
Targeting the Proteasome in Advanced Renal Cell Carcinoma: Complexity and Limitations of Patient-Individualized Preclinical Drug Discovery

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
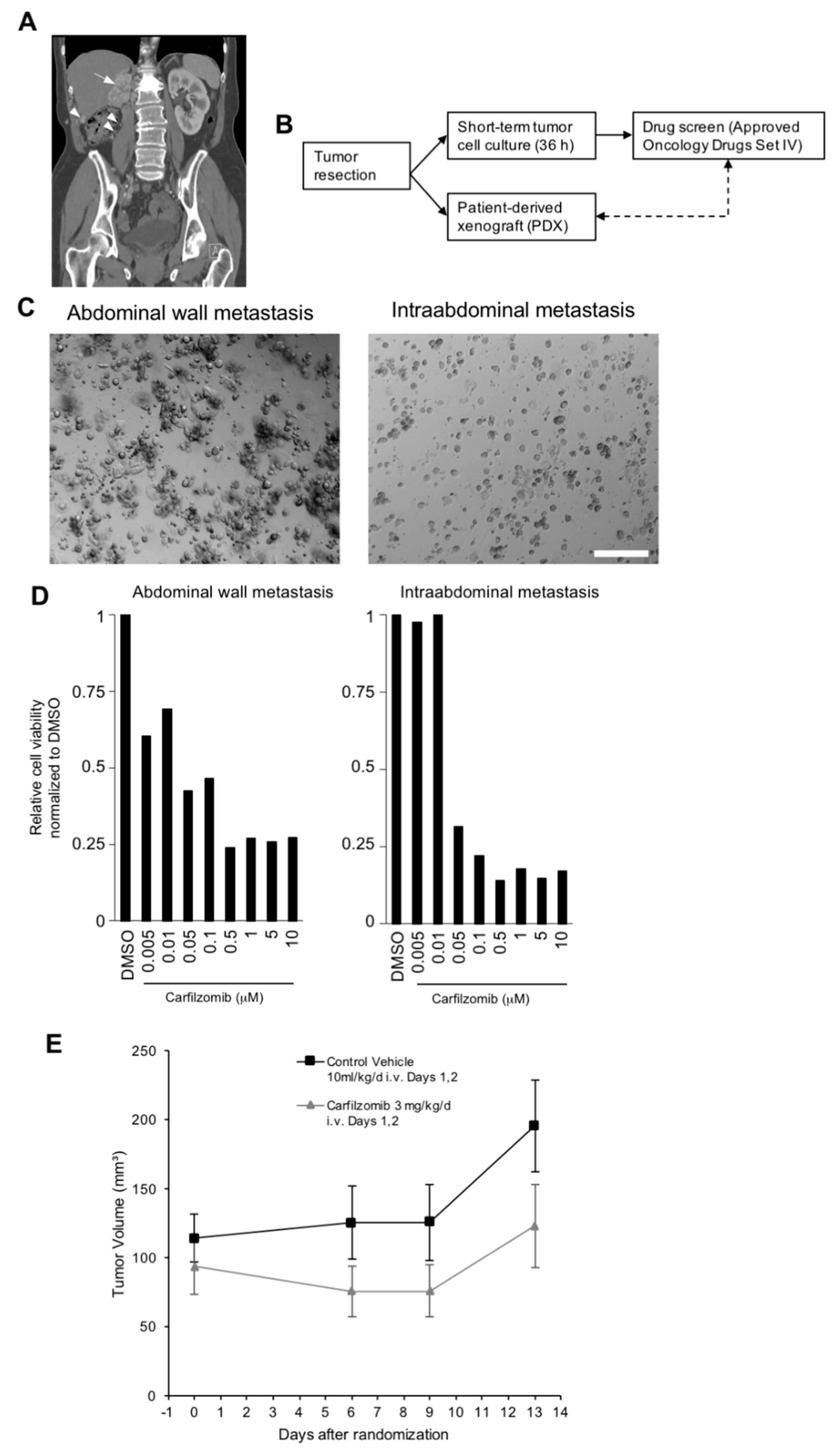
2.1. Short-Term Tumor Cell Cultures
2.2. In Vitro Drug Screening, Inhibitor Treatment and Long-Term Tumor Cell Growth Assays
2.3. PDX Models and In Vivo Drug Testing
2.4. Analysis of Ubiquitinated Proteins
2.5. Statistical Analysis
3. Results
3.1. Carfilzomib Has Potent Antitumoral Activities in RCC Cells
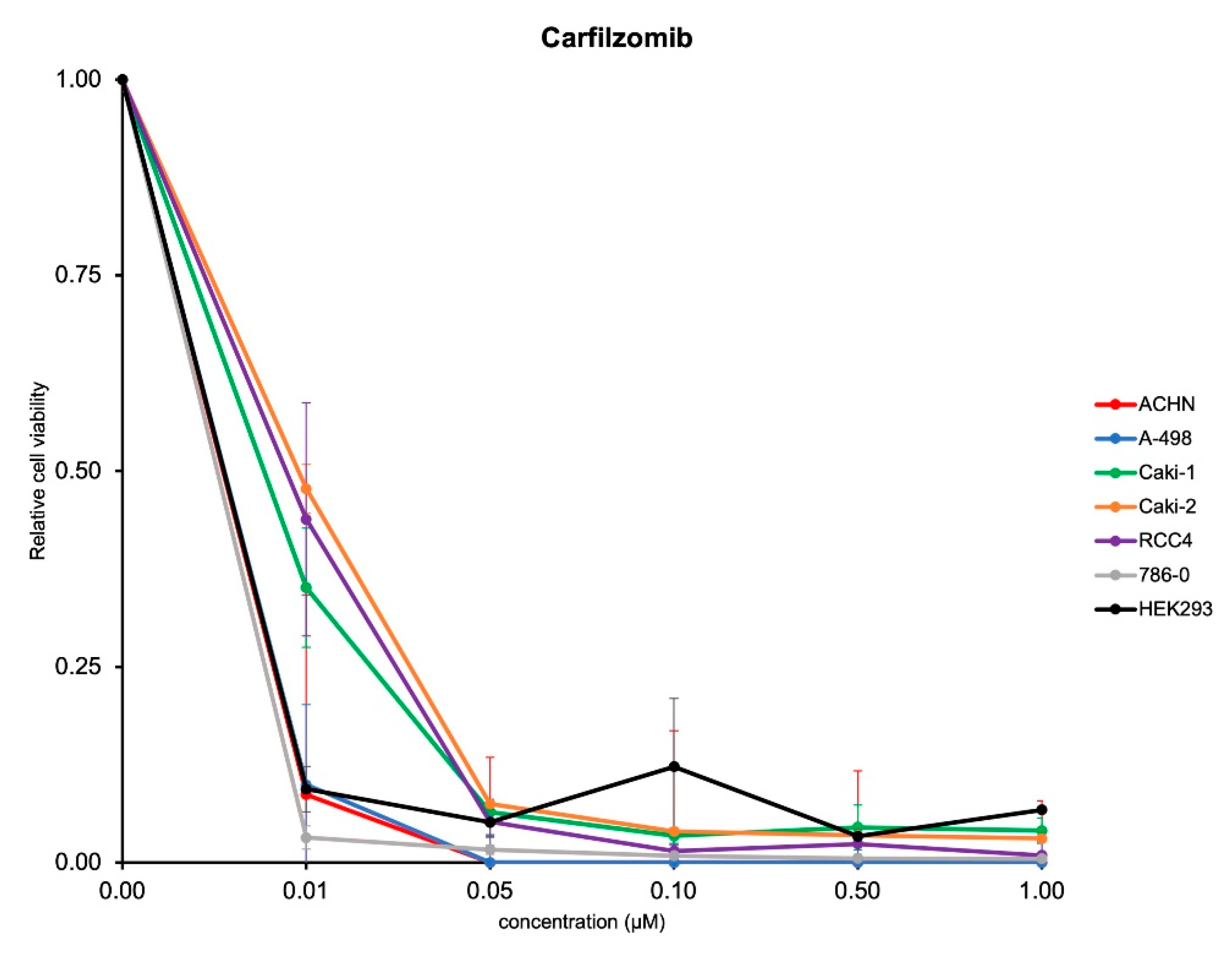
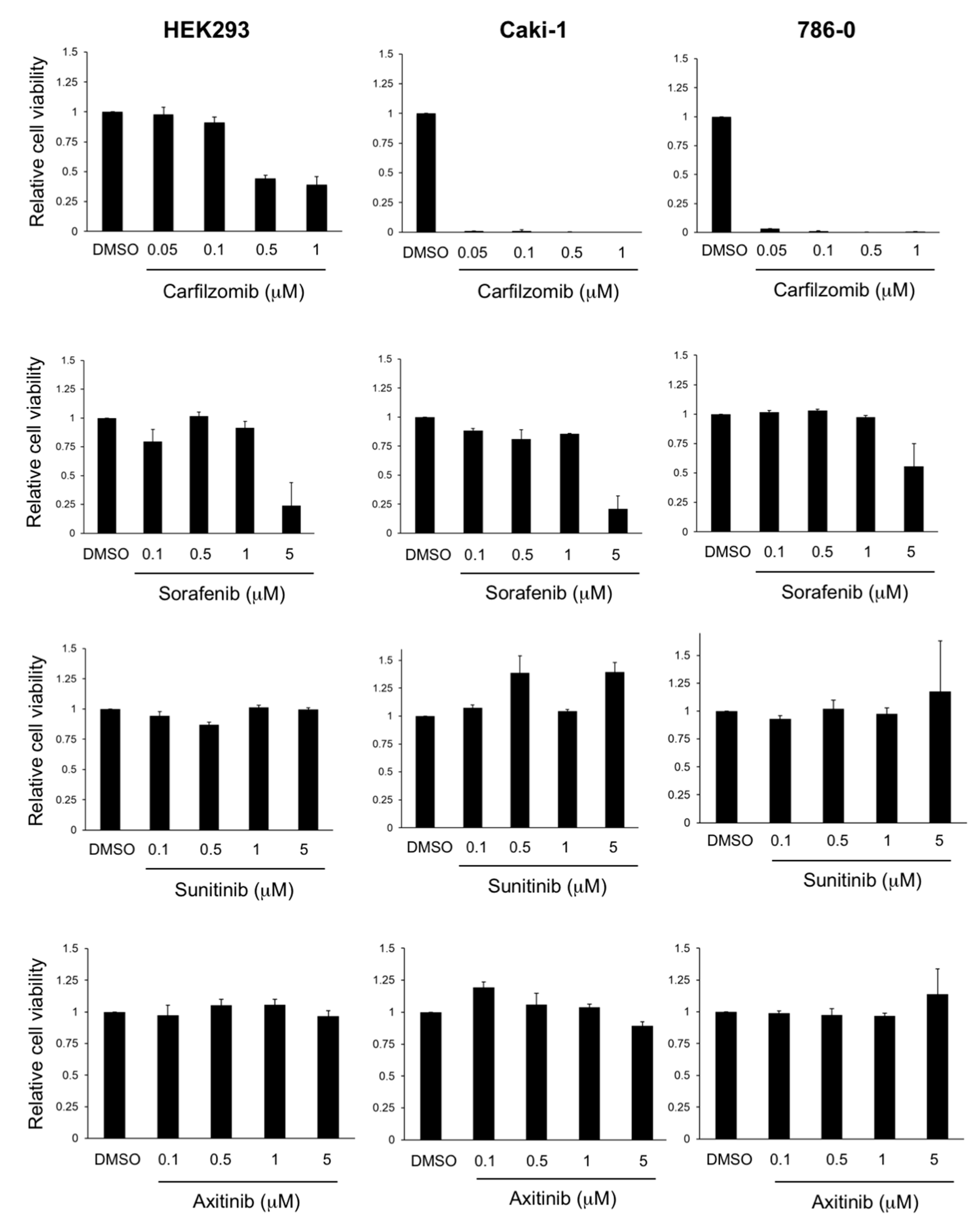
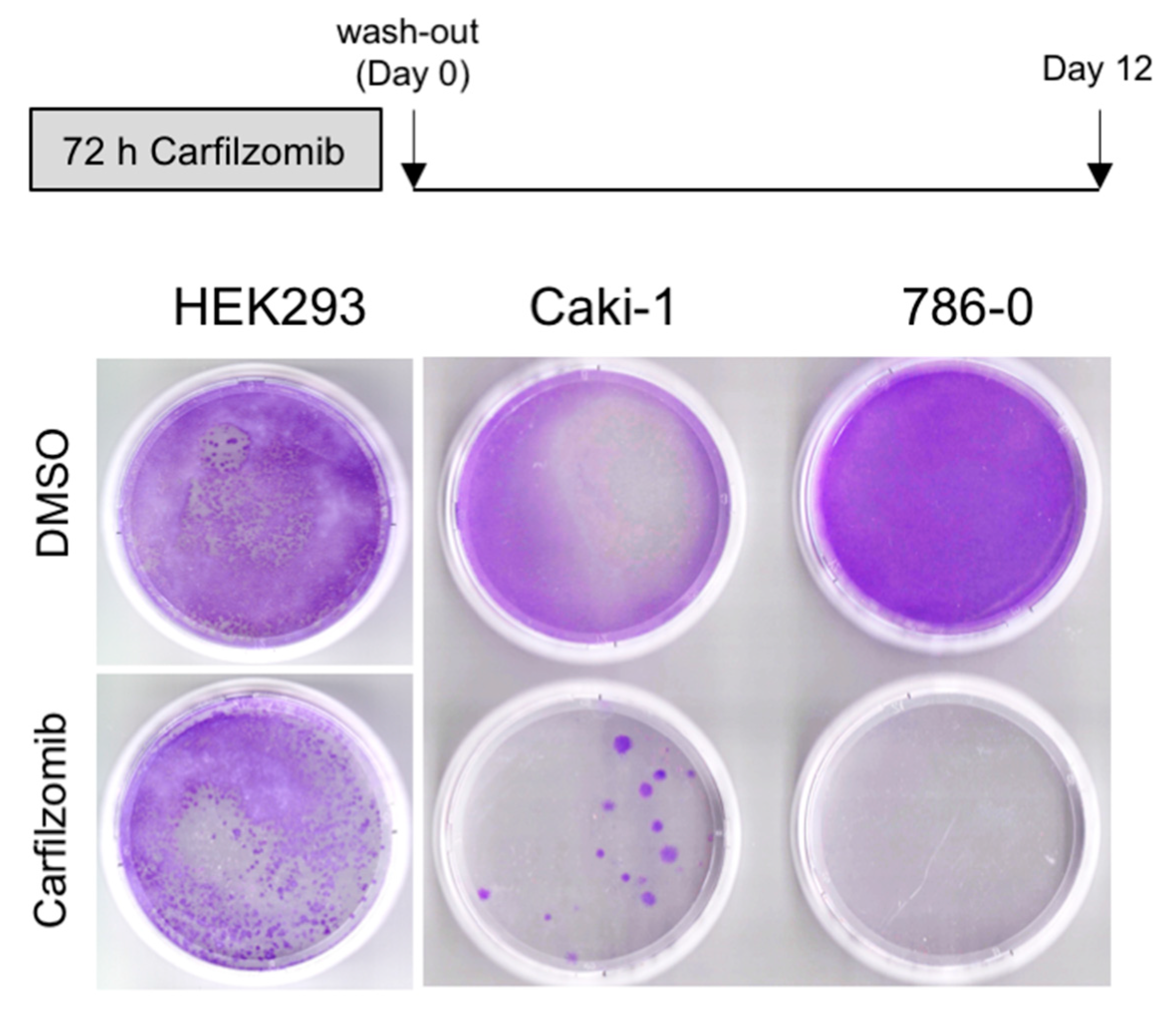
3.2. Antitumoral Activity of Carfilzomib in Established RCC Cell Lines
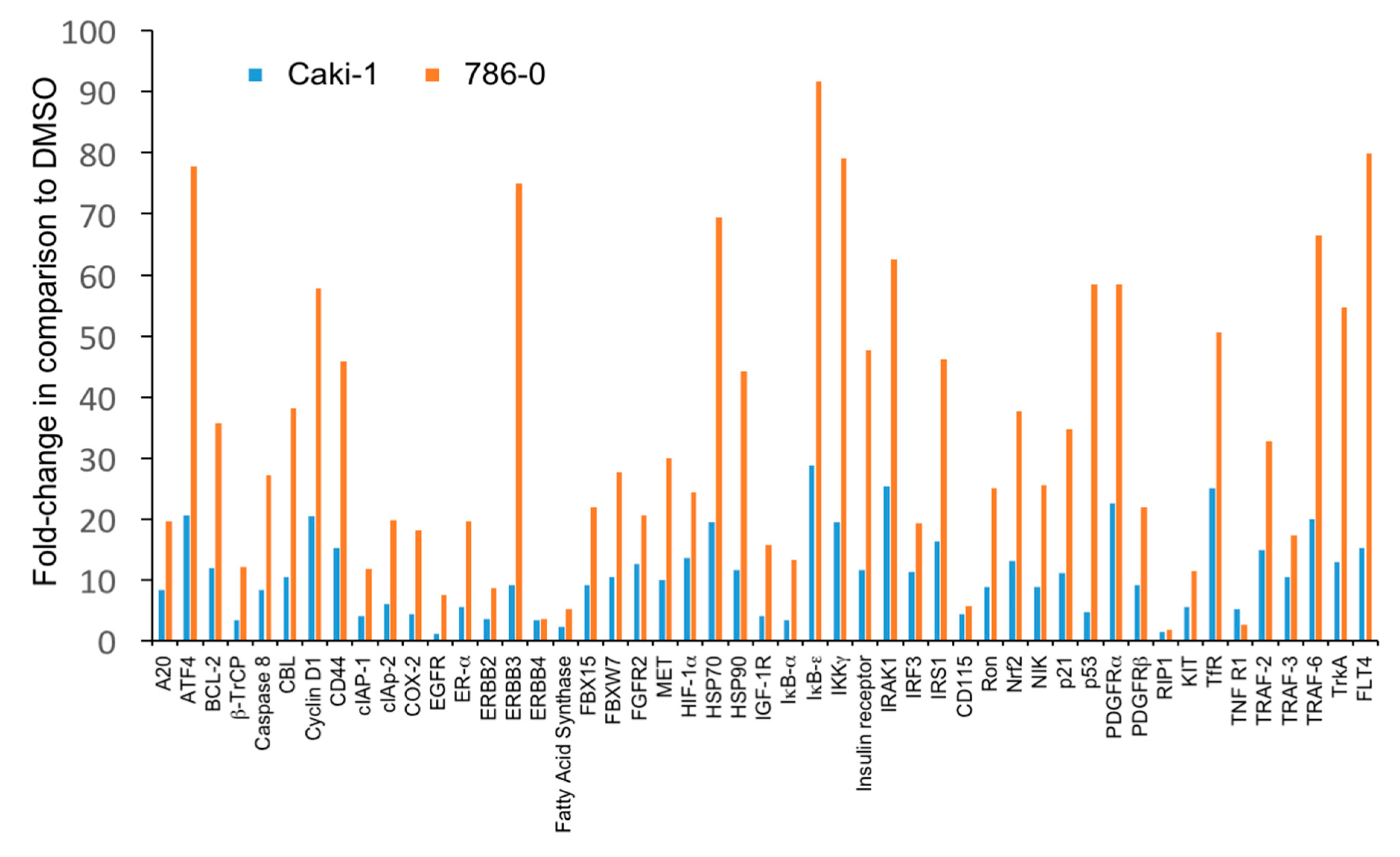
3.3. The Level of Accumulation of Ubiquitinated Proteins Correlates with the Antineoplastic Activity of Carfilzomib
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Choueiri, T.K.; Motzer, R.J. Systemic Therapy for Metastatic Renal-Cell Carcinoma. N. Engl. J. Med. 2017, 376, 354–366. [Google Scholar] [CrossRef]
- Huang, J.J.; Hsieh, J.J. The Therapeutic Landscape of Renal Cell Carcinoma: From the Dark Age to the Golden Age. Semin. Nephrol. 2020, 40, 28–41. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Tannir, N.M.; McDermott, D.F.; Frontera, O.A.; Melichar, B.; Choueiri, T.K.; Plimack, E.R.; Barthélémy, P.; Porta, C.; George, S.; et al. Nivolumab plus Ipilimumab versus Sunitinib in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2018, 378, 1277–1290. [Google Scholar] [CrossRef] [PubMed]
