
Non-Immunotherapy Application of LNP-mRNA: Maximizing Efficacy and Safety


Abstract
1. Introduction
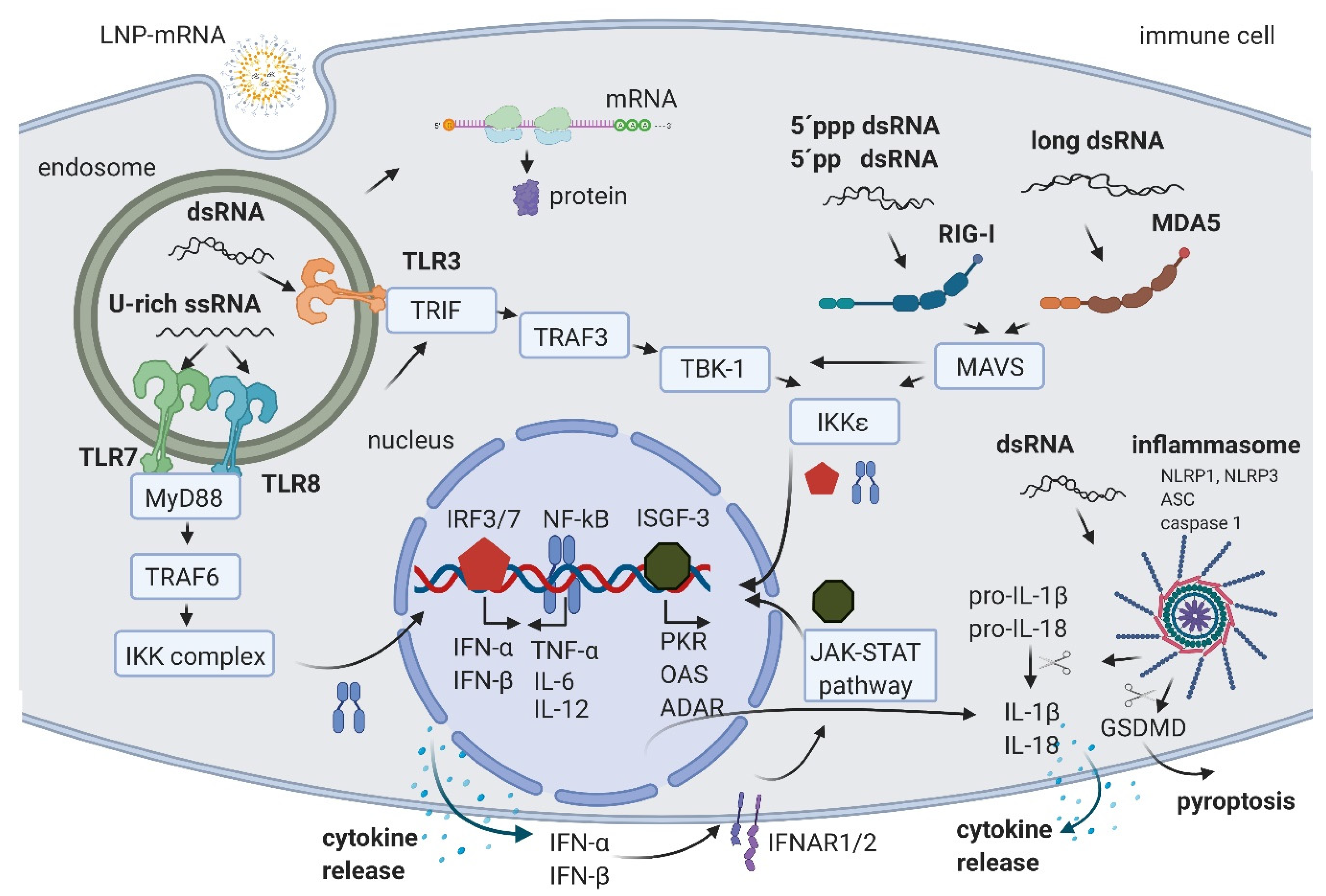
2. IVT mRNA and Byproducts Induced Immune Activation
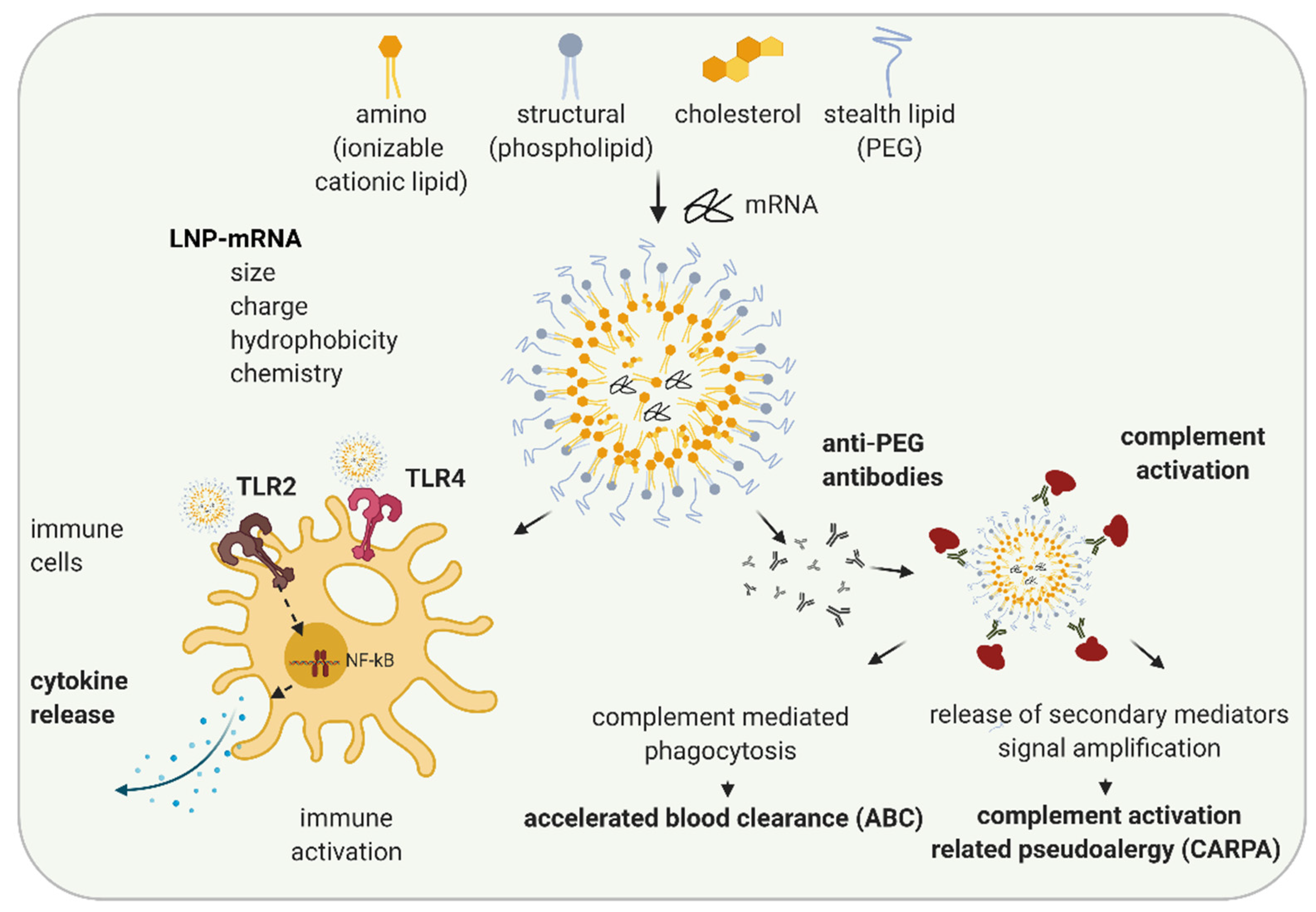
3. LNP Induced Immune Activation
4. LNP-mRNA in RNA Protein Replacement and Other Non-Immunotherapy Applications
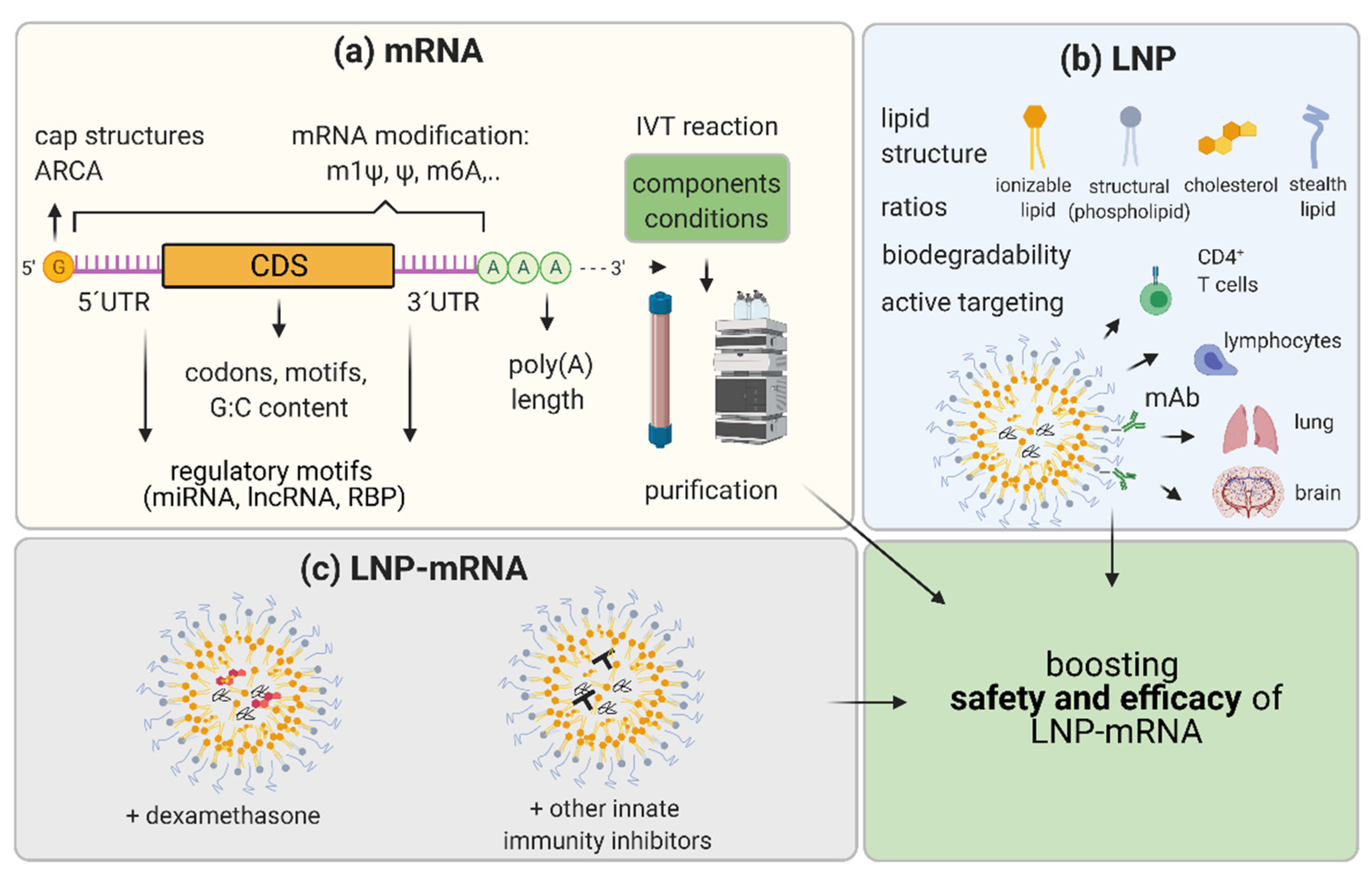
5. Boosting the Efficacy and Safety of LNP-mRNA Applications
5.1. mRNA Nucleoside Modification
5.2. mRNA Sequence and Structure Optimization
5.3. IVT mRNA Production and Purification Methods
5.4. LNP Optimization
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sahin, U.; Karikó, K.; Türeci, Ö. mRNA-based therapeutics—Developing a new class of drugs. Nat. Rev. Drug Discov. 2014, 13, 759–780. [Google Scholar] [CrossRef] [PubMed]
- Devoldere, J.; Dewitte, H.; De Smedt, S.C.; Remaut, K. Evading innate immunity in nonviral mRNA delivery: Don’t shoot the messenger. Drug Discov. Today 2016, 21, 11–25. [Google Scholar] [CrossRef]
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. mRNA vaccines—A new era in vaccinology. Nat. Rev. Drug Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef] [PubMed]
- Pardi, N.; Hogan, M.J.; Weissman, D. Recent advances in mRNA vaccine technology. Curr. Opin. Immunol. 2020, 65, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Kowalski, P.S.; Rudra, A.; Miao, L.; Anderson, D.G. Delivering the Messenger: Advances in Technologies for Therapeutic mRNA Delivery. Mol. Ther. 2019, 27, 710–728. [Google Scholar] [CrossRef] [PubMed]
- Mitragotri, S.; Burke, P.A.; Langer, R. Overcoming the challenges in administering biopharmaceuticals: Formulation and delivery strategies. Nat. Rev. Drug Discov. 2014, 13, 655–672. [Google Scholar] [CrossRef] [PubMed]
- Wolff, J.A.; Malone, R.W.; Williams, P.; Chong, W.; Acsadi, G.; Jani, A.; Felgner, P.L. Direct gene transfer into mouse muscle in vivo. Science 1990, 247, 1465–1468. [Google Scholar] [CrossRef] [PubMed]
- Thi, T.; Suys, E.; Lee, J.; Nguyen, D.; Park, K.; Truong, N. Lipid-Based Nanoparticles in the Clinic and Clinical Trials: From Cancer Nanomedicine to COVID-19 Vaccines. Vaccines 2021, 9, 359. [Google Scholar] [CrossRef]
- Sahin, U.; Muik, A.; Derhovanessian, E.; Vogler, I.; Kranz, L.M.; Vormehr, M.; Baum, A.; Pascal, K.; Quandt, J.; Maurus, D.; et al. COVID-19 vaccine BNT162b1 elicits human antibody and TH1 T cell responses. Nature 2020, 586, 594–599. [Google Scholar] [CrossRef]
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Marc, G.P.; Moreira, E.D.; Zerbini, C.; et al. Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine. N. Engl. J. Med. 2020, 383, 2603–2615. [Google Scholar] [CrossRef]
- Anderson, E.J.; Rouphael, N.G.; Widge, A.T.; Jackson, L.A.; Roberts, P.C.; Makhene, M.; Chappell, J.D.; Denison, M.R.; Stevens, L.J.; Pruijssers, A.J.; et al. Safety and Immunogenicity of SARS-CoV-2 mRNA-1273 Vaccine in Older Adults. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef]
- Oostvogels, L.; Kremsner, P.; Kreidenweiss, A.; Leroux-Roels, I.; Leroux-Roels, G.; Kroidl, A.; Schunk, M.; Schindler, C.; Bosch, J.; Fendel, R.; et al. Phase 1 Assessment of the Safety and Immunogenicity of an mRNA- Lipid Nanoparticle Vaccine Candidate Against SARS-CoV-2 in Human Volunteers. medRxiv 2020. [Google Scholar] [CrossRef]
- Karikó, K.; Buckstein, M.; Ni, H.; Weissman, D. Suppression of RNA Recognition by Toll-like Receptors: The Impact of Nucleoside Modification and the Evolutionary Origin of RNA. Immunity 2005, 23, 165–175. [Google Scholar] [CrossRef]
- Pardi, N.; Hogan, M.J.; Pelc, R.S.; Muramatsu, H.; Andersen, H.; DeMaso, C.R.; Dowd, K.A.; Sutherland, L.L.; Scearce, R.M.; Parks, R.; et al. Zika virus protection by a single low-dose nucleoside-modified mRNA vaccination. Nature 2017, 543, 248–251. [Google Scholar] [CrossRef] [PubMed]
- Bourquin, C.; Schmidt, L.; Hornung, V.; Wurzenberger, C.; Anz, D.; Sandholzer, N.; Schreiber, S.; Voelkl, A.; Hartmann, G.; Endres, S. Immunostimulatory RNA oligonucleotides trigger an antigen-specific cytotoxic T-cell and IgG2a response. Blood 2006, 109, 2953–2960. [Google Scholar] [CrossRef]
- Weissman, D.; Ni, H.; Scales, D.; Dude, A.; Capodici, J.; McGibney, K.; Abdool, A.; Isaacs, S.N.; Cannon, G.; Karikó, K. HIV Gag mRNA Transfection of Dendritic Cells (DC) Delivers Encoded Antigen to MHC Class I and II Molecules, Causes DC Maturation, and Induces a Potent Human In Vitro Primary Immune Response. J. Immunol. 2000, 165, 4710–4717. [Google Scholar] [CrossRef] [PubMed]
- Baiersdörfer, M.; Boros, G.; Muramatsu, H.; Mahiny, A.; Vlatkovic, I.; Sahin, U.; Karikó, K. A Facile Method for the Removal of dsRNA Contaminant from In Vitro-Transcribed mRNA. Mol. Ther. Nucleic Acids 2019, 15, 26–35. [Google Scholar] [CrossRef]
- Wang, Y.; Ludwig, J.; Schuberth, C.; Goldeck, M.; Schlee, M.; Li, H.; Juranek, S.; Sheng, G.; Micura, R.; Tuschl, T.; et al. Structural and functional insights into pattern recognition by the innate immune receptor RIG-I. Nat. Struct. Mol. Biol. 2010, 17, 781–787. [Google Scholar] [CrossRef] [PubMed]
- Mu, X.; Greenwald, E.; Ahmad, S.; Hur, S. An origin of the immunogenicity of in vitro transcribed RNA. Nucleic Acids Res. 2018, 46, 5239–5249. [Google Scholar] [CrossRef] [PubMed]
