
Peptide-Based Nanoparticles for Therapeutic Nucleic Acid Delivery




Abstract
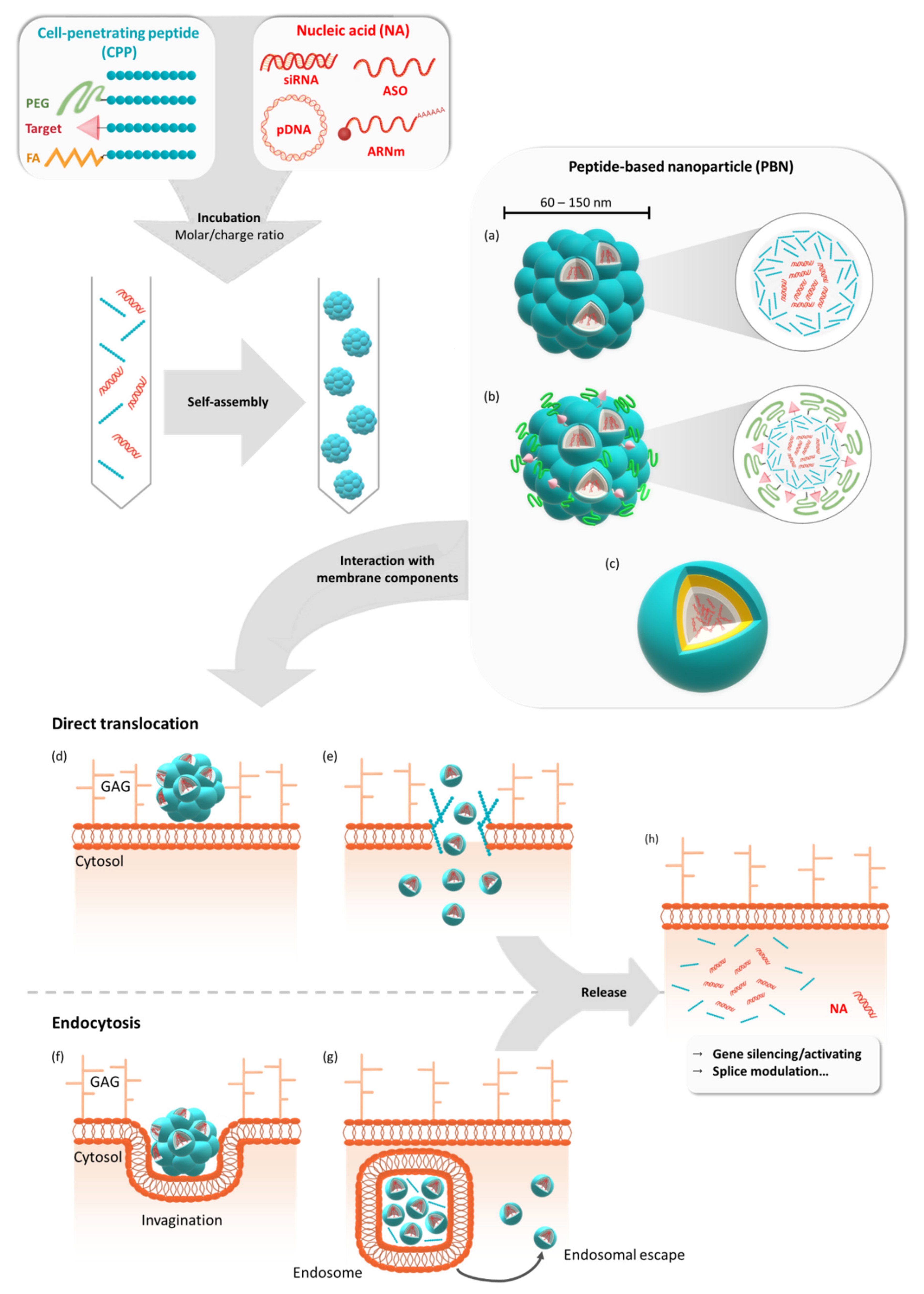
1. Introduction
2. Poly-Cationic Family
3. GALA/KALA/RALA Family
4. PepFect/NickFect Family
5. CADY-K/RICK Family
6. WRAP Family
7. C6 Family
8. Mgpe Family
9. Other CPPs Forming PBNs
9.1. MPG
9.2. CHAT
9.3. StA-TH
9.4. T9(dR)
9.5. p5RHH
9.6. BR2
9.7. S4(13)-PV
9.8. StA-SPA
9.9. KL4
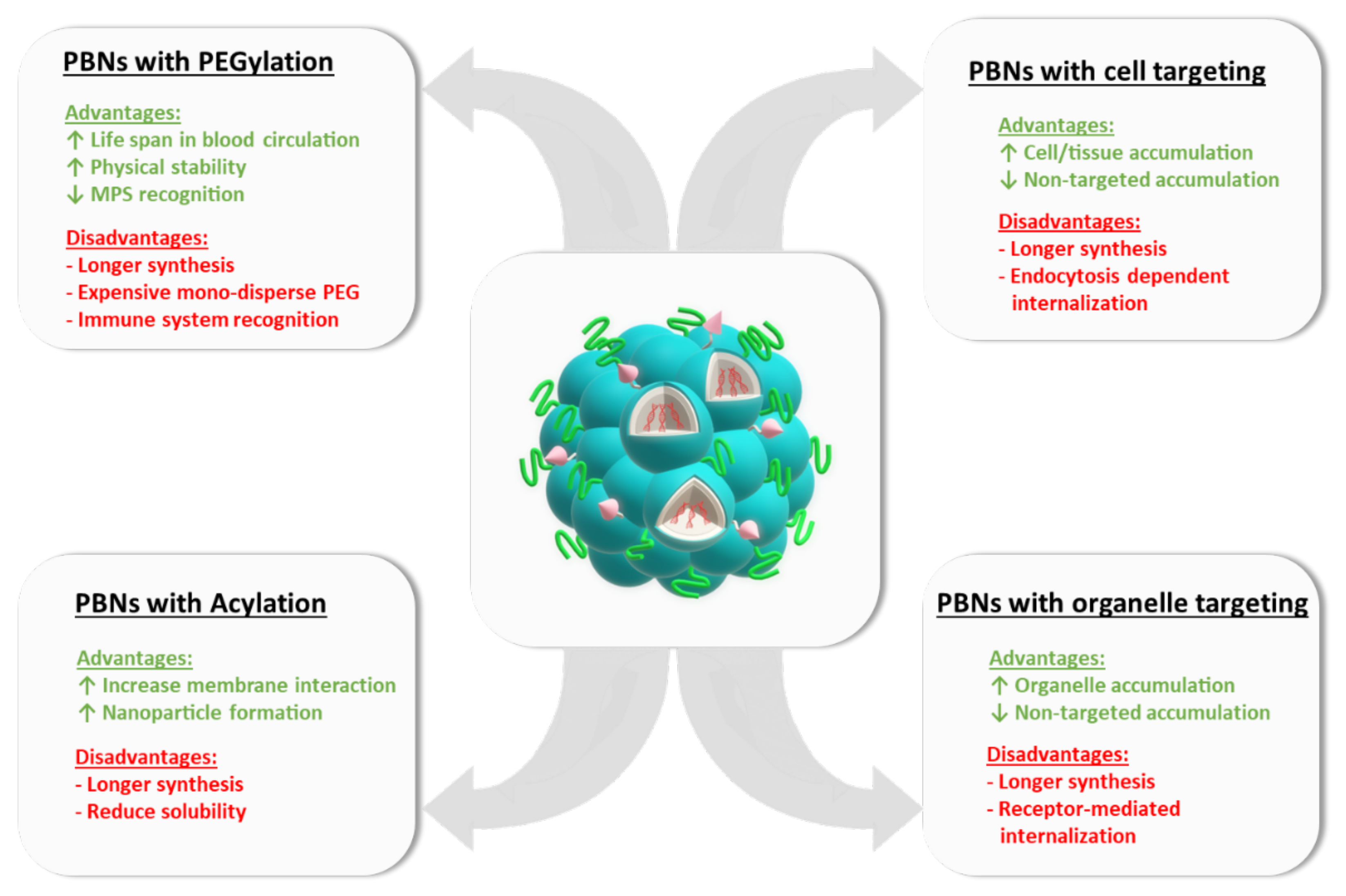
10. Functionalized PBNs
10.1. PEGylation
10.2. Cell/Organ Targeting
10.3. Organelle Targeting
11. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hoy, S.M. Patisiran: First Global Approval. Drugs 2018, 78, 1625–1631. [Google Scholar] [CrossRef] [PubMed]
- Scott, L.J. Givosiran: First Approval. Drugs 2020, 80, 335–339. [Google Scholar] [CrossRef] [PubMed]
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Pérez Marc, G.; Moreira, E.D.; Zerbini, C.; et al. Safety and Efficacy of the BNT162b2 MRNA Covid-19 Vaccine. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Ledford, H. Moderna COVID Vaccine Becomes Second to Get US Authorization. Nature 2020. [Google Scholar] [CrossRef]
- Crooke, S.T. Antisense Strategies. Curr. Mol. Med. 2004, 4, 465–487. [Google Scholar] [CrossRef]
- Tolia, N.H.; Joshua-Tor, L. Slicer and the Argonautes. Nat. Chem. Biol. 2007, 3, 36–43. [Google Scholar] [CrossRef]
- Whitehead, K.A.; Langer, R.; Anderson, D.G. Knocking down Barriers: Advances in SiRNA Delivery. Nat. Rev. Drug Discov. 2009, 8, 129–138. [Google Scholar] [CrossRef]
- Reschke, C.R.; Henshall, D.C. MicroRNA and Epilepsy. Adv. Exp. Med. Biol. 2015, 888, 41–70. [Google Scholar] [CrossRef]
- Gaspar, R.; Coelho, F.; Silva, B.F.B. Lipid-Nucleic Acid Complexes: Physicochemical Aspects and Prospects for Cancer Treatment. Molecules 2020, 25, 5006. [Google Scholar] [CrossRef]
- Ginn, S.L.; Amaya, A.K.; Alexander, I.E.; Edelstein, M.; Abedi, M.R. Gene Therapy Clinical Trials Worldwide to 2017: An Update. J. Gene Med. 2018, 20, e3015. [Google Scholar] [CrossRef]
- Pearson, S.; Jia, H.; Kandachi, K. China Approves First Gene Therapy. Nat. Biotechnol. 2004, 22, 3–4. [Google Scholar] [CrossRef]
- Waldrop, M.A.; Kolb, S.J. Current Treatment Options in Neurology—SMA Therapeutics. Curr. Treat. Options Neurol. 2019, 21, 25. [Google Scholar] [CrossRef]
- Langel, Ü. (Ed.) Cell-Penetrating Peptides; Methods in Molecular Biology; Springer New York: New York, NY, USA, 2015; Volume 1324, ISBN 978-1-4939-2805-7. [Google Scholar]
- Ramsey, J.D.; Flynn, N.H. Cell-Penetrating Peptides Transport Therapeutics into Cells. Pharmacol. Ther. 2015, 154, 78–86. [Google Scholar] [CrossRef]
- Kauffman, W.B.; Fuselier, T.; He, J.; Wimley, W.C. Mechanism Matters: A Taxonomy of Cell Penetrating Peptides. Trends Biochem. Sci. 2015, 40, 749–764. [Google Scholar] [CrossRef]
- Roberts, T.C.; Langer, R.; Wood, M.J.A. Advances in Oligonucleotide Drug Delivery. Nat. Rev. Drug Discov. 2020, 19, 673–694. [Google Scholar] [CrossRef]
- Boisguérin, P.; Deshayes, S.; Gait, M.J.; O’Donovan, L.; Godfrey, C.; Betts, C.A.; Wood, M.J.A.; Lebleu, B. Delivery of Therapeutic Oligonucleotides with Cell Penetrating Peptides. Adv. Drug Deliv. Rev. 2015, 87, 52–67. [Google Scholar] [CrossRef]
- Lehto, T.; Ezzat, K.; Wood, M.J.A.; El Andaloussi, S. Peptides for Nucleic Acid Delivery. Adv. Drug Deliv. Rev. 2016, 106, 172–182. [Google Scholar] [CrossRef]
- Kurrikoff, K.; Langel, Ü. Recent CPP-Based Applications in Medicine. Expert Opin. Drug Deliv. 2019, 16, 1183–1191. [Google Scholar] [CrossRef]
- Morris, M.C.; Vidal, P.; Chaloin, L.; Heitz, F.; Divita, G. A New Peptide Vector for Efficient Delivery of Oligonucleotides into Mammalian Cells. Nucleic Acids Res. 1997, 25, 2730–2736. [Google Scholar] [CrossRef]
- Sharma, R.; Shivpuri, S.; Anand, A.; Kulshreshtha, A.; Ganguli, M. Insight into the Role of Physicochemical Parameters in a Novel Series of Amphipathic Peptides for Efficient DNA Delivery. Mol. Pharm. 2013, 10, 2588–2600. [Google Scholar] [CrossRef]
- Konate, K.; Crombez, L.; Deshayes, S.; Decaffmeyer, M.; Thomas, A.; Brasseur, R.; Aldrian, G.; Heitz, F.; Divita, G. Insight into the Cellular Uptake Mechanism of a Secondary Amphipathic Cell-Penetrating Peptide for SiRNA Delivery. Biochemistry 2010, 49, 3393–3402. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; Nisakar, D.; Shivpuri, S.; Ganguli, M. Contrasting Effects of Cysteine Modification on the Transfection Efficiency of Amphipathic Peptides. Biomaterials 2014, 35, 6563–6575. [Google Scholar] [CrossRef] [PubMed]
- Andaloussi, S.E.L.; Lehto, T.; Mäger, I.; Rosenthal-Aizman, K.; Oprea, I.I.; Simonson, O.E.; Sork, H.; Ezzat, K.; Copolovici, D.M.; Kurrikoff, K.; et al. Design of a Peptide-Based Vector, PepFect6, for Efficient Delivery of SiRNA in Cell Culture and Systemically in Vivo. Nucleic Acids Res. 2011, 39, 3972–3987. [Google Scholar] [CrossRef] [PubMed]
- Vaissière, A.; Aldrian, G.; Konate, K.; Lindberg, M.F.; Jourdan, C.; Telmar, A.; Seisel, Q.; Fernandez, F.; Viguier, V.; Genevois, C.; et al. A Retro-Inverso Cell-Penetrating Peptide for SiRNA Delivery. J. Nanobiotechnology 2017, 15, 34. [Google Scholar] [CrossRef]
- Konate, K.; Dussot, M.; Aldrian, G.; Vaissière, A.; Viguier, V.; Neira, I.F.; Couillaud, F.; Vivès, E.; Boisguerin, P.; Deshayes, S. Peptide-Based Nanoparticles to Rapidly and Efficiently “Wrap ’n Roll” SiRNA into Cells. Bioconjug. Chem. 2019, 30, 592–603. [Google Scholar] [CrossRef]
- Kurrikoff, K.; Gestin, M.; Langel, Ü. Recent in Vivo Advances in Cell-Penetrating Peptide-Assisted Drug Delivery. Expert Opin. Drug Deliv. 2016, 13, 373–387. [Google Scholar] [CrossRef]
- Konate, K.; Lindberg, M.F.; Vaissiere, A.; Jourdan, C.; Aldrian, G.; Margeat, E.; Deshayes, S.; Boisguerin, P. Optimisation of Vectorisation Property: A Comparative Study for a Secondary Amphipathic Peptide. Int. J. Pharm. 2016, 509, 71–84. [Google Scholar] [CrossRef]
