
Strategies for the Improvement of Metal-Based Chemotherapeutic Treatments

 ,
,  ,
,  , and
, and 
Abstract
1. Introduction
2. The Strategies
2.1. Drug Repurposing
The Case of Auranofin
2.2. Simple Modification of the Chemical Structures of Approved Metal-Based Drugs
The Case of Cis-PtI2(NH3)2 and Au(PEt3)I
2.3. Testing Novel Drug Combinations
The Case of Cisplatin and Riluzole Combination Therapy
2.4. Newly Synthesized Complexes Coupling Different Anticancer Drugs
The Case of Arsenoplatin-1
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Barry, N.P.E.; Sadler, P.J. Exploration of the medical periodic table: Towards new targets. Chem. Commun. 2013, 49, 5106–5131. [Google Scholar] [CrossRef]
- Lemire, J.A.; Harrison, J.J.; Turner, R.J. Antimicrobial activity of metals: Mechanisms, molecular targets and applications. Nat. Rev. Microbiol. 2013, 11, 371–384. [Google Scholar] [CrossRef]
- Barry, N.P.E.; Sadler, P.J. 100 years of metal coordination chemistry: From Alfred Werner to anticancer metallodrugs. Pure Appl. Chem. 2014, 86, 1897–1910. [Google Scholar] [CrossRef]
- Wang, R.; Lai, T.P.; Gao, P.; Zhang, H.; Ho, P.L.; Woo, P.C.Y.; Ma, G.; Kao, R.Y.T.; Li, H.; Sun, H. Bismuth antimicrobial drugs serve as broad-spectrum metallo-β-lactamase inhibitors. Nat. Commun. 2018. [Google Scholar] [CrossRef]
- Cirri, D.; Pratesi, A.; Marzo, T.; Messori, L. Metallo therapeutics for COVID-19. Exploiting metal-based compounds for the discovery of new antiviral drugs. Expert Opinon Drug Discov. 2021, 16, 39–46. [Google Scholar] [CrossRef]
- Hambley, T.W. Chemistry: Metal-based therapeutics. Science 2007, 318, 1392–1393. [Google Scholar] [CrossRef]
- Johnstone, T.C.; Suntharalingam, K.; Lippard, S.J. The Next Generation of Platinum Drugs: Targeted Pt(II) Agents, Nanoparticle Delivery, and Pt(IV) Prodrugs. Chem. Rev. 2016, 116, 3436–3486. [Google Scholar] [CrossRef]
- Johnstone, T.C.; Park, G.Y.; Lippard, S.J. Understanding and improving platinum anticancer drugs—Phenanthriplatin. Anticancer Res. 2014, 34, 471–476. [Google Scholar] [PubMed]
- Pedrosa, P.; Carvalho, A.; Baptista, P.V.; Fernandes, A.R. Inorganic Coordination Chemistry: Where We Stand in Cancer Treatment? In Basic Concepts Viewed from Frontier in Inorganic Coordination Chemistry; IntechOpen: London, UK, 2018. [Google Scholar]
- Monro, S.; Colón, K.L.; Yin, H.; Roque, J.; Konda, P.; Gujar, S.; Thummel, R.P.; Lilge, L.; Cameron, C.G.; McFarland, S.A. Transition Metal Complexes and Photodynamic Therapy from a Tumor-Centered Approach: Challenges, Opportunities, and Highlights from the Development of TLD1433. Chem. Rev. 2019, 119, 797–828. [Google Scholar] [CrossRef] [PubMed]
- Boros, E.; Packard, A.B. Radioactive Transition Metals for Imaging and Therapy. Chem. Rev. 2019, 119, 870–901. [Google Scholar] [CrossRef] [PubMed]
- Boros, E.; Dyson, P.J.; Gasser, G. Classification of Metal-Based Drugs according to Their Mechanisms of Action. Chem 2020, 6, 41–60. [Google Scholar] [CrossRef] [PubMed]
- Karadağ, A.; Korkmaz, N.; Aydın, A.; Akbaş, H.; Tekin, Ş.; Yerli, Y.; Şen, F. Metalo components exhibiting significant anticancer and antibacterial properties: A novel sandwich-type like polymeric structure. Sci. Rep. 2020. [Google Scholar] [CrossRef] [PubMed]
- Anthony, E.J.; Bolitho, E.M.; Bridgewater, H.E.; Carter, O.W.L.; Donnelly, J.M.; Imberti, C.; Lant, E.C.; Lermyte, F.; Needham, R.J.; Palau, M.; et al. Metallodrugs are unique: Opportunities and challenges of discovery and development. Chem. Sci. 2020, 11, 12888–12917. [Google Scholar] [CrossRef]
- Siddik, Z.H. Cisplatin: Mode of cytotoxic action and molecular basis of resistance. Oncogene 2003, 22, 7265–7279. [Google Scholar] [CrossRef] [PubMed]
- Morrison, C.N.; Prosser, K.E.; Stokes, R.W.; Cordes, A.; Metzler-Nolte, N.; Cohen, S.M. Expanding medicinal chemistry into 3D space: Metallofragments as 3D scaffolds for fragment-based drug discovery. Chem. Sci. 2020. [Google Scholar] [CrossRef]