- Rehwinkel, J.; Gack, M.U. RIG-I-like receptors: Their regulation and roles in RNA sensing. Nat. Rev. Immunol. 2020, 20, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Heil, F.; Hemmi, H.; Hochrein, H.; Ampenberger, F.; Kirschning, C.; Akira, S.; Lipford, G.; Wagner, H.; Bauer, S. Species-Specific Recognition of Single-Stranded RNA via Toll-like Receptor 7 and 8. Science 2004, 303, 1526–1529. [Google Scholar] [CrossRef]
- Diebold, S.S.; Massacrier, C.; Akira, S.; Paturel, C.; Morel, Y.; Sousa, C.R.E. Nucleic acid agonists for Toll-like receptor 7 are defined by the presence of uridine ribonucleotides. Eur. J. Immunol. 2006, 36, 3256–3267. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Takeda, K. Toll-like receptor signalling. Nat. Rev. Immunol. 2004, 4, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. Innate immune recognition of viral infection. Nat. Immunol. 2006, 7, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Balachandran, S.; Roberts, P.C.; Brown, L.E.; Truong, H.; Pattnaik, A.K.; Archer, D.R.; Barber, G.N. Essential role for the dsRNA-dependent protein kinase PKR in innate immunity to viral infection. Immunity 2000, 13, 129–141. [Google Scholar] [CrossRef]
- Silverman, R.H. Viral Encounters with 2′,5′-Oligoadenylate Synthetase and RNase L during the Interferon Antiviral Response. J. Virol. 2007, 81, 12720–12729. [Google Scholar] [CrossRef] [PubMed]
- Bass, B.L. RNA Editing by Adenosine Deaminases That Act on RNA. Annu. Rev. Biochem. 2002, 71, 817–846. [Google Scholar] [CrossRef] [PubMed]
- Licht, K.; Hartl, M.; Amman, F.; Anrather, D.; Janisiw, M.P.; Jantsch, M.F. Inosine induces context-dependent recoding and translational stalling. Nucleic Acids Res. 2019, 47, 3–14. [Google Scholar] [CrossRef]
- Nishikura, K. A-to-I editing of coding and non-coding RNAs by ADARs. Nat. Rev. Mol. Cell Biol. 2016, 17, 83–96. [Google Scholar] [CrossRef]
- Hartner, J.C.; Walkley, C.R.; Lu, J.; Orkin, S.H. ADAR1 is essential for the maintenance of hematopoiesis and suppression of interferon signaling. Nat. Immunol. 2008, 10, 109–115. [Google Scholar] [CrossRef]
- Liddicoat, B.J.; Piskol, R.; Chalk, A.M.; Ramaswami, G.; Higuchi, M.; Hartner, J.C.; Li, J.B.; Seeburg, P.H.; Walkley, C.R. RNA editing by ADAR1 prevents MDA5 sensing of endogenous dsRNA as nonself. Science 2015, 349, 1115–1120. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Banerjee, S.; Goldstein, S.A.; Dong, B.; Gaughan, C.; Rath, S.; Donovan, J.; Korennykh, A.V.; Silverman, R.H.; Weiss, S.R. Ribonuclease L mediates the cell-lethal phenotype of double-stranded RNA editing enzyme ADAR1 deficiency in a human cell line. eLife 2017, 6, e25687. [Google Scholar] [CrossRef]
- Lamers, M.M.; Hoogen, B.G.V.D.; Haagmans, B.L. ADAR1: “Editor-in-Chief” of Cytoplasmic Innate Immunity. Front. Immunol. 2019, 10, 1763. [Google Scholar] [CrossRef]
- Alexopoulou, L. Recognition of double-stranded RNA and activation of NF-kB by toll-like receptor 3. Nature 2001, 413, 732–738. [Google Scholar] [CrossRef]
- Choe, J.; Kelker, M.S.; Wilson, I.A. Crystal Structure of Human Toll-Like Receptor 3 (TLR3) Ectodomain. Science 2005, 309, 581–585. [Google Scholar] [CrossRef]
- Esaxena, M.; Eyeretssian, G. NOD-Like Receptors: Master Regulators of Inflammation and Cancer. Front. Immunol. 2014, 5, 327. [Google Scholar] [CrossRef]
- Goubau, D.; Schlee, M.; Deddouche, S.; Pruijssers, A.J.; Zillinger, T.; Goldeck, M.; Schuberth, C.; Van Der Veen, A.G.; Fujimura, T.; Rehwinkel, J.; et al. Antiviral immunity via RIG-I-mediated recognition of RNA bearing 5′-diphosphates. Nat. Cell Biol. 2014, 514, 372–375. [Google Scholar] [CrossRef]
- Schmidt, A.; Schwerd, T.; Hamm, W.; Hellmuth, J.C.; Cui, S.; Wenzel, M.; Hoffmann, F.S.; Michallet, M.-C.; Besch, R.; Hopfner, K.-P.; et al. 5′-triphosphate RNA requires base-paired structures to activate antiviral signaling via RIG-I. Proc. Natl. Acad. Sci. USA 2009, 106, 12067–12072. [Google Scholar] [CrossRef] [PubMed]
- Schuberth-Wagner, C.; Ludwig, J.; Bruder, A.K.; Herzner, A.-M.; Zillinger, T.; Goldeck, M.; Schmidt, T.; Schmid-Burgk, J.L.; Kerber, R.; Wolter, S.; et al. A Conserved Histidine in the RNA Sensor RIG-I Controls Immune Tolerance to N1-2′O-Methylated Self RNA. Immunity 2015, 43, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Pichlmair, A.; Schulz, O.; Tan, C.-P.; Rehwinkel, J.; Kato, H.; Takeuchi, O.; Akira, S.; Way, M.; Schiavo, G.; Sousa, C.R.E. Activation of MDA5 Requires Higher-Order RNA Structures Generated during Virus Infection. J. Virol. 2009, 83, 10761–10769. [Google Scholar] [CrossRef]
- Züst, R.; Cervantes-Barragan, L.; Habjan, M.; Maier, R.; Neuman, B.W.; Ziebuhr, J.; Szretter, K.J.; Baker, S.C.; Barchet, W.; Diamond, M.S.; et al. Ribose 2′-O-methylation provides a molecular signature for the distinction of self and non-self mRNA dependent on the RNA sensor Mda5. Nat. Immunol. 2011, 12, 137–143. [Google Scholar] [CrossRef]
- Fang, R.; Jiang, Q.; Zhou, X.; Wang, C.; Guan, Y.; Tao, J.; Xi, J.; Feng, J.-M.; Jiang, Z. MAVS activates TBK1 and IKKε through TRAFs in NEMO dependent and independent manner. PLoS Pathog. 2017, 13, e1006720. [Google Scholar] [CrossRef]
- Andries, O.; De Filette, M.; De Smedt, S.C.; Demeester, J.; Van Poucke, M.; Peelman, L.; Sanders, N.N. Innate immune response and programmed cell death following carrier-mediated delivery of unmodified mRNA to respiratory cells. J. Control. Release 2013, 167, 157–166. [Google Scholar] [CrossRef]