- Tarvirdipour, S.; Huang, X.; Mihali, V.; Schoenenberger, C.-A.; Palivan, C.G. Peptide-Based Nanoassemblies in Gene Therapy and Diagnosis: Paving the Way for Clinical Application. Molecules 2020, 25, 3482. [Google Scholar] [CrossRef]
- Desale, K.; Kuche, K.; Jain, S. Cell-Penetrating Peptides (CPPs): An Overview of Applications for Improving the Potential of Nanotherapeutics. Biomater. Sci. 2021, 9, 1153–1188. [Google Scholar] [CrossRef]
- Li, Y.; Li, Y.; Wang, X.; Lee, R.J.; Teng, L. Fatty Acid Modified Octa-Arginine for Delivery of SiRNA. Int. J. Pharm. 2015, 495, 527–535. [Google Scholar] [CrossRef]
- Alhakamy, N.A.; Ishiguro, S.; Uppalapati, D.; Berkland, C.J.; Tamura, M. AT2R Gene Delivered by Condensed Polylysine Complexes Attenuates Lewis Lung Carcinoma after Intravenous Injection or Intratracheal Spray. Mol. Cancer Ther. 2016, 15, 209–218. [Google Scholar] [CrossRef]
- Alhakamy, N.A.; Dhar, P.; Berkland, C.J. Charge Type, Charge Spacing, and Hydrophobicity of Arginine-Rich Cell-Penetrating Peptides Dictate Gene Transfection. Mol. Pharm. 2016, 13, 1047–1057. [Google Scholar] [CrossRef]
- McCarthy, H.O.; McCaffrey, J.; McCrudden, C.M.; Zholobenko, A.; Ali, A.A.; McBride, J.W.; Massey, A.S.; Pentlavalli, S.; Chen, K.-H.; Cole, G.; et al. Development and Characterization of Self-Assembling Nanoparticles Using a Bio-Inspired Amphipathic Peptide for Gene Delivery. J. Controlled Release 2014, 189, 141–149. [Google Scholar] [CrossRef]
- Bennett, R.; Yakkundi, A.; McKeen, H.D.; McClements, L.; McKeogh, T.J.; McCrudden, C.M.; Arthur, K.; Robson, T.; McCarthy, H.O. RALA-Mediated Delivery of FKBPL Nucleic Acid Therapeutics. Nanomedicine 2015, 10, 2989–3001. [Google Scholar] [CrossRef]
- Mulholland, E.J.; Ali, A.; Robson, T.; Dunne, N.J.; McCarthy, H.O. Delivery of RALA/SiFKBPL Nanoparticles via Electrospun Bilayer Nanofibres: An Innovative Angiogenic Therapy for Wound Repair. J. Controlled Release 2019, 316, 53–65. [Google Scholar] [CrossRef]
- Yan, L.-P.; Castaño, I.M.; Sridharan, R.; Kelly, D.; Lemoine, M.; Cavanagh, B.L.; Dunne, N.J.; McCarthy, H.O.; O’Brien, F.J. Collagen/GAG Scaffolds Activated by RALA-SiMMP-9 Complexes with Potential for Improved Diabetic Foot Ulcer Healing. Mater. Sci. Eng. C Mater. Biol. Appl. 2020, 114, 111022. [Google Scholar] [CrossRef]
- Udhayakumar, V.K.; De Beuckelaer, A.; McCaffrey, J.; McCrudden, C.M.; Kirschman, J.L.; Vanover, D.; Van Hoecke, L.; Roose, K.; Deswarte, K.; De Geest, B.G.; et al. Arginine-Rich Peptide-Based MRNA Nanocomplexes Efficiently Instigate Cytotoxic T Cell Immunity Dependent on the Amphipathic Organization of the Peptide. Adv. Healthc. Mater. 2017, 6. [Google Scholar] [CrossRef]
- Ezzat, K.; Andaloussi, S.E.L.; Zaghloul, E.M.; Lehto, T.; Lindberg, S.; Moreno, P.M.D.; Viola, J.R.; Magdy, T.; Abdo, R.; Guterstam, P.; et al. PepFect 14, a Novel Cell-Penetrating Peptide for Oligonucleotide Delivery in Solution and as Solid Formulation. Nucleic Acids Res. 2011, 39, 5284–5298. [Google Scholar] [CrossRef]
- Van den Brand, D.; Gorris, M.A.J.; van Asbeck, A.H.; Palmen, E.; Ebisch, I.; Dolstra, H.; Hällbrink, M.; Massuger, L.F.A.G.; Brock, R. Peptide-Mediated Delivery of Therapeutic MRNA in Ovarian Cancer. Eur. J. Pharm. Biopharm. 2019, 141, 180–190. [Google Scholar] [CrossRef]
- Gestin, M.; Helmfors, H.; Falato, L.; Lorenzon, N.; Michalakis, F.I.; Langel, Ü. Effect of Small Molecule Signaling in PepFect14 Transfection. PLoS ONE 2020, 15, e0228189. [Google Scholar] [CrossRef]
- Kurrikoff, K.; Veiman, K.-L.; Künnapuu, K.; Peets, E.M.; Lehto, T.; Pärnaste, L.; Arukuusk, P.; Langel, Ü. Effective in Vivo Gene Delivery with Reduced Toxicity, Achieved by Charge and Fatty Acid -Modified Cell Penetrating Peptide. Sci. Rep. 2017, 7, 17056. [Google Scholar] [CrossRef]
- Arukuusk, P.; Pärnaste, L.; Oskolkov, N.; Copolovici, D.-M.; Margus, H.; Padari, K.; Möll, K.; Maslovskaja, J.; Tegova, R.; Kivi, G.; et al. New Generation of Efficient Peptide-Based Vectors, NickFects, for the Delivery of Nucleic Acids. Biochim. Biophys. Acta BBA - Biomembr. 2013, 1828, 1365–1373. [Google Scholar] [CrossRef]
- Freimann, K.; Arukuusk, P.; Kurrikoff, K.; Vasconcelos, L.D.F.; Veiman, K.-L.; Uusna, J.; Margus, H.; Garcia-Sosa, A.T.; Pooga, M.; Langel, Ü. Optimization of in Vivo DNA Delivery with NickFect Peptide Vectors. J. Controlled Release 2016, 241, 135–143. [Google Scholar] [CrossRef]
- Freimann, K.; Arukuusk, P.; Kurrikoff, K.; Pärnaste, L.; Raid, R.; Piirsoo, A.; Pooga, M.; Langel, Ü. Formulation of Stable and Homogeneous Cell-Penetrating Peptide NF55 Nanoparticles for Efficient Gene Delivery In Vivo. Mol. Ther. Nucleic Acids 2018, 10, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Aldrian, G.; Vaissière, A.; Konate, K.; Seisel, Q.; Vivès, E.; Fernandez, F.; Viguier, V.; Genevois, C.; Couillaud, F.; Démèné, H.; et al. PEGylation Rate Influences Peptide-Based Nanoparticles Mediated SiRNA Delivery in Vitro and in Vivo. J. Controlled Release 2017, 256, 79–91. [Google Scholar] [CrossRef]
- Soussan, E.; Cassel, S.; Blanzat, M.; Rico-Lattes, I. Drug Delivery by Soft Matter: Matrix and Vesicular Carriers. Angew. Chem. Int. Ed Engl. 2009, 48, 274–288. [Google Scholar] [CrossRef] [PubMed]
- Jafari, M.; Xu, W.; Naahidi, S.; Chen, B.; Chen, P. A New Amphipathic, Amino-Acid-Pairing (AAP) Peptide as SiRNA Delivery Carrier: Physicochemical Characterization and in Vitro Uptake. J. Phys. Chem. B 2012, 116, 13183–13191. [Google Scholar] [CrossRef] [PubMed]
- Jafari, M.; Karunaratne, D.N.; Sweeting, C.M.; Chen, P. Modification of a Designed Amphipathic Cell-Penetrating Peptide and Its Effect on Solubility, Secondary Structure, and Uptake Efficiency. Biochemistry 2013, 52, 3428–3435. [Google Scholar] [CrossRef] [PubMed]
