Cell. Mol. Physiol. 2010, 298, L15–L22. [Google Scholar] [CrossRef]
- Meldrum, O.W.; Yakubov, G.E.; Bonilla, M.R.; Deshmukh, O.; McGuckin, M.A.; Gidley, M.J. Mucin gel assembly is controlled by a collective action of non-mucin proteins, disulfide bridges, Ca2+-mediated links, and hydrogen bonding. Sci. Rep. 2018, 8, 5802. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, K.A.; Chen, A.C.H.; Radicioni, G.; Lourie, R.; Martin, M.; Broomfield, A.; Sheng, Y.H.; Hasnain, S.Z.; Radford-Smith, G.; Simms, L.A.; et al. Airway Mucus Hyperconcentration in Non-Cystic Fibrosis Bronchiectasis. Am. J. Respir. Crit. Care Med. 2020, 201, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Hill, D.B.; Vasquez, P.A.; Mellnik, J.; McKinley, S.A.; Vose, A.; Mu, F.; Henderson, A.G.; Donaldson, S.H.; Alexis, N.E.; Boucher, R.C.; et al. A Biophysical Basis for Mucus Solids Concentration as a Candidate Biomarker for Airways Disease. PLoS ONE 2014, 9, e87681. [Google Scholar] [CrossRef]
- Patarin, J.; Ghiringhelli, É.; Darsy, G.; Obamba, M.; Bochu, P.; Camara, B.; Quétant, S.; Cracowski, J.-L.; Cracowski, C.; de Saint Vincent, M.R. Rheological analysis of sputum from patients with chronic bronchial diseases. Sci. Rep. 2020, 10, 15685. [Google Scholar] [CrossRef]
- Ermund, A.; Meiss, L.N.; Rodriguez-Pineiro, A.M.; Bähr, A.; Nilsson, H.E.; Trillo-Muyo, S.; Ridley, C.; Thornton, D.J.; Wine, J.J.; Hebert, H.; et al. The normal trachea is cleaned by MUC5B mucin bundles from the submucosal glands coated with the MUC5AC mucin. Biochem. Biophys. Res. Commun. 2017, 492, 331–337. [Google Scholar] [CrossRef]
- Button, B.; Cai, L.-H.; Ehre, C.; Kesimer, M.; Hill, D.B.; Sheehan, J.K.; Boucher, R.C.; Rubinstein, M. A Periciliary Brush Promotes the Lung Health by Separating the Mucus Layer from Airway Epithelia. Science 2012, 337, 937–941. [Google Scholar] [CrossRef] [PubMed]
- Khelloufi, M.-K.; Loiseau, E.; Jaeger, M.; Molinari, N.; Chanez, P.; Gras, D.; Viallat, A. Spatiotemporal organization of cilia drives multiscale mucus swirls in model human bronchial epithelium. Sci. Rep. 2018, 8, 2447. [Google Scholar] [CrossRef]
- Gsell, S.; Loiseau, E.; D’Ortona, U.; Viallat, A.; Favier, J. Hydrodynamic model of directional ciliary-beat organization in human airways. Sci. Rep. 2020, 10, 8405. [Google Scholar] [CrossRef]
- Abdullah, L.H.; Coakley, R.; Webster, M.J.; Zhu, Y.; Tarran, R.; Radicioni, G.; Kesimer, M.; Boucher, R.C.; Davis, C.W.; Ribeiro, C.M.P. Mucin Production and Hydration Responses to Mucopurulent Materials in Normal versus Cystic Fibrosis Airway Epithelia. Am. J. Respir. Crit. Care Med. 2018, 197, 481–491. [Google Scholar] [CrossRef]
- Roy, M.G.; Livraghi-Butrico, A.; Fletcher, A.A.; McElwee, M.M.; Evans, S.E.; Boerner, R.M.; Alexander, S.N.; Bellinghausen, L.K.; Song, A.S.; Petrova, Y.M.; et al. Muc5b is required for airway defence. Nature 2014, 505, 412–416. [Google Scholar] [CrossRef]
- Ostedgaard, L.S.; Moninger, T.O.; McMenimen, J.D.; Sawin, N.M.; Parker, C.P.; Thornell, I.M.; Powers, L.S.; Gansemer, N.D.; Bouzek, D.C.; Cook, D.P.; et al. Gel-forming mucins form distinct morphologic structures in airways. Proc. Natl. Acad. Sci. USA 2017, 114, 6842–6847. [Google Scholar] [CrossRef] [PubMed]
- Park, J.A.; Tschumperlin, D.J. Chronic intermittent mechanical stress increases MUC5AC protein expression. Am. J. Respir. Cell Mol. Biol. 2009, 41, 459–466. [Google Scholar] [CrossRef]
- Young, H.W.; Williams, O.W.; Chandra, D.; Bellinghausen, L.K.; Pérez, G.; Suárez, A.; Tuvim, M.J.; Roy, M.G.; Alexander, S.N.; Moghaddam, S.J.; et al. Central role of Muc5ac expression in mucous metaplasia and its regulation by conserved 5’ elements. Am. J. Respir. Cell Mol. Biol. 2007, 37, 273–290. [Google Scholar] [CrossRef] [PubMed]
- Ehre, C.; Worthington, E.N.; Liesman, R.M.; Grubb, B.R.; Barbier, D.; O’Neal, W.K.; Sallenave, J.M.; Pickles, R.J.; Boucher, R.C. Overexpressing mouse model demonstrates the protective role of Muc5ac in the lungs. Proc. Natl. Acad. Sci. USA 2012, 109, 16528–16533. [Google Scholar] [CrossRef] [PubMed]