- Komeda, S.; Casini, A. Next-Generation Anticancer Metallodrugs. Curr. Top. Med. Chem. 2012, 12, 219–235. [Google Scholar] [CrossRef]
- Ott, I. On the medicinal chemistry of gold complexes as anticancer drugs. Coord. Chem. Rev. 2009, 253, 1670–1681. [Google Scholar] [CrossRef]
- Barresi, E.; Tolbatov, I.; Pratesi, A.; Notarstefano, V.; Baglini, E.; Daniele, S.; Taliani, S.; Re, N.; Giorgini, E.; Martini, C.; et al. A mixed-valence diruthenium(II,III) complex endowed with high stability: From experimental evidence to theoretical interpretation. Dalton Trans. 2020, 49, 14520–14527. [Google Scholar] [CrossRef] [PubMed]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2018, 18, 41–58. [Google Scholar] [CrossRef]
- Roder, C.; Thomson, M.J. Auranofin: Repurposing an Old Drug for a Golden New Age. Drugs R D 2015, 15, 13–20. [Google Scholar] [CrossRef]
- Giorgio, A.; Merlino, A. Gold metalation of proteins: Structural studies. Coord. Chem. Rev. 2020, 407, 213175. [Google Scholar] [CrossRef]
- Pratesi, A.; Cirri, D.; Ciofi, L.; Messori, L. Reactions of Auranofin and Its Pseudohalide Derivatives with Serum Albumin Investigated through ESI-Q-TOF MS. Inorg. Chem. 2018, 57, 10507–10510. [Google Scholar] [CrossRef] [PubMed]
- Zoppi, C.; Messori, L.; Pratesi, A. ESI MS studies highlight the selective interaction of Auranofin with protein free thiols. Dalton Trans. 2020, 49, 5906–5913. [Google Scholar] [CrossRef]
- Pratesi, A.; Gabbiani, C.; Michelucci, E.; Ginanneschi, M.; Papini, A.M.; Rubbiani, R.; Ott, I.; Messori, L. Insights on the mechanism of thioredoxin reductase inhibition by Gold N-heterocyclic carbene compounds using the synthetic linear Selenocysteine containing C-terminal peptide hTrxR(488-499): An ESI-MS investigation. J. Inorg. Biochem. 2014, 136, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Selvaraju, K.; Saei, A.A.; D’Arcy, P.; Zubarev, R.A.; Arnér, E.S.; Linder, S. Repurposing of auranofin: Thioredoxin reductase remains a primary target of the drug. Biochimie 2019, 162, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Fabbrini, M.G.; Cirri, D.; Pratesi, A.; Ciofi, L.; Marzo, T.; Guerri, A.; Nistri, S.; Dell’Accio, A.; Gamberi, T.; Severi, M.; et al. A Fluorescent Silver(I) Carbene Complex with Anticancer Properties: Synthesis, Characterization, and Biological Studies. ChemMedChem 2019, 14, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Marzo, T.; Cirri, D.; Gabbiani, C.; Gamberi, T.; Magherini, F.; Pratesi, A.; Guerri, A.; Biver, T.; Binacchi, F.; Stefanini, M.; et al. Auranofin, Et3PAuCl, and Et3PAuI Are Highly Cytotoxic on Colorectal Cancer Cells: A Chemical and Biological Study. ACS Med. Chem. Lett. 2017, 8, 997–1001. [Google Scholar] [CrossRef] [PubMed]
- Pratesi, A.; Gabbiani, C.; Ginanneschi, M.; Messori, L. Reactions of medicinally relevant gold compounds with the C-terminal motif of thioredoxin reductase elucidated by MS analysis. Chem. Commun. 2010, 46, 7001–7003. [Google Scholar] [CrossRef]
- Ventola, C.L. The antibiotic resistance crisis: Part 1: Causes and threats. Phys. Ther. 2015, 40, 277–283. [Google Scholar]
- Pricker, S.P. Medical uses of gold compounds: Past, present and future. Gold Bull. 1996, 29, 53–60. [Google Scholar] [CrossRef]
- Corti, C.; Holliday, R.; Holliday, R. Gold—Science and Applications; Corti, C., Holliday, R., Eds.; CRC Press: Boca Raton, FL, USA, 2009; ISBN 9780429141614. [Google Scholar]
- Madeira, J.M.; Gibson, D.L.; Kean, W.F.; Klegeris, A. The biological activity of auranofin: Implications for novel treatment of diseases. Inflammopharmacology 2012, 20, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Cassetta, M.I.; Marzo, T.; Fallani, S.; Novelli, A.; Messori, L. Drug repositioning: Auranofin as a prospective antimicrobial agent for the treatment of severe staphylococcal infections. BioMetals 2014, 27, 787–791. [Google Scholar] [CrossRef]