- Jafari, M.; Xu, W.; Pan, R.; Sweeting, C.M.; Karunaratne, D.N.; Chen, P. Serum Stability and Physicochemical Characterization of a Novel Amphipathic Peptide C6M1 for SiRNA Delivery. PLoS ONE 2014, 9, e97797. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Jafari, M.; Yuan, F.; Pan, R.; Chen, B.; Ding, Y.; Sheinin, T.; Chu, D.; Lu, S.; Yuan, Y.; et al. In Vitro and in Vivo Therapeutic SiRNA Delivery Induced by a Tryptophan-Rich Endosomolytic Peptide. J. Mater. Chem. B 2014, 2, 6010–6019. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Pan, R.; Zhao, D.; Chu, D.; Wu, Y.; Wang, R.; Chen, B.; Ding, Y.; Sadatmousavi, P.; Yuan, Y.; et al. Design and Evaluation of Endosomolytic Biocompatible Peptides as Carriers for SiRNA Delivery. Mol. Pharm. 2015, 12, 56–65. [Google Scholar] [CrossRef]
- Pan, R.; Xu, W.; Yuan, F.; Chu, D.; Ding, Y.; Chen, B.; Jafari, M.; Yuan, Y.; Chen, P. A Novel Peptide for Efficient SiRNA Delivery in Vitro and Therapeutics in Vivo. Acta Biomater. 2015, 21, 74–84. [Google Scholar] [CrossRef]
- Nisakar, D.; Vij, M.; Pandey, T.; Natarajan, P.; Sharma, R.; Mishra, S.; Ganguli, M. Deciphering the Role of Chondroitin Sulfate in Increasing the Transfection Efficiency of Amphipathic Peptide-Based Nanocomplexes. ACS Biomater. Sci. Eng. 2019, 5, 45–55. [Google Scholar] [CrossRef]
- Subia, B.; Reinisalo, M.; Dey, N.; Tavakoli, S.; Subrizi, A.; Ganguli, M.; Ruponen, M. Nucleic Acid Delivery to Differentiated Retinal Pigment Epithelial Cells Using Cell-Penetrating Peptide as a Carrier. Eur. J. Pharm. Biopharm. 2019, 140, 91–99. [Google Scholar] [CrossRef]
- McErlean, E.M.; Ziminska, M.; McCrudden, C.M.; McBride, J.W.; Loughran, S.P.; Cole, G.; Mulholland, E.J.; Kett, V.; Buckley, N.E.; Robson, T.; et al. Rational Design and Characterisation of a Linear Cell Penetrating Peptide for Non-Viral Gene Delivery. J. Controlled Release 2020. [Google Scholar] [CrossRef]
- Ji, K.; Xiao, Y.; Zhang, W. Acid-Activated Nonviral Peptide Vector for Gene Delivery. J. Pept. Sci. Off. Publ. Eur. Pept. Soc. 2020, 26, e3230. [Google Scholar] [CrossRef]
- Zhang, C.; Ren, W.; Liu, Q.; Tan, Z.; Li, J.; Tong, C. Transportan-Derived Cell-Penetrating Peptide Delivers SiRNA to Inhibit Replication of Influenza Virus in Vivo. Drug Des. Devel. Ther. 2019, 13, 1059–1068. [Google Scholar] [CrossRef]
- Yan, H.; Duan, X.; Pan, H.; Holguin, N.; Rai, M.F.; Akk, A.; Springer, L.E.; Wickline, S.A.; Sandell, L.J.; Pham, C.T.N. Suppression of NF-ΚB Activity via Nanoparticle-Based SiRNA Delivery Alters Early Cartilage Responses to Injury. Proc. Natl. Acad. Sci. USA 2016, 113, E6199–E6208. [Google Scholar] [CrossRef]
- Yan, H.; Duan, X.; Pan, H.; Akk, A.; Sandell, L.J.; Wickline, S.A.; Rai, M.F.; Pham, C.T.N. Development of a Peptide-SiRNA Nanocomplex Targeting NF- ΚB for Efficient Cartilage Delivery. Sci. Rep. 2019, 9, 442. [Google Scholar] [CrossRef]
- Mills, K.A.; Quinn, J.M.; Roach, S.T.; Palisoul, M.; Nguyen, M.; Noia, H.; Guo, L.; Fazal, J.; Mutch, D.G.; Wickline, S.A.; et al. P5RHH Nanoparticle-Mediated Delivery of AXL SiRNA Inhibits Metastasis of Ovarian and Uterine Cancer Cells in Mouse Xenografts. Sci. Rep. 2019, 9, 4762. [Google Scholar] [CrossRef]
- Lockhart, J.H.; VanWye, J.; Banerjee, R.; Wickline, S.A.; Pan, H.; Totary-Jain, H. Self-Assembled MiRNA-Switch Nanoparticles Target Denuded Regions and Prevent Restenosis. Mol. Ther. J. Am. Soc. Gene Ther. 2021. [Google Scholar] [CrossRef]
- Lim, K.J.; Sung, B.H.; Shin, J.R.; Lee, Y.W.; Kim, D.J.; Yang, K.S.; Kim, S.C. A Cancer Specific Cell-Penetrating Peptide, BR2, for the Efficient Delivery of an ScFv into Cancer Cells. PLoS ONE 2013, 8, e66084. [Google Scholar] [CrossRef]
- Morais, C.M.; Cardoso, A.M.; Cunha, P.P.; Aguiar, L.; Vale, N.; Lage, E.; Pinheiro, M.; Nunes, C.; Gomes, P.; Reis, S.; et al. Acylation of the S413-PV Cell-Penetrating Peptide as a Means of Enhancing Its Capacity to Mediate Nucleic Acid Delivery: Relevance of Peptide/Lipid Interactions. Biochim. Biophys. Acta Biomembr. 2018, 1860, 2619–2634. [Google Scholar] [CrossRef]
- Morais, C.M.; Cardoso, A.M.; Aguiar, L.; Vale, N.; Nóbrega, C.; Zuzarte, M.; Gomes, P.; Pedroso de Lima, M.C.; Jurado, A.S. Lauroylated Histidine-Enriched S413-PV Peptide as an Efficient Gene Silencing Mediator in Cancer Cells. Pharm. Res. 2020, 37, 188. [Google Scholar] [CrossRef]
- Song, J.; Huang, S.; Zhang, Z.; Jia, B.; Xie, H.; Kai, M.; Zhang, W. SPA: A Peptide Antagonist That Acts as a Cell-Penetrating Peptide for Drug Delivery. Drug Deliv. 2019, 27, 91–99. [Google Scholar] [CrossRef]
- Qiu, Y.; Chow, M.Y.T.; Liang, W.; Chung, W.W.Y.; Mak, J.C.W.; Lam, J.K.W. From Pulmonary Surfactant, Synthetic KL4 Peptide as Effective SiRNA Delivery Vector for Pulmonary Delivery. Mol. Pharm. 2017, 14, 4606–4617. [Google Scholar] [CrossRef]
- Nakase, I.; Akita, H.; Kogure, K.; Gräslund, A.; Langel, U.; Harashima, H.; Futaki, S. Efficient Intracellular Delivery of Nucleic Acid Pharmaceuticals Using Cell-Penetrating Peptides. Acc. Chem. Res. 2012, 45, 1132–1139. [Google Scholar] [CrossRef]
- Futaki, S.; Ohashi, W.; Suzuki, T.; Niwa, M.; Tanaka, S.; Ueda, K.; Harashima, H.; Sugiura, Y. Stearylated Arginine-Rich Peptides: A New Class of Transfection Systems. Bioconjug. Chem. 2001, 12, 1005–1011. [Google Scholar] [CrossRef]
- Alizadeh, S.; Irani, S.; Bolhassani, A.; Sadat, S.M. HR9: An Important Cell Penetrating Peptide for Delivery of HCV NS3 DNA into HEK-293T Cells. Avicenna J. Med. Biotechnol. 2020, 12, 44–51. [Google Scholar]
- Torchilin, V.P. Cell Penetrating Peptide-Modified Pharmaceutical Nanocarriers for Intracellular Drug and Gene Delivery. Biopolymers 2008, 90, 604–610. [Google Scholar] [CrossRef]
- Jallouk, A.P.; Palekar, R.U.; Pan, H.; Schlesinger, P.H.; Wickline, S.A. Modifications of Natural Peptides for Nanoparticle and Drug Design. Adv. Protein Chem. Struct. Biol. 2015, 98, 57–91. [Google Scholar] [CrossRef] [PubMed]