- Martens, C.J.; Inglis, S.K.; Valentine, V.G.; Garrison, J.; Conner, G.E.; Ballard, S.T. Mucous solids and liquid secretion by airways: Studies with normal pig, cystic fibrosis human, and non-cystic fibrosis human bronchi. Am. J. Physiol. Lung Cell. Mol. Physiol. 2011, 301, L236–L246. [Google Scholar] [CrossRef] [PubMed]
- Piva, T.C.; Luft, C.; Antunes, K.H.; Marostica, P.J.C.; Pinto, L.A.; Donadio, M.V.F. Extracellular DNA in sputum is associated with pulmonary function and hospitalization in patients with cystic fibrosis. Respir. Med. 2020, 172, 106144. [Google Scholar] [CrossRef]
- Dickey, B.F.; Fahy, J.V.; Kesimer, M.; Boucher, R.C.; Evans, C.M.; Thornton, D. Measuring Airway Mucin 2 in Patients with Severe Chronic Obstructive Pulmonary Disease with Bacterial Colonization. Ann. Am. Thorac. Soc. 2016, 13, 2103–2104. [Google Scholar] [CrossRef]
- Fahy, J.V.; Steiger, D.J.; Liu, J.; Basbaum, C.B.; Finkbeiner, W.E.; Boushey, H.A. Markers of mucus secretion and DNA levels in induced sputum from asthmatic and from healthy subjects. Am. Rev. Respir. Dis. 1993, 147, 1132–1137. [Google Scholar] [CrossRef]
- Welsh, K.G.; Rousseau, K.; Fisher, G.; Bonser, L.R.; Bradding, P.; Brightling, C.E.; Thornton, D.J.; Gaillard, E.A. MUC5AC and a Glycosylated Variant of MUC5B Alter Mucin Composition in Children with Acute Asthma. Chest 2017, 152, 771–779. [Google Scholar] [CrossRef]
- Kirkham, S.; Sheehan, J.K.; Knight, D.; Richardson, P.S.; Thornton, D.J. Heterogeneity of airways mucus: Variations in the amounts and glycoforms of the major oligomeric mucins MUC5AC and MUC5B. Biochem. J. 2002, 361, 537–546. [Google Scholar] [CrossRef]
- Lachowicz-Scroggins, M.E.; Yuan, S.; Kerr, S.C.; Dunican, E.M.; Yu, M.; Carrington, S.D.; Fahy, J.V. Abnormalities in MUC5AC and MUC5B Protein in Airway Mucus in Asthma. Am. J. Respir. Crit. Care Med. 2016, 194, 1296–1299. [Google Scholar] [CrossRef]
- Sibila, O.; Suarez-Cuartin, G.; Rodrigo-Troyano, A.; Fardon, T.C.; Finch, S.; Mateus, E.F.; Garcia-Bellmunt, L.; Castillo, D.; Vidal, S.; Sanchez-Reus, F.; et al. Secreted mucins and airway bacterial colonization in non-CF bronchiectasis. Respirology 2015, 20, 1082–1088. [Google Scholar] [CrossRef]
- Henderson, A.G.; Ehre, C.; Button, B.; Abdullah, L.H.; Cai, L.H.; Leigh, M.W.; DeMaria, G.C.; Matsui, H.; Donaldson, S.H.; Davis, C.W.; et al. Cystic fibrosis airway secretions exhibit mucin hyperconcentration and increased osmotic pressure. J. Clin. Invest. 2014, 124, 3047–3060. [Google Scholar] [CrossRef]
- Hill, D.B.; Long, R.F.; Kissner, W.J.; Atieh, E.; Garbarine, I.C.; Markovetz, M.R.; Fontana, N.C.; Christy, M.; Habibpour, M.; Tarran, R.; et al. Pathological mucus and impaired mucus clearance in cystic fibrosis patients result from increased concentration, not altered pH. Eur. Respir. J. 2018, 52, 1801297. [Google Scholar] [CrossRef]
- Esther, C.R.; Muhlebach, M.S.; Ehre, C.; Hill, D.B.; Wolfgang, M.C.; Kesimer, M.; Ramsey, K.A.; Markovetz, M.R.; Garbarine, I.C.; Forest, M.G.; et al. Mucus accumulation in the lungs precedes structural changes and infection in children with cystic fibrosis. Sci. Transl. Med. 2019, 11, eaav3488. [Google Scholar] [CrossRef]
- Takeyama, K.; Akaba, T.; Kurokawa, A.; Arimura, K.; Kondo, M.; Tagaya, E. Analysis of airway mucin concentration in patients with pulmonary eosinophilia. Eur. Respir. J. 2019, 54, PA4390. [Google Scholar] [CrossRef]
- Takeyama, K.; Kondo, M.; Akaba, T.; Tamaoki, J. Profile of airway mucins in bronchoalveolar lavage fluid of patients with pulmonary alveolar proteinosis. Eur. Respir. J. 2015, 46, PA3870. [Google Scholar] [CrossRef]
- Kesimer, M.; Ford, A.A.; Ceppe, A.; Radicioni, G.; Cao, R.; Davis, C.W.; Doerschuk, C.M.; Alexis, N.E.; Anderson, W.H.; Henderson, A.G.; et al. Airway Mucin Concentration as a Marker of Chronic Bronchitis. N. Engl. J. Med. 2017, 377, 911–922. [Google Scholar] [CrossRef]
- Anderson, W.H.; Coakley, R.D.; Button, B.; Henderson, A.G.; Zeman, K.L.; Alexis, N.E.; Peden, D.B.; Lazarowski, E.R.; Davis, C.W.; Bailey, S.; et al. The Relationship of Mucus Concentration (Hydration) to Mucus Osmotic Pressure and Transport in Chronic Bronchitis. Am. J. Respir. Crit. Care Med. 2015, 192, 182–190. [Google Scholar] [CrossRef]