- Bauernfried, S.; Scherr, M.J.; Pichlmair, A.; Duderstadt, K.E.; Hornung, V. Human NLRP1 is a sensor for double-stranded RNA. Science 2021, 371, eabd0811. [Google Scholar] [CrossRef]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nat. Cell Biol. 2015, 526, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Devarkar, S.C.; Wang, C.; Miller, M.T.; Ramanathan, A.; Jiang, F.; Khan, A.G.; Patel, S.S.; Marcotrigiano, J. Structural basis for m7G recognition and 2′-O-methyl discrimination in capped RNAs by the innate immune receptor RIG-I. Proc. Natl. Acad. Sci. USA 2016, 113, 596–601. [Google Scholar] [CrossRef] [PubMed]
- Diamond, M.S. IFIT1: A dual sensor and effector molecule that detects non-2′-O methylated viral RNA and inhibits its translation. Cytokine Growth Factor Rev. 2014, 25, 543–550. [Google Scholar] [CrossRef]
- Abbas, Y.M.; Laudenbach, B.T.; Martínez-Montero, S.; Cencic, R.; Habjan, M.; Pichlmair, A.; Damha, M.J.; Pelletier, J.; Nagar, B. Structure of human IFIT1 with capped RNA reveals adaptable mRNA binding and mechanisms for sensing N1 and N2 ribose 2′-O methylations. Proc. Natl. Acad. Sci. USA 2017, 114, E2106–E2115. [Google Scholar] [CrossRef] [PubMed]
- Dimitriadis, G.J. Translation of rabbit globin mRNA introduced by liposomes into mouse lymphocytes. Nature 1978, 274, 923–924. [Google Scholar] [CrossRef]
- Kranz, L.M.; Diken, M.; Haas, H.; Kreiter, S.; Loquai, C.; Reuter, K.C.; Meng, M.; Fritz, D.; Vascotto, F.; Hefesha, H.; et al. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature 2016, 534, 396–401. [Google Scholar] [CrossRef]
- Whitehead, K.A.; Langer, R.; Anderson, D.G. Knocking down barriers: Advances in siRNA delivery. Nat. Rev. Drug Discov. 2009, 8, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Semple, S.C.; Akinc, A.; Chen, J.; Sandhu, A.P.; Mui, B.L.; Cho, C.K.; Sah, D.W.Y.; Stebbing, D.; Crosley, E.J.; Yaworski, E.; et al. Rational design of cationic lipids for siRNA delivery. Nat. Biotechnol. 2010, 28, 172–176. [Google Scholar] [CrossRef]
- Committee for Medicinal Products for Human Use (CHMP). Onpattro: International Non-Proprietary Name: Patisiran; EMA/554262/2018; European Medicines Agency: London, UK, 2018. [Google Scholar]
- Butler, J.S.; Chan, A.; Costelha, S.; Fishman, S.; Willoughby, J.L.S.; Borland, T.D.; Milstein, S.; Foster, D.J.; Gonçalves, P.; Chen, Q.; et al. Preclinical evaluation of RNAi as a treatment for transthyretin-mediated amyloidosis. Amyloid 2016, 23, 109–118. [Google Scholar] [CrossRef]
- Nguyen, J.; Szoka, F.C. Nucleic Acid Delivery: The Missing Pieces of the Puzzle? Accounts Chem. Res. 2012, 45, 1153–1162. [Google Scholar] [CrossRef]
- Samaridou, E.; Heyes, J.; Lutwyche, P. Lipid nanoparticles for nucleic acid delivery: Current perspectives. Adv. Drug Deliv. Rev. 2020, 154–155, 37–63. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Ashwanikumar, N.; Robinson, E.; Xia, Y.; Mihai, C.; Iii, J.P.G.; Hou, S.; Esposito, A.A.; Ketova, T.; Welsher, K.; et al. Naturally-occurring cholesterol analogues in lipid nanoparticles induce polymorphic shape and enhance intracellular delivery of mRNA. Nat. Commun. 2020, 11, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Eygeris, Y.; Patel, S.; Jozic, A.; Sahay, G. Deconvoluting Lipid Nanoparticle Structure for Messenger RNA Delivery. Nano Lett. 2020, 20, 4543–4549. [Google Scholar] [CrossRef]
- Nogueira, S.S.; Schlegel, A.; Maxeiner, K.; Weber, B.; Barz, M.; Schroer, M.A.; Blanchet, C.E.; Svergun, D.I.; Ramishetti, S.; Peer, D.; et al. Polysarcosine-Functionalized Lipid Nanoparticles for Therapeutic mRNA Delivery. ACS Appl. Nano Mater. 2020, 3, 10634–10645. [Google Scholar] [CrossRef]
- Suzuki, T.; Suzuki, Y.; Hihara, T.; Kubara, K.; Kondo, K.; Hyodo, K.; Yamazaki, K.; Ishida, T.; Ishihara, H. PEG shedding-rate-dependent blood clearance of PEGylated lipid nanoparticles in mice: Faster PEG shedding attenuates anti-PEG IgM production. Int. J. Pharm. 2020, 588, 119792. [Google Scholar] [CrossRef] [PubMed]
- Pham, C.T.; Mitchell, L.M.; Huang, J.L.; Lubniewski, C.M.; Schall, O.F.; Killgore, J.K.; Pan, D.; Wickline, S.A.; Lanza, G.M.; Hourcade, D.E. Variable Antibody-dependent Activation of Complement by Functionalized Phospholipid Nanoparticle Surfaces. J. Biol. Chem. 2011, 286, 123–130. [Google Scholar] [CrossRef]
- Hassett, K.J.; Benenato, K.E.; Jacquinet, E.; Lee, A.; Woods, A.; Yuzhakov, O.; Himansu, S.; Deterling, J.; Geilich, B.M.; Ketova, T.; et al. Optimization of Lipid Nanoparticles for Intramuscular Administration of mRNA Vaccines. Mol. Ther. Nucleic Acids 2019, 15, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Hardie, J.; Zhang, X.; Rotello, V.M. Effects of engineered nanoparticles on the innate immune system. Semin. Immunol. 2017, 34, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Halamoda-Kenzaoui, B.; Bremer-Hoffmann, S. Main trends of immune effects triggered by nanomedicines in preclinical studies. Int. J. Nanomed. 2018, 13, 5419–5431. [Google Scholar] [CrossRef] [PubMed]