- Váňová, J.; Hejtmánková, A.; Kalbáčová, M.H.; Španielová, H. The Utilization of Cell-Penetrating Peptides in the Intracellular Delivery of Viral Nanoparticles. Materials 2019, 12, 2671. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Nicol, F.; Szoka, F.C. GALA: A Designed Synthetic PH-Responsive Amphipathic Peptide with Applications in Drug and Gene Delivery. Adv. Drug Deliv. Rev. 2004, 56, 967–985. [Google Scholar] [CrossRef] [PubMed]
- Subbarao, N.K.; Fielding, C.J.; Hamilton, R.L.; Szoka, F.C. Lecithin:Cholesterol Acyltransferase Activation by Synthetic Amphipathic Peptides. Proteins 1988, 3, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Plank, C.; Oberhauser, B.; Mechtler, K.; Koch, C.; Wagner, E. The Influence of Endosome-Disruptive Peptides on Gene Transfer Using Synthetic Virus-like Gene Transfer Systems. J. Biol. Chem. 1994, 269, 12918–12924. [Google Scholar] [CrossRef]
- Wyman, T.B.; Nicol, F.; Zelphati, O.; Scaria, P.V.; Plank, C.; Szoka, F.C. Design, Synthesis, and Characterization of a Cationic Peptide That Binds to Nucleic Acids and Permeabilizes Bilayers. Biochemistry 1997, 36, 3008–3017. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Jeong, J.H.; Park, T.G. PEG Grafted Polylysine with Fusogenic Peptide for Gene Delivery: High Transfection Efficiency with Low Cytotoxicity. J. Controlled Release 2002, 79, 283–291. [Google Scholar] [CrossRef]
- Lee, H.; Jeong, J.H.; Park, T.G. A New Gene Delivery Formulation of Polyethylenimine/DNA Complexes Coated with PEG Conjugated Fusogenic Peptide. J. Controlled Release 2001, 76, 183–192. [Google Scholar] [CrossRef]
- Lim, D.W.; Yeom, Y.I.; Park, T.G. Poly(DMAEMA-NVP)-b-PEG-Galactose as Gene Delivery Vector for Hepatocytes. Bioconjug. Chem. 2000, 11, 688–695. [Google Scholar] [CrossRef]
- Katayama, T.; Kinugawa, S.; Takada, S.; Furihata, T.; Fukushima, A.; Yokota, T.; Anzai, T.; Hibino, M.; Harashima, H.; Yamada, Y. A Mitochondrial Delivery System Using Liposome-Based Nanocarriers That Target Myoblast Cells. Mitochondrion 2019, 49, 66–72. [Google Scholar] [CrossRef]
- McErlean, E.M.; McCrudden, C.M.; McBride, J.W.; Cole, G.; Kett, V.L.; Robson, T.; Dunne, N.J.; McCarthy, H.O. Rational Design and Characterisation of an Amphipathic Cell Penetrating Peptide for Non-Viral Gene Delivery. Int. J. Pharm. 2021, 596, 120223. [Google Scholar] [CrossRef]
- McCrudden, C.M.; McBride, J.W.; McCaffrey, J.; McErlean, E.M.; Dunne, N.J.; Kett, V.L.; Coulter, J.A.; Robson, T.; McCarthy, H.O. Gene Therapy with RALA/INOS Composite Nanoparticles Significantly Enhances Survival in a Model of Metastatic Prostate Cancer. Cancer Nanotechnol. 2018, 9, 5. [Google Scholar] [CrossRef]
- Sousa, Â.; Almeida, A.M.; Faria, R.; Konate, K.; Boisguerin, P.; Queiroz, J.A.; Costa, D. Optimization of Peptide-Plasmid DNA Vectors Formulation for Gene Delivery in Cancer Therapy Exploring Design of Experiments. Colloids Surf. B Biointerfaces 2019, 183, 110417. [Google Scholar] [CrossRef]
- Cole, G.; Ali, A.A.; McErlean, E.; Mulholland, E.J.; Short, A.; McCrudden, C.M.; McCaffrey, J.; Robson, T.; Kett, V.L.; Coulter, J.A.; et al. DNA Vaccination via RALA Nanoparticles in a Microneedle Delivery System Induces a Potent Immune Response against the Endogenous Prostate Cancer Stem Cell Antigen. Acta Biomater. 2019, 96, 480–490. [Google Scholar] [CrossRef]
- Mäe, M.; El Andaloussi, S.; Lundin, P.; Oskolkov, N.; Johansson, H.J.; Guterstam, P.; Langel, U. A Stearylated CPP for Delivery of Splice Correcting Oligonucleotides Using a Non-Covalent Co-Incubation Strategy. J. Controlled Release 2009, 134, 221–227. [Google Scholar] [CrossRef]
- Ezzat, K.; Helmfors, H.; Tudoran, O.; Juks, C.; Lindberg, S.; Padari, K.; El-Andaloussi, S.; Pooga, M.; Langel, Ü. Scavenger Receptor-Mediated Uptake of Cell-Penetrating Peptide Nanocomplexes with Oligonucleotides. FASEB J. 2012, 26, 1172–1180. [Google Scholar] [CrossRef]
- Lindberg, S.; Regberg, J.; Eriksson, J.; Helmfors, H.; Muñoz-Alarcón, A.; Srimanee, A.; Figueroa, R.A.; Hallberg, E.; Ezzat, K.; Langel, Ü. A Convergent Uptake Route for Peptide- and Polymer-Based Nucleotide Delivery Systems. J. Controlled Release 2015, 206, 58–66. [Google Scholar] [CrossRef]
- Kurrikoff, K.; Freimann, K.; Veiman, K.-L.; Peets, E.M.; Piirsoo, A.; Langel, Ü. Effective Lung-Targeted RNAi in Mice with Peptide-Based Delivery of Nucleic Acid. Sci. Rep. 2019, 9, 19926. [Google Scholar] [CrossRef]
- Brugidou, J.; Legrand, C.; Méry, J.; Rabié, A. The Retro-Inverso Form of a Homeobox-Derived Short Peptide Is Rapidly Internalised by Cultured Neurones: A New Basis for an Efficient Intracellular Delivery System. Biochem. Biophys. Res. Commun. 1995, 214, 685–693. [Google Scholar] [CrossRef]
- Aldrian-Herrada, G.; Desarménien, M.G.; Orcel, H.; Boissin-Agasse, L.; Méry, J.; Brugidou, J.; Rabié, A. A Peptide Nucleic Acid (PNA) Is More Rapidly Internalized in Cultured Neurons When Coupled to a Retro-Inverso Delivery Peptide. The Antisense Activity Depresses the Target MRNA and Protein in Magnocellular Oxytocin Neurons. Nucleic Acids Res. 1998, 26, 4910–4916. [Google Scholar] [CrossRef]
- Tünnemann, G.; Martin, R.M.; Haupt, S.; Patsch, C.; Edenhofer, F.; Cardoso, M.C. Cargo-Dependent Mode of Uptake and Bioavailability of TAT-Containing Proteins and Peptides in Living Cells. FASEB J. 2006, 20, 1775–1784. [Google Scholar] [CrossRef]
- Chen, Z.; Nie, D.; Hu, Y.; Li, M.; Hou, Z.; Mao, X.; Luo, X.; Xue, X. Efficient Delivery of Antisense Oligonucleotides by an Amphipathic Cell-Penetrating Peptide in Acinetobacter Baumannii. Curr. Drug Deliv. 2019, 16, 728–736. [Google Scholar] [CrossRef]