- Evans, C.M.; Raclawska, D.S.; Ttofali, F.; Liptzin, D.R.; Fletcher, A.A.; Harper, D.N.; McGing, M.A.; McElwee, M.M.; Williams, O.W.; Sanchez, E.; et al. The polymeric mucin Muc5ac is required for allergic airway hyperreactivity. Nat. Commun. 2015, 6, 6281. [Google Scholar] [CrossRef]
- Dickson, R.P.; Huffnagle, G.B. The Lung Microbiome: New Principles for Respiratory Bacteriology in Health and Disease. PLoS Pathog. 2015, 11, e1004923. [Google Scholar] [CrossRef]
- Flynn, J.M.; Niccum, D.; Dunitz, J.M.; Hunter, R.C. Evidence and Role for Bacterial Mucin Degradation in Cystic Fibrosis Airway Disease. PLoS Pathog. 2016, 12, e1005846. [Google Scholar] [CrossRef]
- Tanaka, M.; Nakayama, J. Development of the gut microbiota in infancy and its impact on health in later life. Allergol. Int. 2017, 66, 515–522. [Google Scholar] [CrossRef] [PubMed]
- Fung, T.C.; Olson, C.A.; Hsiao, E.Y. Interactions between the microbiota, immune and nervous systems in health and disease. Nat. Neurosci. 2017, 20, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Gollwitzer, E.S.; Saglani, S.; Trompette, A.; Yadava, K.; Sherburn, R.; McCoy, K.D.; Nicod, L.P.; Lloyd, C.M.; Marsland, B.J. Lung microbiota promotes tolerance to allergens in neonates via PD-L1. Nat. Med. 2014, 20, 642–647. [Google Scholar] [CrossRef] [PubMed]
- Scher, J.U.; Joshua, V.; Artacho, A.; Abdollahi-Roodsaz, S.; Öckinger, J.; Kullberg, S.; Sköld, M.; Eklund, A.; Grunewald, J.; Clemente, J.C.; et al. The lung microbiota in early rheumatoid arthritis and autoimmunity. Microbiome 2016, 4, 60. [Google Scholar] [CrossRef]
- Mathieu, E.; Escribano-Vazquez, U.; Descamps, D.; Cherbuy, C.; Langella, P.; Riffault, S.; Remot, A.; Thomas, M. Paradigms of Lung Microbiota Functions in Health and Disease, Particularly, in Asthma. Front. Physiol. 2018, 9, 1168. [Google Scholar] [CrossRef]
- Segal, L.N.; Clemente, J.C.; Wu, B.G.; Wikoff, W.R.; Gao, Z.; Li, Y.; Ko, J.P.; Rom, W.N.; Blaser, M.J.; Weiden, M.D. Randomised, double-blind, placebo-controlled trial with azithromycin selects for anti-inflammatory microbial metabolites in the emphysematous lung. Thorax 2017, 72, 13–22. [Google Scholar] [CrossRef]
- Crouch, L.I.; Liberato, M.V.; Urbanowicz, P.A.; Baslé, A.; Lamb, C.A.; Stewart, C.J.; Cooke, K.; Doona, M.; Needham, S.; Brady, R.R.; et al. Prominent members of the human gut microbiota express endo-acting O-glycanases to initiate mucin breakdown. bioRxiv 2019. [Google Scholar] [CrossRef]
- Robbe, C.; Capon, C.; Coddeville, B.; Michalski, J.-C. Structural diversity and specific distribution of O-glycans in normal human mucins along the intestinal tract. Biochem. J. 2004, 384, 307–316. [Google Scholar] [CrossRef]
- Tailford, L.E.; Crost, E.H.; Kavanaugh, D.; Juge, N. Mucin glycan foraging in the human gut microbiome. Front. Genet. 2015, 6, 81. [Google Scholar] [CrossRef]
- Echlin, H.; Frank, M.; Rock, C.; Rosch, J.W. Role of the pyruvate metabolic network on carbohydrate metabolism and virulence in Streptococcus pneumoniae. Mol. Microbiol. 2020, 114, 536–552. [Google Scholar] [CrossRef]
- Hobbs, J.K.; Pluvinage, B.; Boraston, A.B. Glycan-metabolizing enzymes in microbe–host interactions: The Streptococcus pneumoniae paradigm. FEBS Lett. 2018, 592, 3865–3897. [Google Scholar] [CrossRef]
- Leonard, A.; Lalk, M. Infection and metabolism—Streptococcus pneumoniae metabolism facing the host environment. Cytokine 2018, 112, 75–86. [Google Scholar] [CrossRef]
- Josenhans, C.; Müthing, J.; Elling, L.; Bartfeld, S.; Schmidt, H. How bacterial pathogens of the gastrointestinal tract use the mucosal glyco-code to harness mucus and microbiota: New ways to study an ancient bag of tricks. Int. J. Med. Microbiol. 2020, 310, 151392. [Google Scholar] [CrossRef]
- DeLeon, S.; Clinton, A.; Fowler, H.; Everett, J.; Horswill, A.R.; Rumbaugh, K.P. Synergistic interactions of Pseudomonas aeruginosa and Staphylococcus aureus in an in vitro wound model. Infect. Immun. 2014, 82, 4718–4728. [Google Scholar] [CrossRef]
- Limoli, D.H.; Yang, J.; Khansaheb, M.K.; Helfman, B.; Peng, L.; Stecenko, A.A.; Goldberg, J.B. Staphylococcus aureus and Pseudomonas aeruginosa co-infection is associated with cystic fibrosis-related diabetes and poor clinical outcomes. Eur. J. Clin. Microbiol. Infect. Dis. 2016, 35, 947–953. [Google Scholar] [CrossRef]