- Rini, B.I.; Plimack, E.R.; Stus, V.; Gafanov, R.; Hawkins, R.; Nosov, D.; Pouliot, F.; Alekseev, B.; Soulières, D.; Melichar, B. Pembrolizumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2019, 380, 1116–1127. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Penkov, K.; Haanen, J.; Rini, B.; Albiges, L.; Campbell, M.T.; Venugopal, B.; Kollmannsberger, C.; Negrier, S.; Uemura, M. Avelumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2019, 380, 1103–1115. [Google Scholar] [CrossRef] [PubMed]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; Tarpey, P.; et al. Intratumor Heterogeneity and Branched Evolution Revealed by Multiregion Sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef] [PubMed]
- Hoefflin, R.; Lahrmann, B.; Warsow, G.; Huebschmann, D.; Spath, C.; Walter, B.; Chen, X.; Hofer, L.; Macher-Goeppinger, S.; Tolstov, Y.; et al. Spatial niche formation but not malignant progression is a driving force for intratumoural heterogeneity. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef]
- Cahill, D.P.; Kinzler, K.W.; Vogelstein, B.; Lengauer, C. Genetic instability and darwinian selection in tumours. Trends Cell Biol. 1999, 9, M57–M60. [Google Scholar] [CrossRef]
- Dietz, S.; Sültmann, H.; Du, Y.; Reisinger, E.; Riediger, A.L.; Volckmar, A.-L.; Stenzinger, A.; Schlesner, M.; Jäger, D.; Hohenfellner, M.; et al. Patient-specific molecular alterations are associated with metastatic clear cell renal cell cancer progressing under tyrosine kinase inhibitor therapy. Oncotarget 2017, 8, 74049–74057. [Google Scholar] [CrossRef]
- Abbosh, C.; Birkbak, N.J.; Wilson, G.A.; Jamal-Hanjani, M.; Constantin, T.; Salari, R.; Le Quesne, J.; Moore, D.A.; Veeriah, S.; Rosenthal, R.; et al. Phylogenetic ctDNA analysis depicts early-stage lung cancer evolution. Nature 2017, 545, 446–451. [Google Scholar] [CrossRef]