- Bechara, C.; Pallerla, M.; Burlina, F.; Illien, F.; Cribier, S.; Sagan, S. Massive Glycosaminoglycan-Dependent Entry of Trp-Containing Cell-Penetrating Peptides Induced by Exogenous Sphingomyelinase or Cholesterol Depletion. Cell. Mol. Life Sci. CMLS 2015, 72, 809–820. [Google Scholar] [CrossRef] [PubMed]
- Jobin, M.-L.; Blanchet, M.; Henry, S.; Chaignepain, S.; Manigand, C.; Castano, S.; Lecomte, S.; Burlina, F.; Sagan, S.; Alves, I.D. The Role of Tryptophans on the Cellular Uptake and Membrane Interaction of Arginine-Rich Cell Penetrating Peptides. Biochim. Biophys. Acta 2015, 1848, 593–602. [Google Scholar] [CrossRef]
- Walrant, A.; Bauzá, A.; Girardet, C.; Alves, I.D.; Lecomte, S.; Illien, F.; Cardon, S.; Chaianantakul, N.; Pallerla, M.; Burlina, F.; et al. Ionpair-π Interactions Favor Cell Penetration of Arginine/Tryptophan-Rich Cell-Penetrating Peptides. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183098. [Google Scholar] [CrossRef]
- Deshayes, S.; Konate, K.; Dussot, M.; Chavey, B.; Vaissière, A.; Van, T.N.N.; Aldrian, G.; Padari, K.; Pooga, M.; Vivès, E.; et al. Deciphering the Internalization Mechanism of WRAP:SiRNA Nanoparticles. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183252. [Google Scholar] [CrossRef]
- Steinhauer, D.A.; Wharton, S.A.; Skehel, J.J.; Wiley, D.C. Studies of the Membrane Fusion Activities of Fusion Peptide Mutants of Influenza Virus Hemagglutinin. J. Virol. 1995, 69, 6643–6651. [Google Scholar] [CrossRef]
- Chen, B.; Yoo, K.; Xu, W.; Pan, R.; Han, X.X.; Chen, P. Characterization and Evaluation of a Peptide-Based SiRNA Delivery System in Vitro. Drug Deliv. Transl. Res. 2017, 7, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Rajpal; Mann, A.; Khanduri, R.; Naik, R.J.; Ganguli, M. Structural Rearrangements and Chemical Modifications in Known Cell Penetrating Peptide Strongly Enhance DNA Delivery Efficiency. J. Controlled Release 2012, 157, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Sims, L.B.; Curry, K.C.; Parupalli, S.; Horner, G.; Frieboes, H.B.; Steinbach-Rankins, J.M. Efficacy of Surface-Modified PLGA Nanoparticles as a Function of Cervical Cancer Type. Pharm. Res. 2019, 36, 66. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Liu, J.; Liu, D.; Han, Y.; Xu, H.; Liu, L.; Leng, X.; Kong, D. A Cell-Penetrating Peptide-Assisted Nanovaccine Promotes Antigen Cross-Presentation and Anti-Tumor Immune Response. Biomater. Sci. 2019, 7, 5516–5527. [Google Scholar] [CrossRef] [PubMed]
- Mehrlatifan, S.; Mirnurollahi, S.M.; Motevalli, F.; Rahimi, P.; Soleymani, S.; Bolhassani, A. The Structural HCV Genes Delivered by MPG Cell Penetrating Peptide Are Directed to Enhance Immune Responses in Mice Model. Drug Deliv. 2016, 23, 2852–2859. [Google Scholar] [CrossRef] [PubMed]
- Rostami, B.; Irani, S.; Bolhassani, A.; Cohan, R.A. Gene and Protein Delivery Using Four Cell Penetrating Peptides for HIV-1 Vaccine Development. IUBMB Life 2019, 71, 1619–1633. [Google Scholar] [CrossRef]
- Hou, K.K.; Pan, H.; Lanza, G.M.; Wickline, S.A. Melittin Derived Peptides for Nanoparticle Based SiRNA Transfection. Biomaterials 2013, 34, 3110–3119. [Google Scholar] [CrossRef] [PubMed]
- Hariton-Gazal, E.; Feder, R.; Mor, A.; Graessmann, A.; Brack-Werner, R.; Jans, D.; Gilon, C.; Loyter, A. Targeting of Nonkaryophilic Cell-Permeable Peptides into the Nuclei of Intact Cells by Covalently Attached Nuclear Localization Signals. Biochemistry 2002, 41, 9208–9214. [Google Scholar] [CrossRef] [PubMed]
- Trabulo, S.; Cardoso, A.L.; Mano, M.; de Lima, M.C.P. Cell-Penetrating Peptides—Mechanisms of Cellular Uptake and Generation of Delivery Systems. Pharmaceuticals 2010, 3, 961–993. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Kai, M.; Zhang, W.; Zhang, J.; Liu, L.; Zhang, B.; Liu, X.; Wang, R. Cellular Uptake of Transportan 10 and Its Analogs in Live Cells: Selectivity and Structure-Activity Relationship Studies. Peptides 2011, 32, 1934–1941. [Google Scholar] [CrossRef]
- Cochrane, C.G.; Revak, S.D.; Merritt, T.A.; Heldt, G.P.; Hallman, M.; Cunningham, M.D.; Easa, D.; Pramanik, A.; Edwards, D.K.; Alberts, M.S. The Efficacy and Safety of KL4-Surfactant in Preterm Infants with Respiratory Distress Syndrome. Am. J. Respir. Crit. Care Med. 1996, 153, 404–410. [Google Scholar] [CrossRef]
- Qiu, Y.; Lo, J.C.K.; Kwok, K.C.W.; Mason, A.J.; Lam, J.K.W. Modification of KL4 Peptide Revealed the Importance of Alpha-Helical Structure for Efficient Small Interfering RNA Delivery. Nucleic Acid Ther. 2020. [Google Scholar] [CrossRef]
- Gulati, N.M.; Stewart, P.L.; Steinmetz, N.F. Bioinspired Shielding Strategies for Nanoparticle Drug Delivery Applications. Mol. Pharm. 2018, 15, 2900–2909. [Google Scholar] [CrossRef]
- Lee, H. Molecular Simulations of PEGylated Biomolecules, Liposomes, and Nanoparticles for Drug Delivery Applications. Pharmaceutics 2020, 12, 533. [Google Scholar] [CrossRef] [PubMed]
- Buschmann, M.D.; Carrasco, M.J.; Alishetty, S.; Paige, M.; Alameh, M.G.; Weissman, D. Nanomaterial Delivery Systems for MRNA Vaccines. Vaccines 2021, 9, 65. [Google Scholar] [CrossRef] [PubMed]
- Osman, G.; Rodriguez, J.; Chan, S.Y.; Chisholm, J.; Duncan, G.; Kim, N.; Tatler, A.L.; Shakesheff, K.M.; Hanes, J.; Suk, J.S.; et al. PEGylated Enhanced Cell Penetrating Peptide Nanoparticles for Lung Gene Therapy. J. Controlled Release 2018, 285, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Man, R.C.H.; Liao, Q.; Kung, K.L.K.; Chow, M.Y.T.; Lam, J.K.W. Effective MRNA Pulmonary Delivery by Dry Powder Formulation of PEGylated Synthetic KL4 Peptide. J. Controlled Release 2019, 314, 102–115. [Google Scholar] [CrossRef]
- Leucuta, S.E. Systemic and Biophase Bioavailability and Pharmacokinetics of Nanoparticulate Drug Delivery Systems. Curr. Drug Deliv. 2013, 10, 208–240. [Google Scholar] [CrossRef]