- Jackson-Rosario, S.E.; Self, W.T. Targeting selenium metabolism and selenoproteins: Novel avenues for drug discovery. Metallomics 2010, 2, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Jackson-Rosario, S.; Cowart, D.; Myers, A.; Tarrien, R.; Levine, R.L.; Scott, R.A.; Self, W.T. Auranofin disrupts selenium metabolism in Clostridium difficile by forming a stable Au-Se adduct. J. Biol. Inorg. Chem. 2009, 14, 507–519. [Google Scholar] [CrossRef]
- Thangamani, S.; Mohammad, H.; Abushahba, M.F.N.; Sobreira, T.J.P.; Hedrick, V.E.; Paul, L.N.; Seleem, M.N. Antibacterial activity and mechanism of action of auranofin against multi-drug resistant bacterial pathogens. Sci. Rep. 2016, 6, 1–13. [Google Scholar] [CrossRef]
- Marzo, T.; Cirri, D.; Pollini, S.; Prato, M.; Fallani, S.; Cassetta, M.I.; Novelli, A.; Rossolini, G.M.; Messori, L. Auranofin and its Analogues Show Potent Antimicrobial Activity against Multidrug-Resistant Pathogens: Structure-Activity Relationships. ChemMedChem 2018, 13, 2448–2454. [Google Scholar] [CrossRef] [PubMed]
- Jackson-Rosario, S.; Self, W.T. Inhibition of selenium metabolism in the oral pathogen Treponema denticola. J. Bacteriol. 2009, 191, 4035–4040. [Google Scholar] [CrossRef] [PubMed]
- Tejman-Yarden, N.; Miyamoto, Y.; Leitsch, D.; Santini, J.; Debnath, A.; Gut, J.; McKerrow, J.H.; Reed, S.L.; Eckmann, L. A reprofiled drug, auranofin, is effective against metronidazole-resistant Giardia lamblia. Antimicrob. Agents Chemother. 2013, 57, 2029–2035. [Google Scholar] [CrossRef]
- Savarino, A.; Shytaj, I.L. Chloroquine and beyond: Exploring anti-rheumatic drugs to reduce immune hyperactivation in HIV/AIDS. Retrovirology 2015, 12, 51. [Google Scholar] [CrossRef]
- Marzo, T.; Messori, L. A Role for Metal-Based Drugs in Fighting COVID-19 Infection? The Case of Auranofin. ACS Med. Chem. Lett. 2020, 11, 1067–1068. [Google Scholar] [CrossRef]
- Guarra, F.; Pratesi, A.; Gabbiani, C.; Biver, T. A focus on the biological targets for coinage metal-NHCs as potential anticancer complexes. J. Inorg. Biochem. 2021, 217, 111355. [Google Scholar] [CrossRef]
- Binacchi, F.; Guarra, F.; Cirri, D.; Marzo, T.; Pratesi, A.; Messori, L.; Gabbiani, C.; Biver, T. On the Different Mode of Action of Au(I)/Ag(I)-NHC Bis-Anthracenyl Complexes Towards Selected Target Biomolecules. Molecules 2020, 25, 5446. [Google Scholar] [CrossRef] [PubMed]
- Dilruba, S.; Kalayda, G.V. Platinum-based drugs: Past, present and future. Cancer Chemother. Pharmacol. 2016, 77, 1103–1124. [Google Scholar] [CrossRef]
- Messori, L.; Marzo, T.; Merlino, A. Interactions of carboplatin and oxaliplatin with proteins: Insights from X-ray structures and mass spectrometry studies of their ribonuclease A adducts. J. Inorg. Biochem. 2015, 153, 136–142. [Google Scholar] [CrossRef]
- Yeo, C.; Ooi, K.; Tiekink, E. Gold-Based Medicine: A Paradigm Shift in Anti-Cancer Therapy? Molecules 2018, 23, 1410. [Google Scholar] [CrossRef]
- Marzo, T.; Ferraro, G.; Merlino, A.; Messori, L. Protein Metalation by Inorganic Anticancer Drugs. In Encyclopedia of Inorganic and Bioinorganic Chemistry; John Wiley & Sons: Hoboken, NJ, USA, 2020; pp. 1–17. [Google Scholar]
- Ludwig, T.; Riethmüller, C.; Gekle, M.; Schwerdt, G.; Oberleithner, H. Nephrotoxicity of platinum complexes is related to basolateral organic cation transport. Kidney Int. 2004, 66, 196–202. [Google Scholar] [CrossRef]
- Bruno, P.M.; Liu, Y.; Park, G.Y.; Murai, J.; Koch, C.E.; Eisen, T.J.; Pritchard, J.R.; Pommier, Y.; Lippard, S.J.; Hemann, M.T. A subset of platinum-containing chemotherapeutic agents kills cells by inducing ribosome biogenesis stress. Nat. Med. 2017, 23, 461–471. [Google Scholar] [CrossRef]
- Marzo, T.; Pratesi, A.; Cirri, D.; Pillozzi, S.; Petroni, G.; Guerri, A.; Arcangeli, A.; Messori, L.; Gabbiani, C. Chlorido and bromido oxaliplatin analogues as potential agents for CRC treatment: Solution behavior, protein binding and cytotoxicity evaluation. Inorg. Chim. Acta 2018, 470, 318–324. [Google Scholar] [CrossRef]