- Turajlic, S.; Xu, H.; Litchfield, K.; Rowan, A.; Horswell, S.; Chambers, T.; O’Brien, T.; Lopez, J.I.; Watkins, T.B.; Nicol, D.; et al. Deterministic Evolutionary Trajectories Influence Primary Tumor Growth: TRACERx Renal. Cell 2018, 173, 595–610. [Google Scholar] [CrossRef]
- Navone, N.M.; Labanca, E. Modeling Cancer Metastasis. In Patient-Derived Xenograft Models of Human Cancer; Springer International Publishing: Cham, Germany, 2017; pp. 93–114. [Google Scholar]
- Gao, H.; Korn, J.M.; Ferretti, S.; Monahan, J.E.; Wang, Y.; Singh, M.; Zhang, C.; Schnell, C.; Yang, G.; Zhang, Y.; et al. High-throughput screening using patient-derived tumor xenografts to predict clinical trial drug response. Nat. Med. 2015, 21, 1318–1325. [Google Scholar] [CrossRef]
- Pauli, C.; Hopkins, B.D.; Prandi, D.; Shaw, R.; Fedrizzi, T.; Sboner, A.; Sailer, V.; Augello, M.; Puca, L.; Rosati, R.; et al. Personalized In Vitro and In Vivo Cancer Models to Guide Precision Medicine. Cancer Discov. 2017, 7, 462–477. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Ellis, M.J.; Li, S.; Larson, D.E.; Chen, K.; Wallis, J.W.; Harris, C.C.; McLellan, M.D.; Fulton, R.S.; Fulton, L.L.; et al. Genome remodelling in a basal-like breast cancer metastasis and xenograft. Nat. Cell Biol. 2010, 464, 999–1005. [Google Scholar] [CrossRef]
- Lin, D.; Wyatt, A.W.; Xue, H.; Wang, Y.; Dong, X.; Haegert, A.; Wu, R.; Brahmbhatt, S.; Mo, F.; Jong, L.; et al. High Fidelity Patient-Derived Xenografts for Accelerating Prostate Cancer Discovery and Drug Development. Cancer Res. 2014, 74, 1272–1283. [Google Scholar] [CrossRef]
- Wegner, C.S.; Hauge, A.; Andersen, L.M.K.; Huang, R.; Simonsen, T.G.; Gaustad, J.-V.; Rofstad, E.K. Increasing aggressiveness of patient-derived xenograft models of cervix carcinoma during serial transplantation. Oncotarget 2018, 9, 21036–21051. [Google Scholar] [CrossRef]