- Hoffman, C.L.; Lalsiamthara, J.; Aballay, A. Host Mucin Is Exploited by Pseudomonas aeruginosa To Provide Monosaccharides Required for a Successful Infection. mBio 2020, 11, e00060-20. [Google Scholar] [CrossRef]
- Wang, B.X.; Wu, C.M.; Ribbeck, K. Home, sweet home: How mucus accommodates our microbiota. FEBS J. 2020, 288, 1789–1799. [Google Scholar] [CrossRef]
- Chakraborty, P.; Bajeli, S.; Kaushal, D.; Radotra, B.D.; Kumar, A. Biofilm formation in the lung contributes to virulence and drug tolerance of Mycobacterium tuberculosis. Nat. Commun. 2021, 12, 1606. [Google Scholar] [CrossRef]
- Nash, E.F.; Coonar, A.; Kremer, R.; Tullis, E.; Hutcheon, M.; Singer, L.G.; Keshavjee, S.; Chaparro, C. Survival of Burkholderia cepacia sepsis following lung transplantation in recipients with cystic fibrosis. Transpl. Infect. Dis. 2010, 12, 551–554. [Google Scholar] [CrossRef]
- Schwab, U.; Abdullah, L.H.; Perlmutt, O.S.; Albert, D.; Davis, C.W.; Arnold, R.R.; Yankaskas, J.R.; Gilligan, P.; Neubauer, H.; Randell, S.H.; et al. Localization of Burkholderia cepacia complex bacteria in cystic fibrosis lungs and interactions with Pseudomonas aeruginosa in hypoxic mucus. Infect. Immun. 2014, 82, 4729–4745. [Google Scholar] [CrossRef]
- Folescu, T.W.; da Costa, C.H.; Cohen, R.W.F.; Neto, O.C.d.C.; Albano, R.M.; Marques, E.A. Burkholderia cepacia complex: Clinical course in cystic fibrosis patients. BMC Pulm. Med. 2015, 15, 158. [Google Scholar] [CrossRef] [PubMed]
- Kavanaugh, N.L.; Zhang, A.Q.; Nobile, C.J.; Johnson, A.D.; Ribbeck, K. Mucins Suppress Virulence Traits of Candida albicans. mBio 2014, 5, e01911–e01914. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, K.M.; Cárcamo-Oyarce, G.; Turner, B.S.; Dellos-Nolan, S.; Co, J.Y.; Lehoux, S.; Cummings, R.D.; Wozniak, D.J.; Ribbeck, K. Mucin glycans attenuate the virulence of Pseudomonas aeruginosa in infection. Nat. Microbiol. 2019, 4, 2146–2154. [Google Scholar] [CrossRef] [PubMed]
- Frenkel, E.S.; Ribbeck, K. Salivary Mucins Protect Surfaces from Colonization by Cariogenic Bacteria. Appl. Environ. Microbiol. 2015, 81, 332–338. [Google Scholar] [CrossRef]
- Sperandio, B.; Fischer, N.; Sansonetti, P.J. Mucosal physical and chemical innate barriers: Lessons from microbial evasion strategies. Semin. Immunol. 2015, 27, 111–118. [Google Scholar] [CrossRef]
- Ohneck, E.J.; Arivett, B.A.; Fiester, S.E.; Wood, C.R.; Metz, M.L.; Simeone, G.M.; Actis, L.A. Mucin acts as a nutrient source and a signal for the differential expression of genes coding for cellular processes and virulence factors in Acinetobacter baumannii. PLoS ONE 2018, 13, e0190599. [Google Scholar] [CrossRef]
- Yesilkaya, H.; Manco, S.; Kadioglu, A.; Terra, V.S.; Andrew, P.W. The ability to utilize mucin affects the regulation of virulence gene expression in Streptococcus pneumoniae. FEMS Microbiol. Lett. 2008, 278, 231–235. [Google Scholar] [CrossRef]
- Co, J.Y.; Cárcamo-Oyarce, G.; Billings, N.; Wheeler, K.M.; Grindy, S.C.; Holten-Andersen, N.; Ribbeck, K. Mucins trigger dispersal of Pseudomonas aeruginosa biofilms. NPJ Biofilms Microbiomes 2018, 4, 23. [Google Scholar] [CrossRef]
- Xia, B.; Royall, J.A.; Damera, G.; Sachdev, G.P.; Cummings, R.D. Altered O-glycosylation and sulfation of airway mucins associated with cystic fibrosis. Glycobiology 2005, 15, 747–775. [Google Scholar] [CrossRef]
- Schulz, B.L.; Sloane, A.J.; Robinson, L.J.; Prasad, S.S.; Lindner, R.A.; Robinson, M.; Bye, P.T.; Nielson, D.W.; Harry, J.L.; Packer, N.H.; et al. Glycosylation of sputum mucins is altered in cystic fibrosis patients. Glycobiology 2007, 17, 698–712. [Google Scholar] [CrossRef]
- Caballero, I.; Ringot-Destrez, B.; Si-Tahar, M.; Barbry, P.; Guillon, A.; Lantier, I.; Berri, M.; Chevaleyre, C.; Fleurot, I.; Barc, C.; et al. Evidence of early increased sialylation of airway mucins and defective mucociliary clearance in CFTR-deficient piglets. J. Cyst. Fibros. 2020, 20, 173–182. [Google Scholar] [CrossRef]