- Cirri, D.; Pillozzi, S.; Gabbiani, C.; Tricomi, J.; Bartoli, G.; Stefanini, M.; Michelucci, E.; Arcangeli, A.; Messori, L.; Marzo, T. PtI2(DACH), the iodido analogue of oxaliplatin as a candidate for colorectal cancer treatment: Chemical and biological features. Dalton Trans. 2017, 46, 3311–3317. [Google Scholar] [CrossRef]
- Dhara, S.C. A rapid method for the synthesis of cis-[Pt(NH3)2Cl2]. Indian J. Chem. 1970, 8, 193–194. [Google Scholar]
- Marzo, T.; Pillozzi, S.; Hrabina, O.; Kasparkova, J.; Brabec, V.; Arcangeli, A.; Bartoli, G.; Severi, M.; Lunghi, A.; Totti, F.; et al. Cis-Pt I2(NH3)2: A reappraisal. Dalton Trans. 2015, 44, 14896–14905. [Google Scholar] [CrossRef]
- Dabrowiak, J.C. Metals in Medicine, 2nd ed.; Atwood, D.A., Meyer, G., Crabtree, R.H., Woollins, J.D., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2017; ISBN 9781119191377. [Google Scholar]
- Messori, L.; Marzo, T.; Gabbiani, C.; Valdes, A.A.; Quiroga, A.G.; Merlino, A. Peculiar features in the crystal structure of the adduct formed between cis-PtI2(NH3)2 and hen egg white lysozyme. Inorg. Chem. 2013, 52, 13827–13829. [Google Scholar] [CrossRef]
- Marzo, T.; Navas, F.; Cirri, D.; Merlino, A.; Ferraro, G.; Messori, L.; Quiroga, A.G. Reactions of a tetranuclear Pt-thiosemicarbazone complex with model proteins. J. Inorg. Biochem. 2018, 181, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Merlino, A.; Marzo, T.; Messori, L. Protein Metalation by Anticancer Metallodrugs: A Joint ESI MS and XRD Investigative Strategy. Chem. Eur. J. 2017, 23, 6942–6947. [Google Scholar] [CrossRef] [PubMed]
- Tolbatov, I.; Marzo, T.; Cirri, D.; Gabbiani, C.; Coletti, C.; Marrone, A.; Paciotti, R.; Messori, L.; Re, N. Reactions of cisplatin and cis-[PtI2(NH3)2] with molecular models of relevant protein sidechains: A comparative analysis. J. Inorg. Biochem. 2020, 209, 111096. [Google Scholar] [CrossRef]
- Musumeci, D.; Platella, C.; Riccardi, C.; Merlino, A.; Marzo, T.; Massai, L.; Messori, L.; Montesarchio, D. A first-in-class and a fished out anticancer platinum compound: Cis-[PtCl2(NH3)2] and cis-[PtI2(NH3)2] compared for their reactivity towards DNA model systems. Dalton Trans. 2016, 45, 8587–8600. [Google Scholar] [CrossRef] [PubMed]
- Marzo, T.; Massai, L.; Pratesi, A.; Stefanini, M.; Cirri, D.; Magherini, F.; Becatti, M.; Landini, I.; Nobili, S.; Mini, E.; et al. Replacement of the Thiosugar of Auranofin with Iodide Enhances the Anticancer Potency in a Mouse Model of Ovarian Cancer. ACS Med. Chem. Lett. 2019, 10, 656–660. [Google Scholar] [CrossRef]
- Loree, J.M.; Sha, A.; Soleimani, M.; Kennecke, H.F.; Ho, M.Y.; Cheung, W.Y.; Mulder, K.E.; Abadi, S.; Spratlin, J.L.; Gill, S. Survival Impact of CAPOX Versus FOLFOX in the Adjuvant Treatment of Stage III Colon Cancer. Clin. Colorectal Cancer 2018, 17, 156–163. [Google Scholar] [CrossRef]
- Li, X.; Ng, A.S.N.; Mak, V.C.Y.; Chan, K.K.L.; Cheung, A.N.Y.; Cheung, L.W.T. Strategic Combination Therapies for Ovarian Cancer. Curr. Cancer Drug Targets 2020, 20, 573–585. [Google Scholar] [CrossRef]
- Lee, M.-W.; Ryu, H.; Song, I.-C.; Yun, H.-J.; Jo, D.-Y.; Ko, Y.B.; Lee, H.-J. Efficacy of cisplatin combined with topotecan in patients with advanced or recurrent ovarian cancer as second- or higher-line palliative chemotherapy. Medicine 2020, 99, e19931. [Google Scholar] [CrossRef]
- Pignata, S.; Scambia, G.; Katsaros, D.; Gallo, C.; Pujade-Lauraine, E.; De Placido, S.; Bologna, A.; Weber, B.; Raspagliesi, F.; Panici, P.B.; et al. Carboplatin plus paclitaxel once a week versus every 3 weeks in patients with advanced ovarian cancer (MITO-7): A randomised, multicentre, open-label, phase 3 trial. Lancet Oncol. 2014, 15, 396–405. [Google Scholar] [CrossRef]