- Ren, Y.; Cheung, H.W.; von Maltzhan, G.; Agrawal, A.; Cowley, G.S.; Weir, B.A.; Boehm, J.S.; Tamayo, P.; Karst, A.M.; Liu, J.F.; et al. Targeted Tumor-Penetrating SiRNA Nanocomplexes for Credentialing the Ovarian Cancer Oncogene ID4. Sci. Transl. Med. 2012, 4, 147ra112. [Google Scholar] [CrossRef]
- Ren, Y.; Hauert, S.; Lo, J.H.; Bhatia, S.N. Identification and Characterization of Receptor-Specific Peptides for SiRNA Delivery. ACS Nano 2012, 6, 8620–8631. [Google Scholar] [CrossRef]
- Lo, J.H.; Hao, L.; Muzumdar, M.D.; Raghavan, S.; Kwon, E.J.; Pulver, E.M.; Hsu, F.; Aguirre, A.J.; Wolpin, B.M.; Fuchs, C.S.; et al. IRGD-Guided Tumor-Penetrating Nanocomplexes for Therapeutic SiRNA Delivery to Pancreatic Cancer. Mol. Cancer Ther. 2018, 17, 2377–2388. [Google Scholar] [CrossRef]
- Jain, P.K.; Lo, J.H.; Rananaware, S.; Downing, M.; Panda, A.; Tai, M.; Raghavan, S.; Fleming, H.E.; Bhatia, S.N. Non-Viral Delivery of CRISPR/Cas9 Complex Using CRISPR-GPS Nanocomplexes. Nanoscale 2019, 11, 21317–21323. [Google Scholar] [CrossRef]
- Cerrato, C.P.; Künnapuu, K.; Langel, Ü. Cell-Penetrating Peptides with Intracellular Organelle Targeting. Expert Opin. Drug Deliv. 2017, 14, 245–255. [Google Scholar] [CrossRef]
- Wang, H.-Y.; Chen, J.-X.; Sun, Y.-X.; Deng, J.-Z.; Li, C.; Zhang, X.-Z.; Zhuo, R.-X. Construction of Cell Penetrating Peptide Vectors with N-Terminal Stearylated Nuclear Localization Signal for Targeted Delivery of DNA into the Cell Nuclei. J. Controlled Release 2011, 155, 26–33. [Google Scholar] [CrossRef]
- Yameen, B.; Choi, W.I.; Vilos, C.; Swami, A.; Shi, J.; Farokhzad, O.C. Insight into Nanoparticle Cellular Uptake and Intracellular Targeting. J. Controlled Release 2014, 190, 485–499. [Google Scholar] [CrossRef]
- Bonifacino, J.S.; Dell’Angelica, E.C. Molecular Bases for the Recognition of Tyrosine-Based Sorting Signals. J. Cell Biol. 1999, 145, 923–926. [Google Scholar] [CrossRef]
- Dekiwadia, C.D.; Lawrie, A.C.; Fecondo, J.V. Peptide-Mediated Cell Penetration and Targeted Delivery of Gold Nanoparticles into Lysosomes. J. Pept. Sci. 2012, 18, 527–534. [Google Scholar] [CrossRef]
- Horton, K.L.; Stewart, K.M.; Fonseca, S.B.; Guo, Q.; Kelley, S.O. Mitochondria-Penetrating Peptides. Chem. Biol. 2008, 15, 375–382. [Google Scholar] [CrossRef]
- Kelley, S.O.; Stewart, K.M.; Mourtada, R. Development of Novel Peptides for Mitochondrial Drug Delivery: Amino Acids Featuring Delocalized Lipophilic Cations. Pharm. Res. 2011, 28, 2808–2819. [Google Scholar] [CrossRef]
- Yousif, L.F.; Stewart, K.M.; Horton, K.L.; Kelley, S.O. Mitochondria-Penetrating Peptides: Sequence Effects and Model Cargo Transport. Chembiochem Eur. J. Chem. Biol. 2009, 10, 2081–2088. [Google Scholar] [CrossRef]
- Zhao, K.; Luo, G.; Zhao, G.-M.; Schiller, P.W.; Szeto, H.H. Transcellular Transport of a Highly Polar 3+ Net Charge Opioid Tetrapeptide. J. Pharmacol. Exp. Ther. 2003, 304, 425–432. [Google Scholar] [CrossRef]
- Zhao, K.; Zhao, G.-M.; Wu, D.; Soong, Y.; Birk, A.V.; Schiller, P.W.; Szeto, H.H. Cell-Permeable Peptide Antioxidants Targeted to Inner Mitochondrial Membrane Inhibit Mitochondrial Swelling, Oxidative Cell Death, and Reperfusion Injury. J. Biol. Chem. 2004, 279, 34682–34690. [Google Scholar] [CrossRef]
- Cerrato, C.P.; Pirisinu, M.; Vlachos, E.N.; Langel, Ü. Novel Cell-Penetrating Peptide Targeting Mitochondria. FASEB J. 2015, 29, 4589–4599. [Google Scholar] [CrossRef]
- Cerrato, C.P.; Kivijärvi, T.; Tozzi, R.; Lehto, T.; Gestin, M.; Langel, Ü. Intracellular Delivery of Therapeutic Antisense Oligonucleotides Targeting MRNA Coding Mitochondrial Proteins by Cell-Penetrating Peptides. J. Mater. Chem. B 2020, 8, 10825–10836. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Sun, A.; Xu, R.; Tao, X.; Dong, Y.; Lv, X.; Wei, D. Cell-Penetrating and Endoplasmic Reticulum-Locating TAT-IL-24-KDEL Fusion Protein Induces Tumor Apoptosis. J. Cell. Physiol. 2016, 231, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, S. DLS and Zeta Potential – What They Are and What They Are Not? J. Controlled Release 2016, 235, 337–351. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Ukidve, A.; Krishnan, V.; Mitragotri, S. Effect of Physicochemical and Surface Properties on in Vivo Fate of Drug Nanocarriers. Adv. Drug Deliv. Rev. 2019, 143, 3–21. [Google Scholar] [CrossRef] [PubMed]
- Nie, S. Understanding and Overcoming Major Barriers in Cancer Nanomedicine. Nanomedicine 2010, 5, 523–528. [Google Scholar] [CrossRef] [PubMed]
- Roser, M.; Fischer, D.; Kissel, T. Surface-Modified Biodegradable Albumin Nano- and Microspheres. II: Effect of Surface Charges on in Vitro Phagocytosis and Biodistribution in Rats. Eur. J. Pharm. Biopharm. Off. J. Arbeitsgemeinschaft Pharm. Verfahrenstechnik EV 1998, 46, 255–263. [Google Scholar] [CrossRef]
- Lingasamy, P.; Teesalu, T. Homing Peptides for Cancer Therapy. Adv. Exp. Med. Biol. 2021, 1295, 29–48. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Liu, E.; Yu, Z.; Pei, X.; Chen, S.; Zhang, P.; Shin, M.-C.; Gong, J.; He, H.; Yang, V.C. CPP-Assisted Intracellular Drug Delivery, What Is Next? Int. J. Mol. Sci. 2016, 17, 1892. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Name | Sequence | Cargo | Ratio | Size (nm) | PdI | ZF (mV) | In Vitro Activity | In Vivo Activity | Ref | |
|---|---|---|---|---|---|---|---|---|---|---|
| Cells | Effect | |||||||||
| Poly-Cationic Familly | ||||||||||
| StA-R8 | Stearyl-RRRRRRRR | siRNA | CR—4:1 | 185.2 | n.d. | 15.6 | HepG2, A549 | 60% survivin KD | n.d. | [31] |
| OA-R8 | Oleyl-RRRRRRRR | 191.9 | 13.2 | |||||||
| K9[Ca2+] | KKKKKKKKK | pDNA | N/P—10 | 200–400 | n.d. | ~ +20 (1 mM KCl) | HeLa, A549, HEK-293, LLC, MDA-MB-231 | Higher pLuc expression as PEI | Mice lung tumor | [32] |
| R9[Ca2+] | RRRRRRRRR | pDNA | N/P—10 | 200 | n.d. | ~ +20 (1 mM KCl) | A549, HEK-293 | Higher or equal pLuc expression as PEI | n.d. | [33] |