- Kozma, G.T.; Shimizu, T.; Ishida, T.; Szebeni, J. Anti-PEG antibodies: Properties, formation, testing and role in adverse immune reactions to PEGylated nano-biopharmaceuticals. Adv. Drug Deliv. Rev. 2020, 154–155, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Szebeni, J. Complement activation-related pseudoallergy: A stress reaction in blood triggered by nanomedicines and biologicals. Mol. Immunol. 2014, 61, 163–173. [Google Scholar] [CrossRef]
- Lonez, C.; Bessodes, M.; Scherman, D.; Vandenbranden, M.; Escriou, V.; Ruysschaert, J. Cationic lipid nanocarriers activate Toll-like receptor 2 and NLRP3 inflammasome pathways. Nanomedicine 2014, 10, 775–782. [Google Scholar] [CrossRef]
- Lonez, C.; Irvine, K.L.; Pizzuto, M.; Schmidt, B.I.; Gay, N.J.; Ruysschaert, J.-M.; Gangloff, M.; Bryant, C.E. Critical residues involved in Toll-like receptor 4 activation by cationic lipid nanocarriers are not located at the lipopolysaccharide-binding interface. Cell Mol. Life Sci. 2015, 72, 3971–3982. [Google Scholar] [CrossRef]
- Kedmi, R.; Ben-Arie, N.; Peer, D. The systemic toxicity of positively charged lipid nanoparticles and the role of Toll-like receptor 4 in immune activation. Biomaterials 2010, 31, 6867–6875. [Google Scholar] [CrossRef]
- Abrams, M.T.; Koser, M.L.; Seitzer, J.; Williams, S.C.; DiPietro, M.A.; Wang, W.; Shaw, A.W.; Mao, X.; Jadhav, V.; Davide, J.P.; et al. Evaluation of Efficacy, Biodistribution, and Inflammation for a Potent siRNA Nanoparticle: Effect of Dexamethasone Co-treatment. Mol. Ther. 2010, 18, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Suk, J.S.; Xu, Q.; Kim, N.; Hanes, J.; Ensign, L.M. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv. Drug Deliv. Rev. 2016, 99, 28–51. [Google Scholar] [CrossRef]
- Richter, A.W.; Åkerblom, E. Antibodies against Polyethylene Glycol Produced in Animals by Immunization with Monomethoxy Polyethylene Glycol Modified Proteins. Int. Arch. Allergy Immunol. 1983, 70, 124–131. [Google Scholar] [CrossRef]
- Shiraishi, K.; Yokoyama, M. Toxicity and immunogenicity concerns related to PEGylated-micelle carrier systems: A review. Sci. Technol. Adv. Mater. 2019, 20, 324–336. [Google Scholar] [CrossRef]
- Dams, E.T.; Laverman, P.; Oyen, W.J.; Storm, G.; Scherphof, G.L.; Van Der Meer, J.W.; Corstens, F.H.; Boerman, O.C. Accelerated blood clearance and altered biodistribution of repeated injections of sterically stabilized liposomes. J. Pharmacol. Exp. Ther. 2000, 292, 1071–1079. [Google Scholar] [PubMed]
- Abu Lila, A.S.; Kiwada, H.; Ishida, T. The accelerated blood clearance (ABC) phenomenon: Clinical challenge and approaches to manage. J. Control. Release 2013, 172, 38–47. [Google Scholar] [CrossRef]
- Mohamed, M.; Abu Lila, A.S.; Shimizu, T.; Alaaeldin, E.; Hussein, A.; Sarhan, H.A.; Szebeni, J.; Ishida, T. PEGylated liposomes: Immunological responses. Sci. Technol. Adv. Mater. 2019, 20, 710–724. [Google Scholar] [CrossRef]
- Kozma, G.T.; Mészáros, T.; Vashegyi, I.; Fülöp, T.; Örfi, E.; Dézsi, L.; Rosivall, L.; Bavli, Y.; Urbanics, R.; Mollnes, T.E.; et al. Pseudo-anaphylaxis to Polyethylene Glycol (PEG)-Coated Liposomes: Roles of Anti-PEG IgM and Complement Activation in a Porcine Model of Human Infusion Reactions. ACS Nano 2019, 13, 9315–9324. [Google Scholar] [CrossRef] [PubMed]
- Stadler, C.R.; Bähr-Mahmud, H.; Celik, L.; Hebich, B.; Roth, A.S.; Roth, R.P.; Karikó, K.; Türeci, Ö.; Sahin, U. Elimination of large tumors in mice by mRNA-encoded bispecific antibodies. Nat. Med. 2017, 23, 815–817. [Google Scholar] [CrossRef]
- Kose, N.; Fox, J.M.; Sapparapu, G.; Bombardi, R.; Tennekoon, R.N.; De Silva, A.D.; Elbashir, S.M.; Theisen, M.A.; Humphris-Narayanan, E.; Ciaramella, G.; et al. A lipid-encapsulated mRNA encoding a potently neutralizing human monoclonal antibody protects against chikungunya infection. Sci. Immunol. 2019, 4, eaaw6647. [Google Scholar] [CrossRef] [PubMed]
- Kormann, M.S.D.; Hasenpusch, G.; Aneja, M.K.; Nica, G.; Flemmer, A.W.; Herber-Jonat, S.; Huppmann, M.; Mays, L.E.; Illenyi, M.C.; Schams, A.; et al. Expression of therapeutic proteins after delivery of chemically modified mRNA in mice. Nat. Biotechnol. 2011, 29, 154–157. [Google Scholar] [CrossRef] [PubMed]
- Nabhan, J.F.; Wood, K.M.; Rao, V.P.; Morin, J.; Bhamidipaty, S.; Labranche, T.P.; Gooch, R.L.; Bozal, F.; Bulawa, C.E.; Guild, B.C. Intrathecal delivery of frataxin mRNA encapsulated in lipid nanoparticles to dorsal root ganglia as a potential therapeutic for Friedreich’s ataxia. Sci. Rep. 2016, 6, 20019. [Google Scholar] [CrossRef] [PubMed]
- DeRosa, F.; Guild, B.; Karve, S.; Smith, L.; Love, K.R.; Dorkin, J.R.; Kauffman, K.J.; Zhang, J.; Yahalom, B.; Anderson, D.G.; et al. Therapeutic efficacy in a hemophilia B model using a biosynthetic mRNA liver depot system. Gene Ther. 2016, 23, 699–707. [Google Scholar] [CrossRef] [PubMed]
- Ramaswamy, S.; Tonnu, N.; Tachikawa, K.; Limphong, P.; Vega, J.B.; Karmali, P.P.; Chivukula, P.; Verma, I.M. Systemic delivery of factor IX messenger RNA for protein replacement therapy. Proc. Natl. Acad. Sci. USA 2017, 114, E1941–E1950. [Google Scholar] [CrossRef]
- Robinson, E.; MacDonald, K.D.; Slaughter, K.; McKinney, M.; Patel, S.; Sun, C.; Sahay, G. Lipid Nanoparticle-Delivered Chemically Modified mRNA Restores Chloride Secretion in Cystic Fibrosis. Mol. Ther. 2018, 26, 2034–2046. [Google Scholar] [CrossRef] [PubMed]