- Mullen, M.M.; Kuroki, L.M.; Thaker, P.H. Novel treatment options in platinum-sensitive recurrent ovarian cancer: A review. Gynecol. Oncol. 2019, 152, 416–425. [Google Scholar] [CrossRef] [PubMed]
- Nishida, N.; Yano, H.; Nishida, T.; Kamura, T.; Kojiro, M. Angiogenesis in cancer. Vasc. Health Risk Manag. 2006, 2, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Kalra, M.; Tong, Y.; Jones, D.R.; Walsh, T.; Danso, M.A.; Ma, C.X.; Silverman, P.; King, M.-C.; Badve, S.S.; Perkins, S.M.; et al. Cisplatin +/− rucaparib after preoperative chemotherapy in patients with triple-negative or BRCA mutated breast cancer. Nat. Partn. J. Breast Cancer 2021, 7, 29. [Google Scholar] [CrossRef]
- Rawla, P.; Sunkara, T.; Barsouk, A. Epidemiology of colorectal cancer: Incidence, mortality, survival, and risk factors. Gastroenterol. Rev. 2019, 14, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Siminoff, L.A.; Rogers, H.L.; Harris-Haywood, S. Missed opportunities for the diagnosis of colorectal cancer. BioMed Res. Int. 2015, 2015. [Google Scholar] [CrossRef] [PubMed]
- Chow, F.C.L.; Chok, K.S.H. Colorectal liver metastases: An update on multidisciplinary approach. World J. Hepatol. 2019, 11, 150–172. [Google Scholar] [CrossRef]
- Sobrero, A.; Lonardi, S.; Rosati, G.; Di Bartolomeo, M.; Ronzoni, M.; Pella, N.; Scartozzi, M.; Banzi, M.; Zampino, M.G.; Pasini, F.; et al. FOLFOX or CAPOX in stage II to III colon cancer: Efficacy results of the italian three or six colon adjuvant trial. J. Clin. Oncol. 2018, 36, 1478–1485. [Google Scholar] [CrossRef]
- Xie, Y.H.; Chen, Y.X.; Fang, J.Y. Comprehensive review of targeted therapy for colorectal cancer. Signal Transduct. Target. Ther. 2020, 5, 1–30. [Google Scholar] [CrossRef]
- Zhang, Q.; Lu, Q. Bin New combination chemotherapy of cisplatin with an electron-donating compound for treatment of multiple cancers. Sci. Rep. 2021. [Google Scholar] [CrossRef]
- Lu, Q.B.; Kalantari, S.; Wang, C.R. Electron transfer reaction mechanism of cisplatin with DNA at the molecular level. Mol. Pharm. 2007. [Google Scholar] [CrossRef] [PubMed]
- D’Amico, M.; Gasparoli, L.; Arcangeli, A. Potassium Channels: Novel Emerging Biomarkers and Targets for Therapy in Cancer. Recent Pat. Anticancer Drug Discov. 2012, 8, 53–65. [Google Scholar] [CrossRef]
- Gasparoli, L.; D’Amico, M.; Masselli, M.; Pillozzi, S.; Caves, R.; Khuwaileh, R.; Tiedke, W.; Mugridge, K.; Pratesi, A.; Mitcheson, J.S.; et al. New pyrimido-indole compound CD-160130 preferentially inhibits the Kv11.1B isoform and produces antileukemic effects without cardiotoxicity. Mol. Pharmacol. 2015, 87, 183–196. [Google Scholar] [CrossRef] [PubMed]
- Muratori, L.; Petroni, G.; Antonuzzo, L.; Boni, L.; Iorio, J.; Lastraioli, E.; Bartoli, G.; Messerini, L.; Di Costanzo, F.; Arcangeli, A. HERG1 positivity and Glut-1 negativity identifies high-risk TNM stage I and II colorectal cancer patients, regardless of adjuvant chemotherapy. OncoTargets Ther. 2016, 9, 6325–6332. [Google Scholar] [CrossRef]
- Pillozzi, S.; D’Amico, M.; Bartoli, G.; Gasparoli, L.; Petroni, G.; Crociani, O.; Marzo, T.; Guerriero, A.; Messori, L.; Severi, M.; et al. The combined activation of K Ca 3.1 and inhibition of K v 11.1/hERG1 currents contribute to overcome Cisplatin resistance in colorectal cancer cells. Br. J. Cancer 2018, 118, 200–212. [Google Scholar] [CrossRef]
- Samuel, P.; Pink, R.C.; Caley, D.P.; Currie, J.M.S.; Brooks, S.A.; Carter, D.R.F. Over-expression of miR-31 or loss of KCNMA1 leads to increased cisplatin resistance in ovarian cancer cells. Tumor Biol. 2016, 37, 2565–2573. [Google Scholar] [CrossRef]
- Leanza, L.; O’Reilly, P.; Doyle, A.; Venturini, E.; Zoratti, M.; Szegezdi, E.; Szabo, I. Correlation between Potassium Channel Expression and Sensitivity to Drug-induced Cell Death in Tumor Cell Lines. Curr. Pharm. Des. 2014, 20, 189–200. [Google Scholar] [CrossRef]