- Hasanov, E.; Tidwell, R.S.; Fernandez, P.; Park, L.; McMichael, C.; Tannir, N.M.; Jonasch, E. Phase II Study of Carfilzomib in Patients With Refractory Renal Cell Carcinoma. Clin. Genitourin. Cancer 2019, 17, 451–456. [Google Scholar] [CrossRef]
- Schneider, M.; Schüler, J.; Höfflin, R.; Korzeniewski, N.; Grüllich, C.; Roth, W.; Teber, D.; Hadaschik, B.; Pahernik, S.; Hohenfellner, M.; et al. Phenotypic drug screening and target validation for improved personalized therapy reveal the complexity of phenotype-genotype correlations in clear cell renal cell carcinoma1Present address: Department of Urology, University Hospital Frankfurt, Germany.2Equal contributions. Urol. Oncol. Semin. Orig. Investig. 2014, 32, 877–884. [Google Scholar] [CrossRef]
- Hughes, J.P.; Rees, S.; Kalindjian, S.B.; Philpott, K.L. Principles of early drug discovery. Br. J. Pharmacol. 2011, 162, 1239–1249. [Google Scholar] [CrossRef] [PubMed]
- Schneider, M.; Korzeniewski, N.; Merkle, K.; Schüler, J.; Grüllich, C.; Hadaschik, B.; Hohenfellner, M.; Duensing, S. The tyrosine kinase inhibitor nilotinib has antineoplastic activity in prostate cancer cells but up-regulates the ERK survival signal—Implications for targeted therapies1Equal contributions. Urol. Oncol. Semin. Orig. Investig. 2015, 33, 72.e1–72.e7. [Google Scholar] [CrossRef]
- Brodaczewska, K.K.; Szczylik, C.; Fiedorowicz, M.; Porta, C.; Czarnecka, A.M. Choosing the right cell line for renal cell cancer research. Mol. Cancer 2016, 15, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Larsson, P.; Engqvist, H.; Biermann, J.; Rönnerman, E.W.; Forssell-Aronsson, E.; Kovács, A.; Karlsson, P.; Helou, K.; Parris, T.Z. Optimization of cell viability assays to improve replicability and reproducibility of cancer drug sensitivity screens. Sci. Rep. 2020, 10, 1–12. [Google Scholar] [CrossRef]