- Allinson, J.P.; Hardy, R.; Donaldson, G.C.; Shaheen, S.O.; Kuh, D.; Wedzicha, J.A. The Presence of Chronic Mucus Hypersecretion across Adult Life in Relation to Chronic Obstructive Pulmonary Disease Development. Am. J. Respir. Crit. Care Med. 2016, 193, 662–672. [Google Scholar] [CrossRef]
- Burgel, P.-R.; Nesme-Meyer, P.; Chanez, P.; Caillaud, D.; Carré, P.; Perez, T.; Roche, N. Cough and Sputum Production Are Associated with Frequent Exacerbations and Hospitalizations in COPD Subjects. Chest 2009, 135, 975–982. [Google Scholar] [CrossRef]
- Vestbo, J.; Prescott, E.; Lange, P. Association of chronic mucus hypersecretion with FEV1 decline and chronic obstructive pulmonary disease morbidity. Copenhagen City Heart Study Group. Am. J. Respir. Crit. Care Med. 1996, 153, 1530–1535. [Google Scholar] [CrossRef]
- Leitao Filho, F.S.; Alotaibi, N.M.; Ngan, D.; Tam, S.; Yang, J.; Hollander, Z.; Chen, V.; FitzGerald, J.M.; Nislow, C.; Leung, J.M.; et al. Sputum Microbiome Is Associated with 1-Year Mortality after Chronic Obstructive Pulmonary Disease Hospitalizations. Am. J. Respir. Crit. Care Med. 2019, 199, 1205–1213. [Google Scholar] [CrossRef]
- Reynolds, J.H.; McDonald, G.; Alton, H.; Gordon, S.B. Pneumonia in the immunocompetent patient. Br. J. Radiol. 2010, 83, 998–1009. [Google Scholar] [CrossRef]
- Garg, M.; Prabhakar, N.; Gulati, A.; Agarwal, R.; Dhooria, S. Spectrum of imaging findings in pulmonary infections. Part 1: Bacterial and viral. Pol. J. Radiol. 2019, 84, e205–e213. [Google Scholar] [CrossRef]
- Farrell, P.M.; White, T.B.; Ren, C.L.; Hempstead, S.E.; Accurso, F.; Derichs, N.; Howenstine, M.; McColley, S.A.; Rock, M.; Rosenfeld, M.; et al. Diagnosis of Cystic Fibrosis: Consensus Guidelines from the Cystic Fibrosis Foundation. J. Pediatrics 2017, 181, S4–S15.e11. [Google Scholar] [CrossRef]
- Birket, S.E.; Chu, K.K.; Houser, G.H.; Liu, L.; Fernandez, C.M.; Solomon, G.M.; Lin, V.; Shastry, S.; Mazur, M.; Sloane, P.A.; et al. Combination therapy with cystic fibrosis transmembrane conductance regulator modulators augment the airway functional microanatomy. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 310, L928–L939. [Google Scholar] [CrossRef]
- Montgomery, S.T.; Dittrich, A.S.; Garratt, L.W.; Turkovic, L.; Frey, D.L.; Stick, S.M.; Mall, M.A.; Kicic, A. Interleukin-1 is associated with inflammation and structural lung disease in young children with cystic fibrosis. J. Cyst. Fibros. 2018, 17, 715–722. [Google Scholar] [CrossRef]
- Fritzsching, B.; Zhou-Suckow, Z.; Trojanek, J.B.; Schubert, S.C.; Schatterny, J.; Hirtz, S.; Agrawal, R.; Muley, T.; Kahn, N.; Sticht, C.; et al. Hypoxic epithelial necrosis triggers neutrophilic inflammation via IL-1 receptor signaling in cystic fibrosis lung disease. Am. J. Respir. Crit. Care Med. 2015, 191, 902–913. [Google Scholar] [CrossRef]
- Button, B.; Anderson, W.H.; Boucher, R.C. Mucus Hyperconcentration as a Unifying Aspect of the Chronic Bronchitic Phenotype. Ann. Am. Thorac. Soc. 2016, 13 (Suppl. 2) (Suppl. 2), S156–S162. [Google Scholar] [CrossRef]
- Rogers, G.B.; Taylor, S.L.; Hoffman, L.R.; Burr, L.D. The impact of CFTR modulator therapies on CF airway microbiology. J. Cyst. Fibros. 2020, 19, 359–364. [Google Scholar] [CrossRef]
- Matsui, H.; Verghese, M.W.; Kesimer, M.; Schwab, U.E.; Randell, S.H.; Sheehan, J.K.; Grubb, B.R.; Boucher, R.C. Reduced Three-Dimensional Motility in Dehydrated Airway Mucus Prevents Neutrophil Capture and Killing Bacteria on Airway Epithelial Surfaces. J. Immunol. 2005, 175, 1090–1099. [Google Scholar] [CrossRef]
- Hoegger, M.J.; Fischer, A.J.; McMenimen, J.D.; Ostedgaard, L.S.; Tucker, A.J.; Awadalla, M.A.; Moninger, T.O.; Michalski, A.S.; Hoffman, E.A.; Zabner, J.; et al. Impaired mucus detachment disrupts mucociliary transport in a piglet model of cystic fibrosis. Science 2014, 345, 818–822. [Google Scholar] [CrossRef]
- Rogers, C.S.; Stoltz, D.A.; Meyerholz, D.K.; Ostedgaard, L.S.; Rokhlina, T.; Taft, P.J.; Rogan, M.P.; Pezzulo, A.A.; Karp, P.H.; Itani, O.A.; et al. Disruption of the CFTR gene produces a model of cystic fibrosis in newborn pigs. Science 2008, 321, 1837–1841. [Google Scholar] [CrossRef]