- Martin, L.P.; Hamilton, T.C.; Schilder, R.J. Platinum resistance: The role of DNA repair pathways. Clin. Cancer Res. 2008, 14, 1291–1295. [Google Scholar] [CrossRef]
- Damia, G.; Broggini, M. Platinum Resistance in Ovarian Cancer: Role of DNA Repair. Cancers 2019, 11, 119. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, J.T.; Lipp, H.P. Toxicity of platinum compounds. Expert Opin. Pharmacother. 2003, 4, 889–901. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Allison, J.P. Immune checkpoint targeting in cancer therapy: Toward combination strategies with curative potential. Cell 2015, 161, 205–214. [Google Scholar] [CrossRef]
- Bang, Y.J.; Van Cutsem, E.; Feyereislova, A.; Chung, H.C.; Shen, L.; Sawaki, A.; Lordick, F.; Ohtsu, A.; Omuro, Y.; Satoh, T.; et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): A phase 3, open-label, randomised controlled trial. Lancet 2010, 376, 687–697. [Google Scholar] [CrossRef]
- Fernández-Moreira, V.; Gimeno, M.C. Heterobimetallic Complexes for Theranostic Applications. Chem. Eur. J. 2018, 24, 3345–3353. [Google Scholar] [CrossRef] [PubMed]
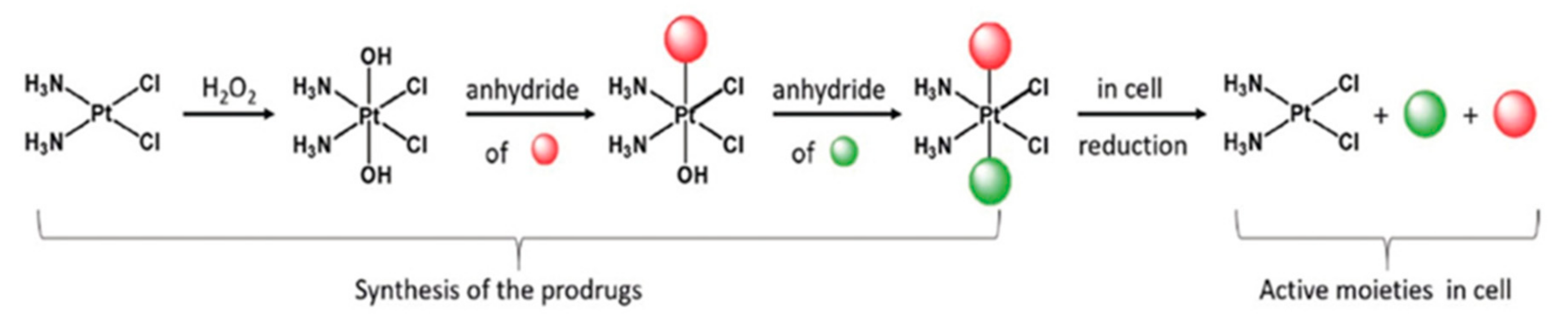
- Petruzzella, E.; Braude, J.P.; Aldrich-Wright, J.R.; Gandin, V.; Gibson, D. A Quadruple-Action Platinum (IV) Prodrug with Anticancer Activity Against KRAS Mutated Cancer Cell Lines. Angew. Chem. 2017, 56, 11539–11544. [Google Scholar] [CrossRef]
- Yempala, T.; Babu, T.; Karmakar, S.; Nemirovski, A.; Ishan, M.; Gandin, V.; Gibson, D. Expanding the Arsenal of Pt IV Anticancer Agents: Multi-action Pt IV Anticancer Agents with Bioactive Ligands Possessing a Hydroxy Functional Group. Angew. Chem. 2019, 131, 18386–18391. [Google Scholar] [CrossRef]
- Gibson, D. Platinum(IV) anticancer prodrugs-hypotheses and facts. Dalton Trans. 2016, 45, 12983–12991. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.Z.; Bonnitcha, P.; Wexselblatt, E.; Klein, A.V.; Najajreh, Y.; Gibson, D.; Hambley, T.W. Facile Preparation of Mono-, Di- and Mixed-Carboxylato Platinum(IV) Complexes for Versatile Anticancer Prodrug Design. Chem. Eur. J. 2013, 19, 1672–1676. [Google Scholar] [CrossRef] [PubMed]
- Song, X.Q.; Ma, Z.Y.; Wu, Y.G.; Dai, M.L.; Wang, D.B.; Xu, J.Y.; Liu, Y. New NSAID-Pt(IV) prodrugs to suppress metastasis and invasion of tumor cells and enhance anti-tumor effect in vitro and in vivo. Eur. J. Med. Chem. 2019, 167, 377–387. [Google Scholar] [CrossRef]
- Barnes, K.R.; Kutikov, A.; Lippard, S.J. Synthesis, characterization, and cytotoxicity of a series of estrogen-tethered platinum(IV) complexes. Chem. Biol. 2004, 11, 557–564. [Google Scholar] [CrossRef]
- Canil, G.; Braccini, S.; Marzo, T.; Marchetti, L.; Pratesi, A.; Biver, T.; Funaioli, T.; Chiellini, F.; Hoeschele, J.D.; Gabbiani, C. Photocytotoxic Pt(iv) complexes as prospective anticancer agents. Dalton Trans. 2019, 48, 10933–10944. [Google Scholar] [CrossRef]
- Ma, L.; Ma, R.; Wang, Z.; Yiu, S.M.; Zhu, G. Heterodinuclear Pt(IV)-Ru(II) anticancer prodrugs to combat both drug resistance and tumor metastasis. Chem. Commun. 2016, 52, 10735–10738. [Google Scholar] [CrossRef]
- Guedes, A.P.M.; Mello-Andrade, F.; Pires, W.C.; De Sousa, M.A.M.; Da Silva, P.F.F.; De Camargo, M.S.; Gemeiner, H.; Amauri, M.A.; Gomes Cardoso, C.; De Melo Reis, P.R.; et al. Heterobimetallic Ru(ii)/Fe(ii) complexes as potent anticancer agents against breast cancer cells, inducing apoptosis through multiple targets. Metallomics 2020, 12, 547–561. [Google Scholar] [CrossRef]
- Nowak-Sliwinska, P.; Segura, T.; Iruela-Arispe, M.L. The chicken chorioallantoic membrane model in biology, medicine and bioengineering. Angiogenesis 2014, 17, 779–804. [Google Scholar] [CrossRef]
- Bravo, C.; Robalo, M.P.; Marques, F.; Fernandes, A.R.; Sequeira, D.A.; Piedade, M.F.M.; Garcia, M.H.; De Brito, M.J.V.; Morais, T.S. First heterobimetallic Cu(i)-dppf complexes designed for anticancer applications: Synthesis, structural characterization and cytotoxicity. New J. Chem. 2019, 43, 12308–12317. [Google Scholar] [CrossRef]