- Heidegger, I.; Pircher, A.; Pichler, R. Targeting the Tumor Microenvironment in Renal Cell Cancer Biology and Therapy. Front. Oncol. 2019, 9, 490. [Google Scholar] [CrossRef] [PubMed]
- Dick, L.R.; Fleming, P.E. Building on bortezomib: Second-generation proteasome inhibitors as anti-cancer therapy. Drug Discov. Today 2010, 15, 243–249. [Google Scholar] [CrossRef]
- McBride, A.; Klaus, J.O.; Stockerl-Goldstein, K. Carfilzomib: A second-generation proteasome inhibitor for the treatment of multiple myeloma. Am. J. Health Pharm. 2015, 72, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.; German, P.; Bai, S.; Reddy, A.S.; Liu, X.-D.; Sun, M.; Zhou, L.; Chen, X.; Zhao, X.; Wu, C.; et al. Genetic and Pharmacological Strategies to Refunctionalize the von Hippel Lindau R167Q Mutant Protein. Cancer Res. 2014, 74, 3127–3136. [Google Scholar] [CrossRef] [PubMed]
- Choueiri, T.K.; Kaelin, W.G. Targeting the HIF2-VEGF axis in renal cell carcinoma. Nat. Med. 2020, 26, 1519–1530. [Google Scholar] [CrossRef]
- Ronnen, E.A.; Kondagunta, G.V.; Motzer, R.J. Medullary Renal Cell Carcinoma and Response to Therapy with Bortezomib. J. Clin. Oncol. 2006, 24, e14. [Google Scholar] [CrossRef]
- Kondagunta, G.V.; Drucker, B.J.; Schwartz, L.H.; Bacik, J.; Marion, S.; Russo, P.; Mazumdar, M.; Motzer, R.J. Phase II Trial of Bortezomib for Patients With Advanced Renal Cell Carcinoma. J. Clin. Oncol. 2004, 22, 3720–3725. [Google Scholar] [CrossRef] [PubMed]
- Davis, N.B.; Taber, D.A.; Ansari, R.H.; Ryan, C.W.; George, C.; Vokes, E.E.; Vogelzang, N.J.; Stadler, W.M. Phase II Trial of PS-341 in Patients With Renal Cell Cancer: A University of Chicago Phase II Consortium Study. J. Clin. Oncol. 2004, 22, 115–119. [Google Scholar] [CrossRef]