- Ash, S.Y.; Harmouche, R.; Putman, R.K.; Ross, J.C.; Martinez, F.J.; Choi, A.M.; Bowler, R.P.; Regan, E.A.; Curtis, J.L.; Han, M.K.; et al. Association between acute respiratory disease events and the MUC5B promoter polymorphism in smokers. Thorax 2018, 73, 1071–1074. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J. Against the Dutch hypothesis: Asthma and chronic obstructive pulmonary disease are distinct diseases. Am. J. Respir. Crit. Care Med. 2006, 174, 240–243. [Google Scholar] [CrossRef]
- van der Pouw Kraan, T.C.T.M.; Küçükaycan, M.; Bakker, A.M.; Baggen, J.M.C.; van der Zee, J.S.; Dentener, M.A.; Wouters, E.F.M.; Verweij, C.L. Chronic obstructive pulmonary disease is associated with the -1055 IL-13 promoter polymorphism. Genes Immun. 2002, 3, 436–439. [Google Scholar] [CrossRef] [PubMed]
- Wills-Karp, M.; Luyimbazi, J.; Xu, X.; Schofield, B.; Neben, T.Y.; Karp, C.L.; Donaldson, D.D. Interleukin-13: Central Mediator of Allergic Asthma. Science 1998, 282, 2258–2261. [Google Scholar] [CrossRef]
- Bonser, L.R.; Zlock, L.; Finkbeiner, W.; Erle, D.J. Epithelial tethering of MUC5AC-rich mucus impairs mucociliary transport in asthma. J. Clin. Invest. 2016, 126, 2367–2371. [Google Scholar] [CrossRef]
- Yuan, S.; Hollinger, M.; Lachowicz-Scroggins, M.E.; Kerr, S.C.; Dunican, E.M.; Daniel, B.M.; Ghosh, S.; Erzurum, S.C.; Willard, B.; Hazen, S.L.; et al. Oxidation increases mucin polymer cross-links to stiffen airway mucus gels. Sci. Transl. Med. 2015, 7, 276ra227. [Google Scholar] [CrossRef] [PubMed]
- Dunican, E.M.; Elicker, B.M.; Gierada, D.S.; Nagle, S.K.; Schiebler, M.L.; Newell, J.D.; Raymond, W.W.; Lachowicz-Scroggins, M.E.; di Maio, S.; Hoffman, E.A.; et al. Mucus plugs in patients with asthma linked to eosinophilia and airflow obstruction. J. Clin. Invest. 2018, 128, 997–1009. [Google Scholar] [CrossRef] [PubMed]
- Dunican, E.M.; Elicker, B.M.; Henry, T.; Gierada, D.S.; Schiebler, M.L.; Anderson, W.; Barjaktarevic, I.; Barr, R.G.; Bleecker, E.R.; Boucher, R.C.; et al. Mucus Plugs and Emphysema in the Pathophysiology of Airflow Obstruction and Hypoxemia in Smokers. Am. J. Respir. Crit. Care Med. 2020, 203, 957–968. [Google Scholar] [CrossRef]
- Dicker, A.J.; Huang, J.T.J.; Lonergan, M.; Keir, H.R.; Fong, C.J.; Tan, B.; Cassidy, A.J.; Finch, S.; Mullerova, H.; Miller, B.E.; et al. The sputum microbiome, airway inflammation, and mortality in chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 2021, 147, 158–167. [Google Scholar] [CrossRef]
- Hufnagl, K.; Pali-Schöll, I.; Roth-Walter, F.; Jensen-Jarolim, E. Dysbiosis of the gut and lung microbiome has a role in asthma. Semin. Immunopathol. 2020, 42, 75–93. [Google Scholar] [CrossRef]
- Mayhew, D.; Devos, N.; Lambert, C.; Brown, J.R.; Clarke, S.C.; Kim, V.L.; Magid-Slav, M.; Miller, B.E.; Ostridge, K.K.; Patel, R.; et al. Longitudinal profiling of the lung microbiome in the AERIS study demonstrates repeatability of bacterial and eosinophilic COPD exacerbations. Thorax 2018, 73, 422–430. [Google Scholar] [CrossRef]
- Jeffries, J.L.; Jia, J.; Choi, W.; Choe, S.; Miao, J.; Xu, Y.; Powell, R.; Lin, J.; Kuang, Z.; Gaskins, H.R.; et al. Pseudomonas aeruginosa pyocyanin modulates mucin glycosylation with sialyl-Lewisx to increase binding to airway epithelial cells. Mucosal Immunol. 2016, 9, 1039–1050. [Google Scholar] [CrossRef]
- Chen, R.; Lim, J.H.; Jono, H.; Gu, X.X.; Kim, Y.S.; Basbaum, C.B.; Murphy, T.F.; Li, J.D. Nontypeable Haemophilus influenzae lipoprotein P6 induces MUC5AC mucin transcription via TLR2-TAK1-dependent p38 MAPK-AP1 and IKKbeta-IkappaBalpha-NF-kappaB signaling pathways. Biochem. Biophys. Res. Commun. 2004, 324, 1087–1094. [Google Scholar] [CrossRef]
- Carnoy, C.; Scharfman, A.; van Brussel, E.; Lamblin, G.; Ramphal, R.; Roussel, P. Pseudomonas aeruginosa outer membrane adhesins for human respiratory mucus glycoproteins. Infect. Immun. 1994, 62, 1896–1900. [Google Scholar] [CrossRef]
- Perez-Vilar, J.; Boucher, R.C. Reevaluating gel-forming mucins’ roles in cystic fibrosis lung disease. Free Radic. Biol. Med. 2004, 37, 1564–1577. [Google Scholar] [CrossRef]
- Rogers, D.F. Mucoactive agents for airway mucus hypersecretory diseases. Respir. Care 2007, 52, 1176–1193. [Google Scholar]