- Porchia, M.; Dolmella, A.; Gandin, V.; Marzano, C.; Pellei, M.; Peruzzo, V.; Refosco, F.; Santini, C.; Tisato, F. Neutral and charged phosphine/scorpionate copper(I) complexes: Effects of ligand assembly on their antiproliferative activity. Eur. J. Med. Chem. 2013, 59, 218–226. [Google Scholar] [CrossRef]
- Gandin, V.; Tisato, F.; Dolmella, A.; Pellei, M.; Santini, C.; Giorgetti, M.; Marzano, C.; Porchia, M. In vitro and in vivo anticancer activity of copper(I) complexes with homoscorpionate tridentate tris(pyrazolyl)borate and auxiliary monodentate phosphine ligands. J. Med. Chem. 2014, 57, 4745–4760. [Google Scholar] [CrossRef]
- Morais, T.S.; Jousseaume, Y.; Piedade, M.F.M.; Roma-Rodrigues, C.; Fernandes, A.R.; Marques, F.; Villa De Brito, M.J.; Helena Garcia, M. Important cytotoxic and cytostatic effects of new copper(i)-phosphane compounds with N,N, N,O and N,S bidentate ligands. Dalton Trans. 2018, 47, 7819–7829. [Google Scholar] [CrossRef] [PubMed]
- Odachowski, M.; Marschner, C.; Blom, B. A review on 1,1-bis(diphenylphosphino)methane bridged homo- and heterobimetallic complexes for anticancer applications: Synthesis, structure, and cytotoxicity. Eur. J. Med. Chem. 2020, 204, 112613. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.M.; Taylor, I.R.; Tibbetts, M.F.; Philpott, J.; Hu, Y.; Tanski, J.M. Hetero-multinuclear ruthenium(III)/platinum(II) complexes that potentially exhibit both antimetastatic and antineoplastic properties. Inorg. Chem. 2012, 51, 12917–12924. [Google Scholar] [CrossRef]
- Jain, S.S.; Anderson, C.M.; DiRienzo, F.; Taylor, I.R.; Jain, K.; Guha, S.; Hoque, N. RNA binding and inhibition of primer extension by a Ru(iii)/Pt(ii) metal complex. Chem. Commun. 2013, 49, 5031–5033. [Google Scholar] [CrossRef]
- Bertrand, B.; Citta, A.; Franken, I.L.; Picquet, M.; Folda, A.; Scalcon, V.; Rigobello, M.P.; Le Gendre, P.; Casini, A.; Bodio, E. Gold(I) NHC-based homo- and heterobimetallic complexes: Synthesis, characterization and evaluation as potential anticancer agents. J. Biol. Inorg. Chem. 2015, 20, 1005–1020. [Google Scholar] [CrossRef]
- Askari, B.; Rudbari, H.A.; Micale, N.; Schirmeister, T.; Maugeri, A.; Navarra, M. Anticancer study of heterobimetallic platinum(II)-ruthenium(II) and platinum(II)-rhodium(III) complexes with bridging dithiooxamide ligand. J. Organomet. Chem. 2019, 900, 120918. [Google Scholar] [CrossRef]
- Lengfelder, E.; Hofmann, W.K.; Nowak, D. Impact of arsenic trioxide in the treatment of acute promyelocytic leukemia. Leukemia 2012, 26, 433–442. [Google Scholar] [CrossRef]
- Adès, L.; Thomas, X.; Bresler, A.G.; Raffoux, E.; Spertini, O.; Vey, N.; Marchand, T.; Récher, C.; Pigneux, A.; Girault, S.; et al. Arsenic trioxide is required in the treatment of newly diagnosed acute promyelocytic leukemia. Analysis of a randomized trial (APL 2006) by the French Belgian Swiss APL group. Haematologica 2018, 103, 2033–2039. [Google Scholar] [CrossRef]
- da Rosa, F.C.; Buque-Pardinho, R.; Schultz-Moreira, M.E.; de Souza, L.G.T.; de Moraes Flores, É.M.; Mortari, S.R.; Dressler, V.L. In vitro stability of arsenic trioxide-liposome encapsulates for acute promyelocytic leukemia treatment. Leuk. Res. 2019, 76, 11–14. [Google Scholar] [CrossRef]
- Coombs, C.C.; Tavakkoli, M.; Tallman, M.S. Acute promyelocytic Leukemia: Where did we start, where are we now, and the future. Blood Cancer J. 2015, 5, e304. [Google Scholar] [CrossRef] [PubMed]
- Subbarayan, P.R.; Ardalan, B. In the war against solid tumors arsenic trioxide needs partners. J. Gastrointest. Cancer 2014, 45, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Das, B.; Rahaman, H.; Ghosh, S.K.; Sengupta, M. Synthesis and Characterization of Arsenic(III) Oxide Nanoparticles as Potent Inhibitors of MCF 7 Cell Proliferation through Proapoptotic Mechanism. Bionanoscience 2020, 10, 420–429. [Google Scholar] [CrossRef]