- Östman, A. The tumor microenvironment controls drug sensitivity. Nat. Med. 2012, 18, 1332–1334. [Google Scholar] [CrossRef] [PubMed]
- Berges, C.; Haberstock, H.; Fuchs, D.; Miltz, M.; Sadeghi, M.; Opelz, G.; Daniel, V.; Naujokat, C. Proteasome inhibition suppresses essential immune functions of human CD4+T cells. Immunology 2008, 124, 234–246. [Google Scholar] [CrossRef] [PubMed]
- Giraldo, N.; Becht, E.; Vano, Y.; Petitprez, F.; Lacroix, L.; Validire, P.; Sanchez-Salas, R.; Ingels, A.; Oudard, S.; Moatti, A.; et al. Tumor-Infiltrating and Peripheral Blood T-cell Immunophenotypes Predict Early Relapse in Localized Clear Cell Renal Cell Carcinoma. Clin. Cancer Res. 2017, 23, 4416–4428. [Google Scholar] [CrossRef] [PubMed]
- Cucchi, D.; Groen, R.; Janssen, J.; Cloos, J. Ex vivo cultures and drug testing of primary acute myeloid leukemia samples: Current techniques and implications for experimental design and outcome. Drug Resist. Updat. 2020, 53, 100730. [Google Scholar] [CrossRef]
- Lee, J.; Choi, A.; Cho, S.; Jun, Y.; Na, D.; Lee, A.; Jang, G.; Kwon, J.Y.; Kim, J.; Lee, S.; et al. Genome-scale CRISPR screening identifies cell cycle and protein ubiquitination processes as druggable targets for erlotinib-resistant lung cancer. Mol. Oncol. 2021, 15, 487–502. [Google Scholar] [CrossRef]
- Takahashi, K.; Inukai, T.; Imamura, T.; Yano, M.; Tomoyasu, C.; Lucas, D.M.; Nemoto, A.; Sato, H.; Huang, M.; Abe, M.; et al. Anti-leukemic activity of bortezomib and carfilzomib on B-cell precursor ALL cell lines. PLoS ONE 2017, 12, e0188680. [Google Scholar] [CrossRef] [PubMed]