- An, D.; Schneller, J.L.; Frassetto, A.; Liang, S.; Zhu, X.; Park, J.-S.; Theisen, M.; Hong, S.-J.; Zhou, J.; Rajendran, R.; et al. Systemic Messenger RNA Therapy as a Treatment for Methylmalonic Acidemia. Cell Rep. 2018, 24, 2520. [Google Scholar] [CrossRef]
- Prieve, M.G.; Harvie, P.; Monahan, S.D.; Roy, D.; Li, A.G.; Blevins, T.L.; Paschal, A.E.; Waldheim, M.; Bell, E.C.; Galperin, A.; et al. Targeted mRNA Therapy for Ornithine Transcarbamylase Deficiency. Mol. Ther. 2018, 26, 801–813. [Google Scholar] [CrossRef]
- Jiang, L.; Berraondo, P.; Jericó, D.; Guey, L.T.; Sampedro, A.; Frassetto, A.; Benenato, K.E.; Burke, K.; Santamaría, E.; Alegre, M.; et al. Systemic messenger RNA as an etiological treatment for acute intermittent porphyria. Nat. Med. 2018, 24, 1899–1909. [Google Scholar] [CrossRef]
- Liu-Chen, S.; Connolly, B.; Cheng, L.; Subramanian, R.R.; Han, Z. mRNA treatment produces sustained expression of enzymatically active human ADAMTS13 in mice. Sci. Rep. 2018, 8, 7859. [Google Scholar] [CrossRef] [PubMed]
- Apgar, J.F.; Tang, J.-P.; Singh, P.; Balasubramanian, N.; Burke, J.; Hodges, M.R.; Lasaro, M.A.; Lin, L.; Miliard, B.L.; Moore, K.; et al. Quantitative Systems Pharmacology Model of hUGT1A1-modRNA Encoding for the UGT1A1 Enzyme to Treat Crigler-Najjar Syndrome Type 1. CPT: Pharmacometrics Syst. Pharmacol. 2018, 7, 404–412. [Google Scholar] [CrossRef] [PubMed]
- Connolly, B.; Isaacs, C.; Cheng, L.; Asrani, K.H.; Subramanian, R.R. SERPINA1 mRNA as a Treatment for Alpha-1 Antitrypsin Deficiency. J. Nucleic Acids 2018, 2018, 1–7. [Google Scholar] [CrossRef]
- Roseman, D.S.; Khan, T.; Rajas, F.; Jun, L.S.; Asrani, K.H.; Isaacs, C.; Farelli, J.D.; Subramanian, R.R. G6PC mRNA Therapy Positively Regulates Fasting Blood Glucose and Decreases Liver Abnormalities in a Mouse Model of Glycogen Storage Disease 1a. Mol. Ther. 2018, 26, 814–821. [Google Scholar] [CrossRef]
- Asrani, K.H.; Cheng, L.; Cheng, C.J.; Subramanian, R.R. Arginase I mRNA therapy—A novel approach to rescue arginase 1 enzyme deficiency. RNA Biol. 2018, 15, 914–922. [Google Scholar] [CrossRef]
- Cao, J.; An, D.; Galduroz, M.; Zhuo, J.; Liang, S.; Eybye, M.; Frassetto, A.; Kuroda, E.; Funahashi, A.; Santana, J.; et al. mRNA Therapy Improves Metabolic and Behavioral Abnormalities in a Murine Model of Citrin Deficiency. Mol. Ther. 2019, 27, 1242–1251. [Google Scholar] [CrossRef]
- DeRosa, F.; Smith, L.; Shen, Y.; Huang, Y.; Pan, J.; Xie, H.; Yahalom, B.; Heartlein, M.W. Improved Efficacy in a Fabry Disease Model Using a Systemic mRNA Liver Depot System as Compared to Enzyme Replacement Therapy. Mol. Ther. 2019, 27, 878–889. [Google Scholar] [CrossRef] [PubMed]
- Hauser, S.; Poenisch, M.; Schelling, Y.; Höflinger, P.; Schuster, S.; Teegler, A.; Betten, R.; Gustafsson, J.-Å.; Hübener-Schmid, J.; Schlake, T.; et al. mRNA as a Novel Treatment Strategy for Hereditary Spastic Paraplegia Type 5. Mol. Ther. Methods Clin. Dev. 2019, 15, 359–370. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Yin, L.; Theisen, M.; Zhuo, J.; Siddiqui, S.; Levy, B.; Presnyak, V.; Frassetto, A.; Milton, J.; Salerno, T.; et al. Systemic mRNA Therapy for the Treatment of Fabry Disease: Preclinical Studies in Wild-Type Mice, Fabry Mouse Model, and Wild-Type Non-human Primates. Am. J. Hum. Genet. 2019, 104, 625–637. [Google Scholar] [CrossRef] [PubMed]
- Truong, B.; Allegri, G.; Liu, X.-B.; Burke, K.E.; Zhu, X.; Cederbaum, S.D.; Häberle, J.; Martini, P.G.V.; Lipshutz, G.S. Lipid nanoparticle-targeted mRNA therapy as a treatment for the inherited metabolic liver disorder arginase deficiency. Proc. Natl. Acad. Sci. USA 2019, 116, 21150–21159. [Google Scholar] [CrossRef]
- An, D.; Frassetto, A.; Jacquinet, E.; Eybye, M.; Milano, J.; DeAntonis, C.; Nguyen, V.; Laureano, R.; Milton, J.; Sabnis, S.; et al. Long-term efficacy and safety of mRNA therapy in two murine models of methylmalonic acidemia. EBioMedicine 2019, 45, 519–528. [Google Scholar] [CrossRef]
- Balakrishnan, B.; An, D.; Nguyen, V.; DeAntonis, C.; Martini, P.G.; Lai, K. Novel mRNA-Based Therapy Reduces Toxic Galactose Metabolites and Overcomes Galactose Sensitivity in a Mouse Model of Classic Galactosemia. Mol. Ther. 2020, 28, 304–312. [Google Scholar] [CrossRef]
- Karadagi, A.; Cavedon, A.G.; Zemack, H.; Nowak, G.; Eybye, M.E.; Zhu, X.; Guadagnin, E.; White, R.A.; Rice, L.M.; Frassetto, A.L.; et al. Systemic modified messenger RNA for replacement therapy in alpha 1-antitrypsin deficiency. Sci. Rep. 2020, 10, 1–11. [Google Scholar] [CrossRef]
- Elsabahy, M.; Wooley, K.L. Cytokines as biomarkers of nanoparticle immunotoxicity. Chem. Soc. Rev. 2013, 42, 5552–5576. [Google Scholar] [CrossRef]
- Breslin, S. Cytokine-Release Syndrome: Overview and Nursing Implications. Clin. J. Oncol. Nurs. 2007, 11, 37–41. [Google Scholar] [CrossRef] [PubMed]
- Fajgenbaum, D.C.; June, C.H. Cytokine Storm. N. Engl. J. Med. 2020, 383, 2255–2273. [Google Scholar] [CrossRef] [PubMed]
- Kleiner, G.; Marcuzzi, A.; Zanin, V.; Monasta, L.; Zauli, G. Cytokine Levels in the Serum of Healthy Subjects. Mediat. Inflamm. 2013, 2013, 1–6. [Google Scholar] [CrossRef]
- U.S. Department of Health and Human Services Food and Drug Administration. Guidance for Industry, Immunogenicity Assessment for Therapeutic Protein Products; FDA-2013-D-0092; Center for Drug Evaluation and Research, Center for Biologics Evaluation and Research: Silver Spring, MD, USA, 2014.