- Hu, J.; Dong, Y.; Ding, L.; Dong, Y.; Wu, Z.; Wang, W.; Shen, M.; Duan, Y. Local delivery of arsenic trioxide nanoparticles for hepatocellular carcinoma treatment. Signal Transduct. Target. Ther. 2019, 4, 1–7. [Google Scholar] [CrossRef]
- Ettlinger, R.; Sönksen, M.; Graf, M.; Moreno, N.; Denysenko, D.; Volkmer, D.; Kerl, K.; Bunzen, H. Metal-organic framework nanoparticles for arsenic trioxide drug delivery. J. Mater. Chem. 2018, 6, 6481–6489. [Google Scholar] [CrossRef]
- Miodragović, D.; Merlino, A.; Swindell, E.P.; Bogachkov, A.; Ahn, R.W.; Abuhadba, S.; Ferraro, G.; Marzo, T.; Mazar, A.P.; Messori, L.; et al. Arsenoplatin-1 Is a Dual Pharmacophore Anticancer Agent. J. Am. Chem. Soc. 2019, 141, 6453–6457. [Google Scholar] [CrossRef]
- Gatti, L.; Cassinelli, G.; Zaffaroni, N.; Lanzi, C.; Perego, P. New mechanisms for old drugs: Insights into DNA-unrelated effects of platinum compounds and drug resistance determinants. Drug Resist. Updates 2015, 20, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Hoonjan, M.; Jadhav, V.; Bhatt, P. Arsenic trioxide: Insights into its evolution to an anticancer agent. J. Biol. Inorg. Chem. 2018, 23, 313–329. [Google Scholar] [CrossRef]
- Miodragović, D.U.; Quentzel, J.A.; Kurutz, J.W.; Stern, C.L.; Ahn, R.W.; Kandela, I.; Mazar, A.; O’Halloran, T.V. Robust Structure and Reactivity of Aqueous Arsenous Acid-Platinum(II) Anticancer Complexes. Angew. Chem. 2013, 52, 10749–10752. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.C. Drug combination studies and their synergy quantification using the chou-talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef]
- Zhou, X.; Cooper, K.L.; Sun, X.; Liu, K.J.; Hudson, L.G. Selective sensitization of zinc finger protein oxidation by reactive oxygen species through arsenic binding. J. Biol. Chem. 2015, 290, 18361–18369. [Google Scholar] [CrossRef]
- Ferraro, G.; Pratesi, A.; Cirri, D.; Imbimbo, P.; Maria-Monti, D.; Messori, L.; Merlino, A. Arsenoplatin-Ferritin Nanocage: Structure and Cytotoxicity. Int. J. Mol. Sci. 2021, 22, 1874. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
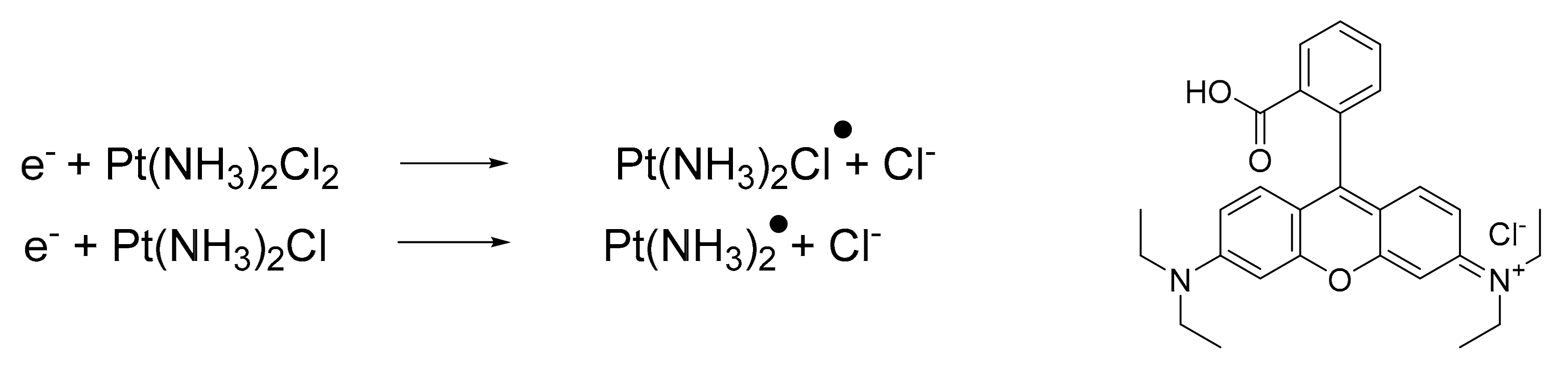{kind=link}
| Name of the Study | Primary Outcome | Study Groups | PFS (Months) | HR (95% CI) | p-Value | OS (Months) | HR (95% CI) | p-Value |
|---|---|---|---|---|---|---|---|---|
| ICON4 | OS | Carbo | 10 | 0.76 (0.66–0.89) | 0.0004 | 24 | 0.82 (0.69–0.97) | 0.02 |
| Carbo/Taxol | 13 | 29 | ||||||
| AGO | PFS | Carbo | 5.8 | 0.72 (0.58–0.90) | 0.003 | 17.3 | 0.96 (0.75–1.23) | 0.73 |
| Carbo/Gem | 8.6 | 18.0 | ||||||
| CALYPSO | PFS | Carbo/Taxol | 9.4 | 0.82 (0.72–0.94) | 0.005 | 30.7 | 0.99 (0.85–1.16) | 0.94 |
| Carbo/PLD | 11.3 | 33.0 | ||||||
| OCEANS | PFS | Carbo/Gem/Placebo | 8.4 | 0.48 (0.39–0.61) | <0.0001 | 32.9 | 0.95 (0.77–1.17) | 0.65 |
| Carbo/Gem/Bev | 12.4 | 33.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cirri, D.; Bartoli, F.; Pratesi, A.; Baglini, E.; Barresi, E.; Marzo, T. Strategies for the Improvement of Metal-Based Chemotherapeutic Treatments. Biomedicines 2021, 9, 504. https://doi.org/10.3390/biomedicines9050504
Cirri D, Bartoli F, Pratesi A, Baglini E, Barresi E, Marzo T. Strategies for the Improvement of Metal-Based Chemotherapeutic Treatments. Biomedicines. 2021; 9(5):504. https://doi.org/10.3390/biomedicines9050504
Chicago/Turabian StyleCirri, Damiano, Francesco Bartoli, Alessandro Pratesi, Emma Baglini, Elisabetta Barresi, and Tiziano Marzo. 2021. "Strategies for the Improvement of Metal-Based Chemotherapeutic Treatments" Biomedicines 9, no. 5: 504. https://doi.org/10.3390/biomedicines9050504
APA StyleCirri, D., Bartoli, F., Pratesi, A., Baglini, E., Barresi, E., & Marzo, T. (2021). Strategies for the Improvement of Metal-Based Chemotherapeutic Treatments. Biomedicines, 9(5), 504. https://doi.org/10.3390/biomedicines9050504