- Ben-David, U.; Ha, G.; Tseng, Y.-Y.; Greenwald, N.F.; Oh, C.; Shih, J.; McFarland, J.M.; Wong, B.; Boehm, J.S.; Beroukhim, R.; et al. Patient-derived xenografts undergo mouse-specific tumor evolution. Nat. Genet. 2017, 49, 1567–1575. [Google Scholar] [CrossRef]
- Shi, J.; Li, Y.; Jia, R.; Fan, X. The fidelity of cancer cells in PDX models: Characteristics, mechanism and clinical significance. Int. J. Cancer 2020, 146, 2078–2088. [Google Scholar] [CrossRef]
- Gorospe, M.; Egan, J.M.; Zbar, B.; Lerman, M.; Geil, L.; Kuzmin, I.; Holbrook, N.J. Protective Function of von Hippel-Lindau Protein against Impaired Protein Processing in Renal Carcinoma Cells. Mol. Cell. Biol. 1999, 19, 1289–1300. [Google Scholar] [CrossRef] [PubMed][Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | Sex/Age at Diagnosis (Years) | RCC Histology | Tumor Stage | Grade | Time to Tumor Progression (Months) | Origin of Specimens Used for Functional Drug Screening | Top Drug Screen Hits (Fold Change in Cell Viability at a 1 μM Concentration Normalized to DMSO) |
|---|---|---|---|---|---|---|---|
| RCC001 | F/58 | clear cell | pT2a, cN0, cM0 | 2 | 37.6 | Abdominal wall metastasis/Intraabdominal metastasis | Carfilzomib (0.19) Dactinomycin (0.24) |
| RCC006 | M/48 | clear cell | pT3a, pN0 (0/6) pM1 (gall bladder) | 3 | n/a | Tumor nephrectomy | Dactinomycin (0.67) Erlotinib (0.68) Carfilzomib (0.72) |
| RCC008 | M/59 | clear cell | pT3, cN1, cM1 (OSS, PUL, BRA, pericardium) | 3–4 | n/a | Tumor nephrectomy | Dabrafenib (0.24) Bortezomib (0.25) Carfilzomib (0.28) |
| RCC014 | M/53 | sarcomatoid (major), clear cell (minor) | pT1a, cN0, cM0 | 4 | 7.7 | Local recurrence | Imatinib (0.62) Capecitabine (0.64) Vorinostat (0.64) |
| RCC016 | M/57 | clear cell | pT2, cN0, cM0 | 2 | 182.5 | Local recurrence/Intraabdominal metastases | Afatenib (0.94) |