- Tam, J.; Nash, E.F.; Ratjen, F.; Tullis, E.; Stephenson, A. Nebulized and oral thiol derivatives for pulmonary disease in cystic fibrosis. Cochrane Database Syst. Rev. 2013. [Google Scholar] [CrossRef]
- Poole, P.; Sathananthan, K.; Fortescue, R. Mucolytic agents versus placebo for chronic bronchitis or chronic obstructive pulmonary disease. Cochrane Database Syst Rev. 2019, 5, Cd001287. [Google Scholar] [CrossRef]
- Suri, R. The Use of Human Deoxyribonuclease (rhDNase) in the Management of Cystic Fibrosis. BioDrugs 2005, 19, 135–144. [Google Scholar] [CrossRef]
- Konstan, M.W.; Ratjen, F. Effect of dornase alfa on inflammation and lung function: Potential role in the early treatment of cystic fibrosis. J. Cyst. Fibros. 2012, 11, 78–83. [Google Scholar] [CrossRef]
- Shak, S. Aerosolized recombinant human DNase I for the treatment of cystic fibrosis. Chest 1995, 107, 65s–70s. [Google Scholar] [CrossRef]
- Conrad, C.; Lymp, J.; Thompson, V.; Dunn, C.; Davies, Z.; Chatfield, B.; Nichols, D.; Clancy, J.; Vender, R.; Egan, M.; et al. Long-term treatment with oral N-acetylcysteine: Affects lung function but not sputum inflammation in cystic fibrosis subjects. A phase II randomized placebo-controlled trial. J. Cyst. Fibros. 2014, 14. [Google Scholar] [CrossRef]
- O’Donnell, A.E.; Barker, A.F.; Ilowite, J.S.; Fick, R.B. Treatment of idiopathic bronchiectasis with aerosolized recombinant human DNase I. rhDNase Study Group. Chest 1998, 113, 1329–1334. [Google Scholar] [CrossRef]
- Dal Negro, R.W.; Wedzicha, J.A.; Iversen, M.; Fontana, G.; Page, C.; Cicero, A.F.; Pozzi, E.; Calverley, P.M.A. Effect of erdosteine on the rate and duration of COPD exacerbations: The RESTORE study. Eur Respir. J. 2017, 50, 1602265. [Google Scholar] [CrossRef] [PubMed]
- James, G.D.; Petersen, I.; Nazareth, I.; Wedzicha, J.A.; Donaldson, G.C. Use of long-term antibiotic treatment in COPD patients in the UK: A retrospective cohort study. Prim. Care Respir. J. 2013, 22, 271–277. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chung, K.F. Potential Role of the Lung Microbiome in Shaping Asthma Phenotypes. Ann. Am. Thorac. Soc. 2017, 14, S326–S331. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Li, L.-Q.; Chen, C.-Y.; Zhang, G.-S.; Cui, W.; Tian, B.-P. Do probiotics help prevent ventilator-associated pneumonia in the critically ill patients? A systematic review with meta-analysis. ERJ Open Res. 2020, 17, 5. [Google Scholar] [CrossRef] [PubMed]
- Azad, M.B.; Coneys, J.G.; Kozyrskyj, A.L.; Field, C.J.; Ramsey, C.D.; Becker, A.B.; Friesen, C.; Abou-Setta, A.M.; Zarychanski, R. Probiotic supplementation during pregnancy or infancy for the prevention of asthma and wheeze: Systematic review and meta-analysis. BMJ 2013, 347, f6471. [Google Scholar] [CrossRef]
- Nunes, C.F.; Nogueira, J.S.; Vianna, P.H.O.; Ciambarella, B.T.; Rodrigues, P.M.; Miranda, K.R.; Lobo, L.A.; Domingues, R.; Busch, M.; Atella, G.C.; et al. Probiotic treatment during neonatal age provides optimal protection against experimental asthma through the modulation of microbiota and T cells. Int. Immunol. 2018, 30, 155–169. [Google Scholar] [CrossRef]
- Arrieta, M.-C.; Stiemsma, L.T.; Dimitriu, P.A.; Thorson, L.; Russell, S.; Yurist-Doutsch, S.; Kuzeljevic, B.; Gold, M.J.; Britton, H.M.; Lefebvre, D.L.; et al. Early infancy microbial and metabolic alterations affect risk of childhood asthma. Sci. Transl. Med. 2015, 7, 307ra152. [Google Scholar] [CrossRef]
- Mendez, R.; Banerjee, S.; Martinez, O.; Perez-Cardona, A.; Abbo, L.M. 615. Can We Restore the Lung Microbiome with Fecal Microbiota Transplant (FMT)? Open Forum Infect. Dis. 2018, 5, S224. [Google Scholar] [CrossRef]
- Spacova, I.; Ceuppens, J.L.; Seys, S.F.; Petrova, M.I.; Lebeer, S. Probiotics against airway allergy: Host factors to consider. Dis. Model. Mech. 2018, 11, dmm034314. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Type of Comparison | Stratification | Mucus Parameters | Reference | ||||
|---|---|---|---|---|---|---|---|
| Mucus Solids Concentration (%) | Total Mucin µg/mL | MUC5B µg/mL or µg/g | MUC5AC µg/mL or µg/g | MUC5B: MUC5AC | |||