| RH9[Ca2+] | RRHHRRHRR | ~ +12 (1 mM KCl) | ||||||||
| RA9[Ca2+] | RRAARRARR | ~ +8 (1 mM KCl) | ||||||||
| RL9[Ca2+] | RRLLRRLRR | |||||||||
| RW9[Ca2+] | RRWWRRWRR | ~ +10 (1 mM KCl) | ||||||||
| GALA/KALA/RALA familly | ||||||||||
| RALA | WEARLARALARALARHLARALARALRACEA-C | pDNA | N/P—10 | 51 | 0.35 | +29 # | ZR-75-1, PC-3, NCTC-929 | eGFP expression not better than Lipofectamin but less toxic | pLuc expression in the lungs and liver of mice | [34] |
| siRNA | N/P—10 | ∼55–65 | <0,60 | ∼ +20–25 # | ZR-75-1 | Equal FKBPL KD compared to Oligofectamin but less toxic | RALA:siFKBPL has no effect on tumor growth | [35] | ||
| siRNA | N/P—6 | 76.6 | n.d. | +16.5 # | HMEC-1 | Efficient FKBPL KD | RALA:siFKBPL in wound patches increases wound healing in mice | [36] | ||
| siRNA | N/P—9 | 100–110 | <0.35 | ~ +38 # | hDF (2D and 3D), THP-1 derived macrophages | Efficient MMP-9 KD | n.d. | [37] | ||
| mRNA | N/P—10 | 91 | n.d. | +26.3 # | DC2.4 | Expression of eGFP | Increased T cell response compared to DOTAP transfection | [38] | ||
| RGSG | WEGRSGRGSGRGSGRHSGRGSGRGSRG-C | mRNA | N/P—10 | 150 | n.d. | +2 # | DC2.4 | No eGFP expression compared to RALA | Less T cell response compared to RALA | [38] |
| RRRR | WEGRRRRRRR-C | mRNA | N/P—10 | 1050 | −5 # | |||||
| PepFect/NickFect familly | ||||||||||
| PF14 | Stearyl- AGYLLGKLLOOLAAAALOOLL | SCO | MR—5:1 | 363 | n.d. | −28.4 (0.01 mM KCl) | HeLa pLuc705, U2OS, mdx mouse myotubes | Equal or better splice correction compared to Lipofectamin | n.d. | [39] |
| mRNA | N/P—3 | 92 | 0.248–0.259 | n.d. | SKOV-3 (2D and 3D) | eGFP expression Lower in 2D but higher in 3D compared to Lipofectamine MessengerMax | mCherry expression in xenografted mice | [40] | ||
| SCO | MR—5:1 | 295.3 * | 0.732 * | n.d. | HeLa pLuc 705 | Internalization in comparison with small molecules | n.d. | [41] | ||
| PF14 | Stearyl- AGYLLGKLLOOLAAAALOOLL | pDNA | N/P—2 | ~150 | n.d. | ~ +35 # | CHO | Dose-dependent pLuc expression | pLuc expression in the lungs and liver of mice | [42] |
| PF14-O | Stearyl-AGYLLGKLLOOLAOOALOOLL | pDNA | N/P—2 | 125 | n.d. | ~ +32 # | CHO | pLuc expression with PF14-O better than PF14-E | pLuc expression in the lungs and liver of mice | |
| PF14-E | Stearyl-AGYLLGKLLEOLAAAALOOLL | 125 | n.d. | ~ +35 # | n.d. | |||||
| C0-PF14 | AGYLLGKLLOOLAAAALOOLL | 1500 | n.d. | ~ +8 # | Nearly no pLuc expression | n.d. | ||||
| C10-PF14 | Decanoyl-AGYLLGKLLOOLAAAALOOLL | 100 | n.d. | ~ + 22 # | pLuc transfection with C10-PF14 lower than C22-PF14 | n.d. | ||||
| C22-PF14 | Docosanoyl-AGYLLGKLLOOLAAAALOOLL | 125 | n.d. | ~ +40 # | pLuc expression in the lungs and liver of mice | |||||
| C22-PF14-O | Docosanoyl-AGYLLGKLLOOLAOOALOOLL | 100 | n.d. | ~ +30 # | pLuc transfection of C22-PF14 and C22-PF14-O equivalent to PF14 and better that C10-PF14 | pLuc expression in the lungs and liver of mice | ||||
| NF53 | (Stearyl-AGYLLG)ε-KINLKALAALAKKIL | pDNA | CR—3:1 | 74.3 | 0.360 | −14.9 (OptiMEM + 10% FBS) | CHO | Equal eGFP expression compared to LF200 | n.d. | [43] |
| SCO | MR—10:1 | 135.3 | 0.459 | −8.8 (OptiMEM + 10% FBS) | HeLa pLuc 705 | Lower splice correction compared to LF200 | ||||
| siRNA | MR—10:1 | 68.6 | 0.529 | −11.9 (OptiMEM + 10% FBS) | EGFP-CHO | Higher eGFP silencing compared to RNAiMax | ||||
| NF61 | Stearyl-AGYLLGKINLKALAALAKKIL | pDNA | CR—3:1 | 68.7 | 0.200 | −17.9 (OptiMEM + 10% FBS) | CHO | Equal eGFP expression compared to LF200 | ||
| SCO | MR—10:1 | 60.5 | 0.286 | −10.2 (OptiMEM + 10% FBS) | HeLa pLuc 705 | Lower splice correction compared to LF200 | ||||
| siRNA | MR—10:1 | 159.4 | 0.348 | −13.6 (OptiMEM + 10% FBS) | EGFP-CHO | Equal eGFP silencing compared to RNAiMax | ||||
| NF51 | (Stearyl-AGYLLG)δ-OINLKALAALAKKIL | pDNA | CR—3:1 | 62 | 0.138 | −11.5 (OptiMEM + 10% FBS) | CHO, MEF, Jurkat, A20 | Higher eGFP expression compared to LF200 | ||
| SCO | MR—10:1 | 86.0 | 0.298 | −11.1 (OptiMEM + 10% FBS) | HeLa pLuc 705 | Higher splice correction compared to LF200 | ||||
| siRNA | MR—10:1 | 74.2 | 0.197 | −11.8 (OptiMEM + 10% FBS) | EGFP-CHO | Higher eGFP silencing compared to RNAiMax | ||||
| NF51 | (Stearyl-AGYLLG)δ-OINLKALAALAKKIL | pDNA | CR—4:1 | n.d. | n.d. | n.d. | HeLa, U87-MG, N2A and HT1080 | eGFP expression comparable to LF200, NF55 better than NF51 and NF54 | pLuc expression in the lung, liver, and brain of healthy mice and those bearing intracranial tumors | [44] |
| NF54 | (Stearyl-AGYLLG)δ-OINLKALAALAAKIL | n.d. | ||||||||
| NF55 | (Stearyl-AGYLLG)δ-OINLKALAALAKAIL | 50–150 | ||||||||
| NF55 | (Stearyl-AGYLLG)δ-OINLKALAALAKAIL | pDNA | CR—4:1 | 85 # | 0.211 | n.d. | CHO | pLuc expression comparable to Freiman 2016 | pLuc lung expression | [45] |
| CADY-K/RICK family | ||||||||||
| CADY-K | GLWRALWRLLRSLWRLLWK | siRNA | MR—20:1 | 116 | 0.30 | +38.0 (5 mM NaCl) | U87, Neuro2A, B16 | Efficient Luciferase and CyclinB1 proteins KD | n.d. | [25,28] |
| d-Cady-k | glwralwrllrslwrllwk | siRNA | MR—20:1 | 90 | 0.30 | +40.0 (5 mM NaCl) | U87 # | |||
| RICK | kwllrwlsrllrwlarwlg | siRNA | MR—20:1 | 92 | 0.24 | +40.0 (5 mM NaCl) | ||||
| PEG-RICK | PEG2000-Ckwllrwlsrllrwlarwlg | siRNA | MR—20:1 | 69 (20% PEG) | 0.29 (20% PEG) | +37.0 (20% PEG) (5 mM NaCl) | U87 | Efficient Luciferase and CDK4 proteins KD for 20% PEG-RICK NPs and less cytotixicity than RNAimax. | 20% PEG-RICK NPs significantly reduce liver and kidney accumulation in mice | [46] |
| WRAP family | ||||||||||
| WRAP1 | LLWRLWRLLWRLWRLL | siRNA | MR—20:1 | 73.3 | 0.38 | +42.2 (5 mM NaCl) | U87, KB, MCF7, HuH7, Neuro2A, MDA-MB-231, CMT93, HT29, RM1, GL261 | Efficient Luciferase or CDK4 KD, fast internalization and less toxic than RNAimax | n.d. | [26] |
| WRAP5 | LLRLLRWWWRLLRLL | siRNA | MR—20:1 | 80.0 | 0.29 | +28.8 (5 mM NaCl) | ||||
| pDNA | N/P—3 | 102 | n.d. | +33 (Tris buffer pH 7) | n.d. | n.d. | n.d. | [47] | ||