| RCC024 | M/67 | clear cell | pT3a, cN1, pM1 (OSS, PUL) | 2 | n/a | Tumor nephrectomy | Decitabine (0.16) Erlotinib (0.17) Carfilzomib (0.23) Estramustine (0.24) |
| RCC025 | M/59 | clear cell | pT3c, cN0, cM0 | unknown | 14.3 | Peritoneal recurrence | Nilotinib (0.38) Ponatinib (0.43) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, J.; Pohl, L.; Schüler, J.; Korzeniewski, N.; Reimold, P.; Kaczorowski, A.; Hou, W.; Zschäbitz, S.; Nientiedt, C.; Jäger, D.; et al. Targeting the Proteasome in Advanced Renal Cell Carcinoma: Complexity and Limitations of Patient-Individualized Preclinical Drug Discovery. Biomedicines 2021, 9, 627. https://doi.org/10.3390/biomedicines9060627
Li J, Pohl L, Schüler J, Korzeniewski N, Reimold P, Kaczorowski A, Hou W, Zschäbitz S, Nientiedt C, Jäger D, et al. Targeting the Proteasome in Advanced Renal Cell Carcinoma: Complexity and Limitations of Patient-Individualized Preclinical Drug Discovery. Biomedicines. 2021; 9(6):627. https://doi.org/10.3390/biomedicines9060627
Chicago/Turabian StyleLi, Jielin, Laura Pohl, Julia Schüler, Nina Korzeniewski, Philipp Reimold, Adam Kaczorowski, Weibin Hou, Stefanie Zschäbitz, Cathleen Nientiedt, Dirk Jäger, and et al. 2021. "Targeting the Proteasome in Advanced Renal Cell Carcinoma: Complexity and Limitations of Patient-Individualized Preclinical Drug Discovery" Biomedicines 9, no. 6: 627. https://doi.org/10.3390/biomedicines9060627
APA StyleLi, J., Pohl, L., Schüler, J., Korzeniewski, N., Reimold, P., Kaczorowski, A., Hou, W., Zschäbitz, S., Nientiedt, C., Jäger, D., Hohenfellner, M., Duensing, A., & Duensing, S. (2021). Targeting the Proteasome in Advanced Renal Cell Carcinoma: Complexity and Limitations of Patient-Individualized Preclinical Drug Discovery. Biomedicines, 9(6), 627. https://doi.org/10.3390/biomedicines9060627