| Asthma | |||||||
| History of sputum production | Normal | - | 800 | - | - | - | [46] |
| Asthma (no phlegm) | - | 1100 | - | - | - | ||
| Asthma (phlegm) | - | 4250 | - | - | - | ||
| Pediatric asthma | Normal | - | 246 | 239 | 7.6 | 7.3:1 | [47] |
| Stable asthma | - | 231 | 208 | 22 | 9.33:1 | ||
| Acute asthma | - | 211 | 166 | 45 | 3.7:1 | ||
| Asthma phenotype | Normal | - | 1597 (3101) | 269 (276) | 1328 (1030) | - | [48] |
| Asthma | - | 4753 (5785) | 1798 (3130) | 2955 (2720) | - | ||
| Effect of asthma exacerbation | Normal | - | - | - | - | 1.8:1 | [49] |
| Stable | - | - | - | - | 0.7:1 | ||
| Exacerbation | - | - | - | - | 0.6:1 | ||
| Non-CF Bronchiectasis | |||||||
| Phenotype | Normal | 1.9 | 1820 | 85 | 3 | 28:1 | [30] |
| Bronchiectasis | 2.7 | 4549 | 550 | 100 | 5.5:1 | ||
| Induction method | Induced | - | 3800 | - | - | - | [30] |
| Spontaneous | - | 4800 | - | - | - | ||
| P. aeruginosa infection | Positive | - | - | Below det. | 6.9 × 10−4 | - | [50] |
| Negative | - | - | Below det. | 4.7 × 10−4 | - | ||
| Cystic fibrosis (CF) | |||||||
| Mucin hyperconcentration | Non-CF | - | 2710 | 7.8 | 1.2 | 6.5:1 | [51] |
| CF | - | 6454 | 1.1 | 1.1 | 1:1 | ||
| Mucus hyperconcentration | Adult CF | 5.2 (2.3) | - | - | - | - | [52] |
| Mechanisms initiating pediatric lung disease | Non-CF pulmonary disease | - | 229 (455) | - | - | 3.7:1 | [53] |
| CF | - | 475 (1089) | - | - | 3.7:1 | ||
| Pulmonary eosinophilia (PE) | |||||||
| Airway mucin concentration | Normal | - | - | - | 8.0 × 10−6 | - | [54] |
| PE | - | - | - | 2.0 × 10−5 | - | ||
| Chronic PE | - | - | - | 2.3 × 10−5 | - | ||
| Pulmonary alveolar proteinosis (PAP) | |||||||
| Airway mucins in PAP | Normal | - | - | 9.0 × 10−5 | 1.0 × 10−5 | - | [55] |
| PAP | - | - | 5.4 × 10−5 | 9.0 × 10−6 | - | ||
| Chronic obstructive pulmonary disease (COPD) | |||||||
| Annualized exacerbation rate | 0 | - | 2200 | - | - | - | [56] |
| > 0 to < 2 | - | 2400 | - | - | - | ||
| ≥ 2 | - | 4200 | - | - | - | ||
| Sputum production | Never smoker | - | 1600 | - | - | - | [56] |
| Ever smoker | - | 2200 | - | - | - | ||
| Ever smoker, phlegm | - | 3100 | - | - | - | ||
| COPD severity (lung function) | Gold Stage 0 | - | 2400 | - | - | - | [56] |
| Gold Stage 1 | - | 2450 | - | - | - | ||
| Gold Stage 2 | - | 3100 | - | - | - | ||
| Gold Stage 3 | - | 3100 | - | - | - | ||
| Effect of smoking and a diagnosis of chronic bronchitis (CB) | Never smoker (age 27) | - | 1603 (1041) | - | - | - | [56] |
| Ever smoker (age 30) | - | 1692 (827) | - | - | - | ||
| CB, ever smoker (age 61) | - | 3868 (2297) | - | - | - | ||
| Current or former smoker | Never smoker | 1.96 | 1.84 | - | - | - | [57] |
| Ever smoker | 1.7 | 1.45 | - | - | - | ||
| CB, ever smoker | 3 | 2.92 | - | - | - | ||
| MUC5B pmol/mL | MUC5AC pmol/mL | ||||||
| COPD symptom severity | Normal, never smoker | - | - | 110 | 15 | 7.3:1 | [56] |
| No COPD, ever smoker | - | - | 220 | 80 | 2.8:1 | ||
| Mild-/Mod COPD, ever smoker | - | - | 170 | 75 | 2.3:1 | ||
| Severe COPD, ever smoker | - | - | 300 | 110 | 2.7:1 | ||
| Diagnostic applicability | No CB | - | 1500 | 95 | 8.6 | 11:1 | [56] |
| Classically defined | - | 3500 | 200 | 52 | 3.8:1 | ||
| SGRQ^ defined | - | 3200 | 230 | 53 | 4.3:1 | ||
| Disease Type | Cumulative Findings |
|---|---|
| Asthma |
|
| Non-CF bronchiectasis |
|
| Cystic fibrosis | |
| Pulmonary eosinophilia |
|
| Pulmonary alveolar proteinosis |
|
| COPD |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meldrum, O.W.; Chotirmall, S.H. Mucus, Microbiomes and Pulmonary Disease. Biomedicines 2021, 9, 675. https://doi.org/10.3390/biomedicines9060675
Meldrum OW, Chotirmall SH. Mucus, Microbiomes and Pulmonary Disease. Biomedicines. 2021; 9(6):675. https://doi.org/10.3390/biomedicines9060675
Chicago/Turabian StyleMeldrum, Oliver W., and Sanjay H. Chotirmall. 2021. "Mucus, Microbiomes and Pulmonary Disease" Biomedicines 9, no. 6: 675. https://doi.org/10.3390/biomedicines9060675
APA StyleMeldrum, O. W., & Chotirmall, S. H. (2021). Mucus, Microbiomes and Pulmonary Disease. Biomedicines, 9(6), 675. https://doi.org/10.3390/biomedicines9060675