- Stebbings, R.; Findlay, L.; Edwards, C.; Eastwood, D.; Bird, C.; North, D.; Mistry, Y.; Dilger, P.; Liefooghe, E.; Cludts, I.; et al. “Cytokine Storm” in the Phase I Trial of Monoclonal Antibody TGN1412: Better Understanding the Causes to Improve PreClinical Testing of Immunotherapeutics. J. Immunol. 2007, 179, 3325–3331. [Google Scholar] [CrossRef] [PubMed]
- Sedic, M.; Senn, J.J.; Lynn, A.; Laska, M.; Smith, M.; Platz, S.J.; Bolen, J.; Hoge, S.; Bulychev, A.; Jacquinet, E.; et al. Safety Evaluation of Lipid Nanoparticle–Formulated Modified mRNA in the Sprague-Dawley Rat and Cynomolgus Monkey. Veter Pathol. 2017, 55, 341–354. [Google Scholar] [CrossRef] [PubMed]
- Sabnis, S.; Kumarasinghe, E.S.; Salerno, T.; Mihai, C.; Ketova, T.; Senn, J.J.; Lynn, A.; Bulychev, A.; McFadyen, I.; Chan, J.; et al. A Novel Amino Lipid Series for mRNA Delivery: Improved Endosomal Escape and Sustained Pharmacology and Safety in Non-human Primates. Mol. Ther. 2018, 26, 1509–1519. [Google Scholar] [CrossRef]
- Maugeri, M.; Nawaz, M.; Papadimitriou, A.; Angerfors, A.; Camponeschi, A.; Na, M.; Hölttä, M.; Skantze, P.; Johansson, S.; Sundqvist, M.; et al. Linkage between endosomal escape of LNP-mRNA and loading into EVs for transport to other cells. Nat. Commun. 2019, 10, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Karikó, K.; Muramatsu, H.; Welsh, F.A.; Ludwig, J.; Kato, H.; Akira, S.; Weissman, D. Incorporation of Pseudouridine Into mRNA Yields Superior Nonimmunogenic Vector With Increased Translational Capacity and Biological Stability. Mol. Ther. 2008, 16, 1833–1840. [Google Scholar] [CrossRef]
- Andries, O.; Mc Cafferty, S.; De Smedt, S.C.; Weiss, R.; Sanders, N.N.; Kitada, T. N1-methylpseudouridine-incorporated mRNA outperforms pseudouridine-incorporated mRNA by providing enhanced protein expression and reduced immunogenicity in mammalian cell lines and mice. J. Control. Release 2015, 217, 337–344. [Google Scholar] [CrossRef]
- Pardi, N.; Secreto, A.J.; Shan, X.; Debonera, F.; Glover, J.; Yi, Y.; Muramatsu, H.; Ni, H.; Mui, B.L.; Tam, Y.K.; et al. Administration of nucleoside-modified mRNA encoding broadly neutralizing antibody protects humanized mice from HIV-1 challenge. Nat. Commun. 2017, 8, 14630. [Google Scholar] [CrossRef]
- Richner, J.M.; Himansu, S.; Dowd, K.A.; Butler, S.L.; Salazar, V.; Fox, J.M.; Julander, J.G.; Tang, W.W.; Shresta, S.; Pierson, T.C.; et al. Modified mRNA Vaccines Protect against Zika Virus Infection. Cell 2017, 168, 1114–1125.e10. [Google Scholar] [CrossRef]
- Corbett, K.S.; Edwards, D.K.; Leist, S.R.; Abiona, O.M.; Boyoglu-Barnum, S.; Gillespie, R.A.; Himansu, S.; Schäfer, A.; Ziwawo, C.T.; DiPiazza, A.T.; et al. SARS-CoV-2 mRNA vaccine design enabled by prototype pathogen preparedness. Nature 2020, 586, 567–571. [Google Scholar] [CrossRef] [PubMed]
- Ramanathan, A.; Robb, G.B.; Chan, S.-H. mRNA capping: Biological functions and applications. Nucleic Acids Res. 2016, 44, 7511–7526. [Google Scholar] [CrossRef] [PubMed]
- Muthukrishnan, S.; Moss, B.; Cooper, J.A.; Maxwell, E.S. Influence of 5′-terminal cap structure on the initiation of translation of vaccinia virus mRNA. J. Biol. Chem. 1978, 253, 1710–1715. [Google Scholar] [CrossRef]
- Gingras, A.-C.; Raught, B.; Sonenberg, N. eIF4 Initiation Factors: Effectors of mRNA Recruitment to Ribosomes and Regulators of Translation. Annu. Rev. Biochem. 1999, 68, 913–963. [Google Scholar] [CrossRef]
- Wells, S.E.; Hillner, P.E.; Vale, R.D.; Sachs, A.B. Circularization of mRNA by Eukaryotic Translation Initiation Factors. Mol. Cell 1998, 2, 135–140. [Google Scholar] [CrossRef]
- Grudzien-Nogalska, E.; Kiledjian, M. New insights into decapping enzymes and selective mRNA decay. Wiley Interdiscip. Rev. RNA 2017, 8, e1379. [Google Scholar] [CrossRef] [PubMed]
- Sikorski, P.J.; Warminski, M.; Kubacka, D.; Ratajczak, T.; Nowis, D.; Kowalska, J.; Jemielity, J. The identity and methylation status of the first transcribed nucleotide in eukaryotic mRNA 5′ cap modulates protein expression in living cells. Nucleic Acids Res. 2020, 48, 1607–1626. [Google Scholar] [CrossRef] [PubMed]
- Grudzien-Nogalska, E.; Jemielity, J.; Kowalska, J.; Darzynkiewicz, E.; Rhoads, R.E. Phosphorothioate cap analogs stabilize mRNA and increase translational efficiency in mammalian cells. RNA 2007, 13, 1745–1755. [Google Scholar] [CrossRef] [PubMed]
- Kowalska, J.; Lewdorowicz, M.; Zuberek, J.; Grudzien-Nogalska, E.; Bojarska, E.; Stepinski, J.; Rhoads, R.E.; Darzynkiewicz, E.; Davis, R.E.; Jemielity, J. Synthesis and characterization of mRNA cap analogs containing phosphorothioate substitutions that bind tightly to eIF4E and are resistant to the decapping pyrophosphatase DcpS. RNA 2008, 14, 1119–1131. [Google Scholar] [CrossRef]
- Kuhn, A.N.; Diken, M.; Kreiter, S.; Selmi, A.; Kowalska, J.; Jemielity, J.; Darzynkiewicz, E.; Huber, C.; Türeci, Ö.; Sahin, U. Phosphorothioate cap analogs increase stability and translational efficiency of RNA vaccines in immature dendritic cells and induce superior immune responses in vivo. Gene Ther. 2010, 17, 961–971. [Google Scholar] [CrossRef]
- Vaidyanathan, S.; Azizian, K.T.; Haque, A.A.; Henderson, J.M.; Hendel, A.; Shore, S.; Antony, J.S.; Hogrefe, R.I.; Kormann, M.S.; Porteus, M.H.; et al. Uridine Depletion and Chemical Modification Increase Cas9 mRNA Activity and Reduce Immunogenicity without HPLC Purification. Mol. Ther. Nucleic Acids 2018, 12, 530–542. [Google Scholar] [CrossRef] [PubMed]
- Sahin, U.; Muik, A.; Vogler, I.; Derhovanessian, E.; Kranz, L.M.; Vormehr, M.; Quandt, J.; Bidmon, N.; Ulges, A.; Baum, A.; et al. BNT162b2 induces SARS-CoV-2-neutralising antibodies and T cells in humans. medRxiv 2020. [Google Scholar] [CrossRef]
- Mayya, V.K.; Duchaine, T.F. Ciphers and Executioners: How 3′-Untranslated Regions Determine the Fate of Messenger RNAs. Front. Genet. 2019, 10, 6. [Google Scholar] [CrossRef]
- Hinnebusch, A.G.; Ivanov, I.P.; Sonenberg, N. Translational control by 5′-untranslated regions of eukaryotic mRNAs. Science 2016, 352, 1413–1416. [Google Scholar] [CrossRef]
- Jain, R.; Frederick, J.P.; Huang, E.Y.; Burke, K.E.; Mauger, D.M.; Andrianova, E.A.; Farlow, S.J.; Siddiqui, S.; Pimentel, J.; Cheung-Ong, K.; et al. MicroRNAs Enable mRNA Therapeutics to Selectively Program Cancer Cells to Self-Destruct. Nucleic Acids Ther. 2018, 28, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Karikó, K.; Kuo, A.; Barnathan, E.S. Overexpression of urokinase receptor in mammalian cells following administration of the in vitro transcribed encoding mRNA. Gene Ther. 1999, 6, 1092–1100. [Google Scholar] [CrossRef]
- Holtkamp, S.; Kreiter, S.; Selmi, A.; Simon, P.; Koslowski, M.; Huber, C.; Türeci, O.; Sahin, U. Modification of antigen-encoding RNA increases stability, translational efficacy, and T-cell stimulatory capacity of dendritic cells. Blood 2006, 108, 4009–4017. [Google Scholar] [CrossRef]
- Asrani, K.H.; Farelli, J.D.; Stahley, M.R.; Miller, R.L.; Cheng, C.J.; Subramanian, R.R.; Brown, J.M. Optimization of mRNA untranslated regions for improved expression of therapeutic mRNA. RNA Biol. 2018, 15, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Sample, P.J.; Wang, B.; Reid, D.W.; Presnyak, V.; McFadyen, I.J.; Morris, D.R.; Seelig, G. Human 5′ UTR design and variant effect prediction from a massively parallel translation assay. Nat. Biotechnol. 2019, 37, 803–809. [Google Scholar] [CrossRef]
- von Niessen, A.G.O.; Poleganov, M.A.; Rechner, C.; Plaschke, A.; Kranz, L.M.; Fesser, S.; Diken, M.; Löwer, M.; Vallazza, B.; Beissert, T.; et al. Improving mRNA-Based Therapeutic Gene Delivery by Expression-Augmenting 3′ UTRs Identified by Cellular Library Screening. Mol. Ther. 2019, 27, 824–836. [Google Scholar] [CrossRef]