| C6 family | ||||||||||
| C6 | RLLRLLLRLWRRLLRLLR | siRNA | MR—40:1 | 150–250 | n.d. | +60.0 # | CHO-K1 | Internalization of fluorescently labelled siRNA without cytotoxicity | n.d. | [48] |
| C6M1 | RLWRLLWRLWRRLWRLLR | siRNA | MR—30:1 | ~70 | n.d. | +31 (HEPES) +5 (PBS) | CHO-K1 | Better internalization of fluorescently labelled siRNA than C6, significant inhibition of GAPDH expression | n.d. | [49,50] |
| siRNA | MR—60:1 | ~100–200 | n.d. | ~ +50 # | CHO-K1 | Significant inhibition of GAPDH expression | Inhibition of tumor growth with Bcl-2 siRNA in A549 cancer cells xenografted in mice | [51] | ||
| C6M3 | RLWHLLWRLWRRLHRLLR | siRNA | MR—40:1 | ~90 | n.d. | +32.0 # | CHO-K1, RAW 264.7 | Strong uptake CHO-K1 cells, significant inhibition of GAPDH expression and no significant cytokine induction | Inhibition of tumor growth with Bcl-2 siRNA in A549 cancer cells xenografted in mice | [52] |
| C6M6 | GLWHLLLHLWRRLLRLLR | siRNA | MR—60:1 | ~110 | n.d. | +36.0 # | ||||
| DM1 | DEG-RLWRLLWRLWRRLWRLLR | siRNA | MR—40:1 | n.d. | n.d. | n.d. | CHO-K1, C166-GFP | Significant inhibition of GAPDH and eGFP expression and DEGylation improves serum resistance | n.d. | [53] |
| Mpge family | ||||||||||
| Mgpe-1 | SRLSHLRHHYSKKWHRFR | pDNA | N/P—10 | 80.13 | 0.151 | +36.5 # | CHO-K1, MCF-7, A549 | Equal pLuc expression as Lipofectamine but less toxic and higher pLuc expression than Cellfectin/Superfect | n.d. | [21] |
| Mgpe-2 | LLYWFSRSHRHHSKKHRR | N/P—10 | 110.25 | 0.141 | +31.95 # | |||||
| Mgpe-3 | RRLRHLRHHYRRRWHRFR | N/P—10 | 63.26 | 0.152 | +33.5 # | |||||
| Mgpe-4 | LLYWFRRRHRHHRRRHRR | N/P—5 | 62.84 | 0.155 | +25.45 # | |||||
| Mgpe-10 | CLLYWFRRRHRHHRRRHRRC | pDNA | N/P—10 | 128.49 | 0.166 | +27.7 # | CHO-K1, MCF-7, A549, B16, B35, H 1299, HEK, Jurkat, MDA-MB-231, RAW, U87, T47D, Hela | Higher transfection efficiency, less toxic than Lipofectamine and Chondroitin sulfate combination | n.d. | [23,54] |
| Mgpe-9 | CRRLRHLRHHYRRRWHRFRC | pDNA | N/P—10 | 85.77 | 0.240 | +35.5 # | ||||
| pDNA | N/P—10 | 50.63 | n.d. | +24.0 # | dividing/differentiated ARPE-19/hfRPE cells | eGFP and Gaussia Luciferase expression | n.d. | [55] | ||
| siRNA | N/P—10 | 173.9 | n.d. | n.d. | Differentiated ARPE-19 | 80% GAPDH knockdown GAPDH for polyplexes at N/P 30 and combined with condroitin sulphate | n.d. | |||
| Other PBN-forming peptides | ||||||||||
| CHAT | CHHHRRRWRRRHHHC | pDNA | N/P—12 | 207 | 0.25 | +29 # | MCF7, MDA-MB-231, DU-145, PC-3 | eGFP expression comparable to RALA | 10-fold increased pLuc expression in lung, liver and kidneys, 5-fold in tumor | [56] |
| StA-TH | Stearyl- AGYLLGHINLHHLAHL (Aib)HHIL | pDNA | N/P—3 | >200 (TEM) | n.d. | n.d. | CHO, U251 | High internalization and p53 activity (pro-apoptotic) at pH 5.5 | n.d. | [57] |
| T9(dR) | GWTLNSAGYLLGKINLKALAALAKKIL-(dR)9 | siRNA | MR 4:1 | 350–550 | n.d. | n.d. | 293T, MDCK, RAW, A549 | Silencing of nucleoprotein expression | Better survival and weight recovery of PR8 influenza viru-infected mice | [58] |
| p5RHH | VLTTGLPALISWIRRRHRRHC | siRNA | MR—100:1 | ~55 (TEM) | n.d. | n.d. | / | / | Silencing NF-kB expression reduced chondrocyte apoptosi in a murine model of controlled knee joint impact injury | [59] |
| ~ +12 # | HUVEC | p65 slencing | n.d. | [60] | ||||||
| n.d. | n.d. | n.d. | ARK1, OVCAR8 | AXL silencing | Reduced tumor nodules and weight | [61] | ||||
| mRNA | 350 ng mRNA:2.0 nmol p5RHH | <200 | n.d. | +6 (OptiMEM) | B16F10, CASMC, HAoEC | RFP, Luc, GFP expression | RFP expression on injured femoral artery. mRNA construct (p27-miRNA-126-3p) prevents restenosis in a femoral artery wire injury mouse model | [62] | ||
| BR2 | RAGLPFQVGRLLRRLLR | siRNA | N/P—8 | 150–200 | n.d. | ~ +10 # | HeLa, HCT116, HaCat, NIH3T3 | GFP nd VEGF silencing comparable to PEI | n.d. | [63] |
| C18-S413-PV | Stearoyl-ALWKTLLKKVLKAPKKKRKVC | siRNA | CR—2 | ~250 | n.d. | ~+12 (HBS-2) | U87 | No significant GFP silencing | n.d. | [64] |
| C16-S413-PV | Palmitoyl-ALWKTLLKKVLKAPKKKRKVC | ~250 | ~+10 (HBS-2) | No significant GFP silencing | ||||||
| C14-S413-PV | Myristoyl-ALWKTLLKKVLKAPKKKRKVC | 350 | ~ +10 (HBS-2) | GFP silencing | ||||||
| C12-S413-PV | Lauroyl-ALWKTLLKKVLKAPKKKRKVC | 750 | +8 (HBS-2) | GFP silencing | ||||||
| siRNA | CR—5 | 192 | 0.44 | +23.6 (HBS) | U87, HeLa | GFP silencing lower than LF2000 | n.d. | [65] | ||
| C12-H5-S413-PV | Lauroyl-HHHHH-ALWKTLLKKVLKAPKKKRKVC | 173 | 0.24 | +22.6 (HBS) | GFP silencing equal to FL2000 | |||||
| H5-S413-PV-C12 | HHHHH-ALWKTLLKKVLKAPKKKRKVC-Lauroyl | 184 | 0.69 | +19.7 (HBS) | ||||||
| StA-SPA | Stearyl-rPKPwQwFwLL | pDNA | N/P—2 | >200 (TEM) | n.d. | n.d. | CHO | Nearly equal pLuc expression as LF2000 | n.d. | [66] |
| KL4 | KLLLLKLLLLKLLLLKLLLLK | siRNA | (w/w)—20:1 | 280 | 0.28 | n.d. | A549, BEAS-2B | Reduced GAPDH expression comparable to Lipofectamin 2000 | n.d. | [67] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boisguérin, P.; Konate, K.; Josse, E.; Vivès, E.; Deshayes, S. Peptide-Based Nanoparticles for Therapeutic Nucleic Acid Delivery. Biomedicines 2021, 9, 583. https://doi.org/10.3390/biomedicines9050583
Boisguérin P, Konate K, Josse E, Vivès E, Deshayes S. Peptide-Based Nanoparticles for Therapeutic Nucleic Acid Delivery. Biomedicines. 2021; 9(5):583. https://doi.org/10.3390/biomedicines9050583
Chicago/Turabian StyleBoisguérin, Prisca, Karidia Konate, Emilie Josse, Eric Vivès, and Sébastien Deshayes. 2021. "Peptide-Based Nanoparticles for Therapeutic Nucleic Acid Delivery" Biomedicines 9, no. 5: 583. https://doi.org/10.3390/biomedicines9050583
APA StyleBoisguérin, P., Konate, K., Josse, E., Vivès, E., & Deshayes, S. (2021). Peptide-Based Nanoparticles for Therapeutic Nucleic Acid Delivery. Biomedicines, 9(5), 583. https://doi.org/10.3390/biomedicines9050583