- Gustafsson, C.; Govindarajan, S.; Minshull, J. Codon bias and heterologous protein expression. Trends Biotechnol. 2004, 22, 346–353. [Google Scholar] [CrossRef] [PubMed]
- Mauro, V.P.; Chappell, S.A. A critical analysis of codon optimization in human therapeutics. Trends Mol. Med. 2014, 20, 604–613. [Google Scholar] [CrossRef]
- Nieuwkoop, T.; Finger-Bou, M.; Van Der Oost, J.; Claassens, N.J. The Ongoing Quest to Crack the Genetic Code for Protein Production. Mol. Cell 2020, 80, 193–209. [Google Scholar] [CrossRef] [PubMed]
- Gallie, D.R. The cap and poly(A) tail function synergistically to regulate mRNA translational efficiency. Genes Dev. 1991, 5, 2108–2116. [Google Scholar] [CrossRef]
- Pardi, N.; Muramatsu, H.; Weissman, D.; Karikó, K. In Vitro Transcription of Long RNA Containing Modified Nucleosides. Methods Mol. Biol. 2013, 969, 29–42. [Google Scholar] [CrossRef]
- Legnini, I.; Alles, J.; Karaiskos, N.; Ayoub, S.; Rajewsky, N. FLAM-seq: Full-length mRNA sequencing reveals principles of poly(A) tail length control. Nat. Methods 2019, 16, 879–886. [Google Scholar] [CrossRef] [PubMed]
- Subtelny, A.O.; Eichhorn, S.W.; Chen, G.R.; Sive, H.L.; Bartel, D.P. Poly(A)-tail profiling reveals an embryonic switch in translational control. Nat. Cell Biol. 2014, 508, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.; Lim, J.; Ha, M.; Kim, V.N. TAIL-seq: Genome-wide Determination of Poly(A) Tail Length and 3′ End Modifications. Mol. Cell 2014, 53, 1044–1052. [Google Scholar] [CrossRef]
- Wu, M.Z.; Asahara, H.; Tzertzinis, G.; Roy, B. Synthesis of low immunogenicity RNA with high-temperature in vitro transcription. RNA 2020, 26, 345–360. [Google Scholar] [CrossRef]
- Karikó, K.; Muramatsu, H.; Ludwig, J.; Weissman, D. Generating the optimal mRNA for therapy: HPLC purification eliminates immune activation and improves translation of nucleoside-modified, protein-encoding mRNA. Nucleic Acids Res. 2011, 39, e142. [Google Scholar] [CrossRef]
- Maier, M.A.; Jayaraman, M.; Matsuda, S.; Liu, J.; Barros, S.; Querbes, W.; Tam, Y.K.; Ansell, S.M.; Kumar, V.; Qin, J.; et al. Biodegradable Lipids Enabling Rapidly Eliminated Lipid Nanoparticles for Systemic Delivery of RNAi Therapeutics. Mol. Ther. 2013, 21, 1570–1578. [Google Scholar] [CrossRef]
- Bahl, K.; Senn, J.J.; Yuzhakov, O.; Bulychev, A.; Brito, L.A.; Hassett, K.J.; Laska, M.E.; Smith, M.; Almarsson, Ö.; Thompson, J.; et al. Preclinical and Clinical Demonstration of Immunogenicity by mRNA Vaccines against H10N8 and H7N9 Influenza Viruses. Mol. Ther. 2017, 25, 1316–1327. [Google Scholar] [CrossRef]
- Kauffman, K.J.; Dorkin, J.R.; Yang, J.H.; Heartlein, M.W.; DeRosa, F.; Mir, F.F.; Fenton, O.S.; Anderson, D.G. Optimization of Lipid Nanoparticle Formulations for mRNA Delivery in Vivo with Fractional Factorial and Definitive Screening Designs. Nano Lett. 2015, 15, 7300–7306. [Google Scholar] [CrossRef] [PubMed]
- Sago, C.D.; Lokugamage, M.P.; Paunovska, K.; Vanover, D.A.; Monaco, C.M.; Shah, N.N.; Castro, M.G.; Anderson, S.E.; Rudoltz, T.G.; Lando, G.N.; et al. High-throughput in vivo screen of functional mRNA delivery identifies nanoparticles for endothelial cell gene editing. Proc. Natl. Acad. Sci. USA 2018, 115, E9944–E9952. [Google Scholar] [CrossRef] [PubMed]
- Rampado, R.; Crotti, S.; Caliceti, P.; Pucciarelli, S.; Agostini, M. Recent Advances in Understanding the Protein Corona of Nanoparticles and in the Formulation of “Stealthy” Nanomaterials. Front. Bioeng. Biotechnol. 2020, 8, 166. [Google Scholar] [CrossRef] [PubMed]
- Saha, K.; Rahimi, M.; Yazdani, M.; Kim, S.T.; Moyano, D.F.; Hou, S.; Das, R.; Mout, R.; Rezaee, F.; Mahmoudi, M.; et al. Regulation of Macrophage Recognition through the Interplay of Nanoparticle Surface Functionality and Protein Corona. ACS Nano 2016, 10, 4421–4430. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Qin, J.; Jiang, Y.; Duncan, R.G.; Brigham, B.; Fishman, S.; Nair, J.K.; Akinc, A.; Barros, S.A.; Kasperkovitz, P.V. Shielding of Lipid Nanoparticles for siRNA Delivery: Impact on Physicochemical Properties, Cytokine Induction, and Efficacy. Mol. Ther. Nucleic Acids 2014, 3, e210. [Google Scholar] [CrossRef]
- Judge, A.; McClintock, K.; Phelps, J.R.; MacLachlan, I. Hypersensitivity and Loss of Disease Site Targeting Caused by Antibody Responses to PEGylated Liposomes. Mol. Ther. 2006, 13, 328–337. [Google Scholar] [CrossRef]
- Akinc, A.; Querbes, W.; De, S.; Qin, J.; Frank-Kamenetsky, M.; Jayaprakash, K.N.; Jayaraman, M.; Rajeev, K.G.; Cantley, W.L.; Dorkin, J.R.; et al. Targeted Delivery of RNAi Therapeutics with Endogenous and Exogenous Ligand-Based Mechanisms. Mol. Ther. 2010, 18, 1357–1364. [Google Scholar] [CrossRef]
- Wisse, E.; Jacobs, F.; Topal, B.; Frederik, P.; De Geest, B. The size of endothelial fenestrae in human liver sinusoids: Implications for hepatocyte-directed gene transfer. Gene Ther. 2008, 15, 1193–1199. [Google Scholar] [CrossRef] [PubMed]
- Gan, Z.; Lokugamage, M.P.; Hatit, M.Z.C.; Loughrey, D.; Paunovska, K.; Sato, M.; Cristian, A.; Dahlman, J.E. Nanoparticles containing constrained phospholipids deliver mRNA to liver immune cells in vivo without targeting ligands. Bioeng. Transl. Med. 2020, 5. [Google Scholar] [CrossRef]
- Zukancic, D.; Suys, E.J.A.; Pilkington, E.H.; Algarni, A.; Al-Wassiti, H.; Truong, N.P. The Importance of Poly(ethylene glycol) and Lipid Structure in Targeted Gene Delivery to Lymph Nodes by Lipid Nanoparticles. Pharmaceutics 2020, 12, 1068. [Google Scholar] [CrossRef]
- Ramishetti, S.; Kedmi, R.; Goldsmith, M.; Leonard, F.; Sprague, A.G.; Godin, B.; Gozin, M.; Cullis, P.R.; Dykxhoorn, D.M.; Peer, D. Systemic Gene Silencing in Primary T Lymphocytes Using Targeted Lipid Nanoparticles. ACS Nano 2015, 9, 6706–6716. [Google Scholar] [CrossRef]
- Veiga, N.; Goldsmith, M.; Granot, Y.; Rosenblum, D.; Dammes, N.; Kedmi, R.; Ramishetti, S.; Peer, D. Cell specific delivery of modified mRNA expressing therapeutic proteins to leukocytes. Nat. Commun. 2018, 9, 1–9. [Google Scholar] [CrossRef]
- Ramishetti, S.; Hazan-Halevy, I.; Palakuri, R.; Chatterjee, S.; Gonna, S.N.; Dammes, N.; Freilich, I.; Shmuel, L.K.; Danino, D.; Peer, D. A Combinatorial Library of Lipid Nanoparticles for RNA Delivery to Leukocytes. Adv. Mater. 2020, 32, e1906128. [Google Scholar] [CrossRef] [PubMed]
- Marcos-Contreras, O.A.; Greineder, C.F.; Kiseleva, R.Y.; Parhiz, H.; Walsh, L.R.; Zuluaga-Ramirez, V.; Myerson, J.W.; Hood, E.D.; Villa, C.H.; Tombacz, I.; et al. Selective targeting of nanomedicine to inflamed cerebral vasculature to enhance the blood–brain barrier. Proc. Natl. Acad. Sci. USA 2020, 117, 3405–3414. [Google Scholar] [CrossRef] [PubMed]
- Parhiz, H.; Shuvaev, V.V.; Pardi, N.; Khoshnejad, M.; Kiseleva, R.Y.; Brenner, J.S.; Uhler, T.; Tuyishime, S.; Mui, B.L.; Tam, Y.K.; et al. PECAM-1 directed re-targeting of exogenous mRNA providing two orders of magnitude enhancement of vascular delivery and expression in lungs independent of apolipoprotein E-mediated uptake. J. Control. Release 2018, 291, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zaifman, J.; Kulkarni, J.A.; Zhigaltsev, I.V.; Tam, Y.K.; Ciufolini, M.A.; Tam, Y.Y.C.; Cullis, P.R. Dexamethasone prodrugs as potent suppressors of the immunostimulatory effects of lipid nanoparticle formulations of nucleic acids. J. Control. Release 2018, 286, 46–54. [Google Scholar] [CrossRef]
- Zhong, Z.; Mc Cafferty, S.; Combes, F.; Huysmans, H.; De Temmerman, J.; Gitsels, A.; Vanrompay, D.; Catani, J.P.; Sanders, N.N. mRNA therapeutics deliver a hopeful message. Nano Today 2018, 23, 16–39. [Google Scholar] [CrossRef]
- Beck, J.D.; Reidenbach, D.; Salomon, N.; Sahin, U.; Türeci, Ö.; Vormehr, M.; Kranz, L.M. mRNA therapeutics in cancer immunotherapy. Mol. Cancer 2021, 20, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Blakney, A.; Ip, S.; Geall, A. An Update on Self-Amplifying mRNA Vaccine Development. Vaccines 2021, 9, 97. [Google Scholar] [CrossRef] [PubMed]
- Bloom, K.; Berg, F.V.D.; Arbuthnot, P. Self-amplifying RNA vaccines for infectious diseases. Gene Ther. 2021, 28, 117–129. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Delivery | mRNA | Disease | Company | Safety Data | Reference |
|---|---|---|---|---|---|
| LNP | Frataxin (FXN) | Friedreich’s ataxia | Pfizer | no | Nabhan et al., Scientific Reports, 2016 [81] |
| LNP | Factor IX | Hemophilia B | Shire | no | DeRosa F. et al., Gene Therapy, 2016 [82] |
| LNP | Factor IX | Hemophilia B | Arcturus | cytokines, liver toxicity, liver histopathology | Ramaswamy S. et al., PNAS 3, 2017 [83] |
| LNP | cystic fibrosis trans-membrane conductance regulator (CFTR) | Cystic Fibrosis | Arcturus, Translate Bio | no | Robinson et al., Mol Therapy, 2018 [84] |
| LNP | methylmalonyl-CoA mutase (MUT) | Methylmalonic Acidemia | Moderna | cytokines, ADA 1, liver toxicity | An D. et al., Cell reports, 2018 [85] |
| LNP/ HMT 2 | ornithine transcarbamylase (OTC) | OTC Deficiency | PhaseRx | cytokines, liver toxicity, liver histopathology | Prieve M. et al., Molecular Therapy, 2018 [86] |
| LNP | porphobilinogen deaminase (PBGD) | Acute intermittent porphyria | Moderna | liver toxicity, ADA 1 | Jiang L. et al., Nature Medicine, 2018 [87] |
| LNP | disintegrin and metalloprotease with thrombospondin type 1 repeats, member 13 (ADAMTS13) | Thrombotic thrombocytopenic purpura | Alexion | no | Liu-Chen S. et al., Scientific Reports, 2018 [88] |
| LNP | uridine-diphosphateglucuronosyltransferase (UGT1A1) | Crigler-Najjar Syndrome Type 1 | Alexion | no | Apgar J. et al., CPT Pharmacometrics Syst. Pharmacol, 2018 [89] |
| LNP | serine protease inhibitor, group A, member 1 (SERPINA1) | Alpha-1 Antitrypsin Deficiency | Alexion | no | Connolly B. et al., Journal of Nucleic Acids, 2018 [90] |
| LNP | glucose-6-phosphatase (G6Pase) | Glycogen storage disease type Ia | Alexion | no | Roseman D. et al., Molecular Therapy, 2018 [91] |
| LNP | arginase I (ARG1) | Arginase I deficiency | Alexion | no | Asrani et al., RNA Biology, 2018 [92] |
| LNP | citrin (aspartate/glutamate transporter) | Citrin deficiency | Moderna | no | Cao J. et al., Molecular Therapy, 2019 [93] |
| LNP | alpha galactosidase A (a-Gal A) | Fabry Disease | Translate Bio, Shire | no | De Rosa et al., Molecular Therapy, 2019 [94] |
| LNP | oxysterol 7-a-hydroxylase (CYP7B1) | Hereditary Spastic Paraplegia Type 5 | CureVac | liver toxicity | Hauser S. et al., Molecular Therapy, 2019 [95] |
| LNP | alpha galactosidase A (a-Gal A) | Fabry Disease | Moderna | liver toxicity, ADA 1 | Zhu et al., The American Journal of Human Genetics, 2019 [96] |
| LNP | arginase 1 (ARG1) | Arginase deficiency | Moderna | liver histopathology | Truong B. et al., PNAS3, 2019 [97] |
| LNP | methylmalonyl-CoA mutase (MUT) | Methylmalonic Acidemia | Moderna | liver toxicity, liver histopathology | An et al., EbioMedicine, 2019 [98] |
| LNP | galactose-1 phosphate uridylyltransferase (GALT) | Galactosemia | Moderna | no | Balakrishnan B. et al., Molecular Therapy, 2020 [99] |
| LNP | serine protease inhibitor, group A, member 1 (SERPINA1) | Alpha-1 Antitrypsin Deficiency | Moderna | liver toxicity, liver histopathology | Karadagi A. et al., Scientific Reports, 2020 [100] |
| Drug Candidate | Animal, Dose, Time | Cytokines/Chemokines | Significant Upregulation Compared to Control | Assay | Reference |
|---|---|---|---|---|---|
| LNP-Factor IX mRNA | mouse, 4 mg/kg i.v.; 4 h, 7 h, 24 h and 48 h after third dose | IL-1α, IL-1β, IL-2, IL-3, IL-4, IL-5, MIP-1α, IL-10, IL-12 p40, IL-12 p70, IL-13, IL-17α, G-CSF, GM-CSF, IFNγ, KC, MCP-1, MIP-1β, RANTES, TNFα, IL-6, Eotaxin | yes (4 h and 7 h): G-CSF, MCP-1, MIP-1β, IL-6, RANTES; no (24 h and 48 h) | Biorad multiplex | Ramaswamy S. et al., PNAS, 2017 [83] |
| LNP-MUT 1 mRNA | mouse, 0.2 mg/kg i.v.; 24 h after third or fifth weekly dose | IL-6, IFNγ, TNFα, IL-1β | no (24 h) | MSD 4 multiplex | An D., et al., Cell reports, 2018 [85] |
| LNP/HMT 2 OTC 3 mRNA | mouse, 3 mg/kg i.v.; 3 h and 24 h after ninth repeat dose | IL-6, IL-12, GM-CSF, IFNγ, TNFα, CXCL10, MCP-1 | yes (3 h and 24 h): IL-12 | Luminex multiplex, ELISA 5 (CXCL10) | Prieve M. et al., Molecular Therapy, 2018 [86] |
| Candidate | Biological Target | Disease | Company | Year Start | Clinical Phase | Number |
|---|---|---|---|---|---|---|
| MRT5005 | CFTR 1 | Cystic Fibrosis | Translate Bio | 2017 | Phase 1/2 | NCT03375047 |
| MRT5201 | OTC 2 | OTC Deficiency | Translate Bio | 2018 | Phase 1/2 | NCT03767270 (program discontinued) |
| mRNA-3704 | MUT 3 | Methylmalonic Acidemia | ModernaTX, Inc. | 2019 | Phase 1/2 | NCT03810690 EU 2019-001061-32 (Terminated due to business decision) |
| mRNA-3927 | PCCA and PCCB 4 | Propionic Acidemia | ModernaTX, Inc. | 2019 (US), 2020 (EU) | Phase 1/2 | NCT04159103 (not yet recruiting) EU 2019-003529-36 |
| ARCT-810 | OTC 2 | OTC Deficiency | Arcturus | 2020 | Phase 1 |
NCT04416126 (completed, healthy adult subjects)
NCT04442347 (recruiting) |
| Drug Candidate | Biological Target | Disease | Company | Website |
|---|---|---|---|---|
| LUNAR-CF | CFTR 1 | Cystic Fibrosis | Arcturus | https://arcturusrx.com/pipeline/ |
| LUNAR-CV | undisclosed | rare cardiovascular disease | Arcturus | https://arcturusrx.com/pipeline/ |
| undisclosed | CFTR 1 | Cystic Fibrosis | Translate Bio | https://translate.bio/pipeline/ |
| undisclosed | undisclosed | Primary Ciliary Dyskinesia | Translate Bio | https://translate.bio/pipeline/ |
| undisclosed | undisclosed | Pulmonary Arterial Hypertension | Translate Bio | https://translate.bio/pipeline/ |
| undisclosed | undisclosed | Idiopathic Pulmonary Fibrosis | Translate Bio | https://translate.bio/pipeline/ |
| undisclosed | undisclosed | Ocular diseases | CureVac | https://www.curevac.com/en/pipeline/ |
| undisclosed | undisclosed | Lung respiratory diseases | CureVac | https://www.curevac.com/en/pipeline/ |
| BNT171 | undisclosed | undisclosed | BioNTech/Genevant | https://biontech.de/de/science/pipeline |
| 4 rare disease indications | undisclosed | undisclosed | BioNTech/Genevant | https://biontech.de/de/science/pipeline |
| mRNA-3283 | PAH 2 | Phenylketonuria | Moderna | https://www.modernatx.com/pipeline |
| mRNA-3745 | G6Pase 3 | Glycogen Storage Disorder Type 1a | Moderna | https://www.modernatx.com/pipeline |
| AZD7970 | Relaxin-2 | Heart Failure | Moderna/AstraZeneca | https://www.modernatx.com/pipeline |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vlatkovic, I. Non-Immunotherapy Application of LNP-mRNA: Maximizing Efficacy and Safety. Biomedicines 2021, 9, 530. https://doi.org/10.3390/biomedicines9050530
Vlatkovic I. Non-Immunotherapy Application of LNP-mRNA: Maximizing Efficacy and Safety. Biomedicines. 2021; 9(5):530. https://doi.org/10.3390/biomedicines9050530
Chicago/Turabian StyleVlatkovic, Irena. 2021. "Non-Immunotherapy Application of LNP-mRNA: Maximizing Efficacy and Safety" Biomedicines 9, no. 5: 530. https://doi.org/10.3390/biomedicines9050530
APA StyleVlatkovic, I. (2021). Non-Immunotherapy Application of LNP-mRNA: Maximizing Efficacy and Safety. Biomedicines, 9(5), 530. https://doi.org/10.3390/biomedicines9050530