
The Evolution of Antisense Oligonucleotide Chemistry—A Personal Journey


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. The Beginning
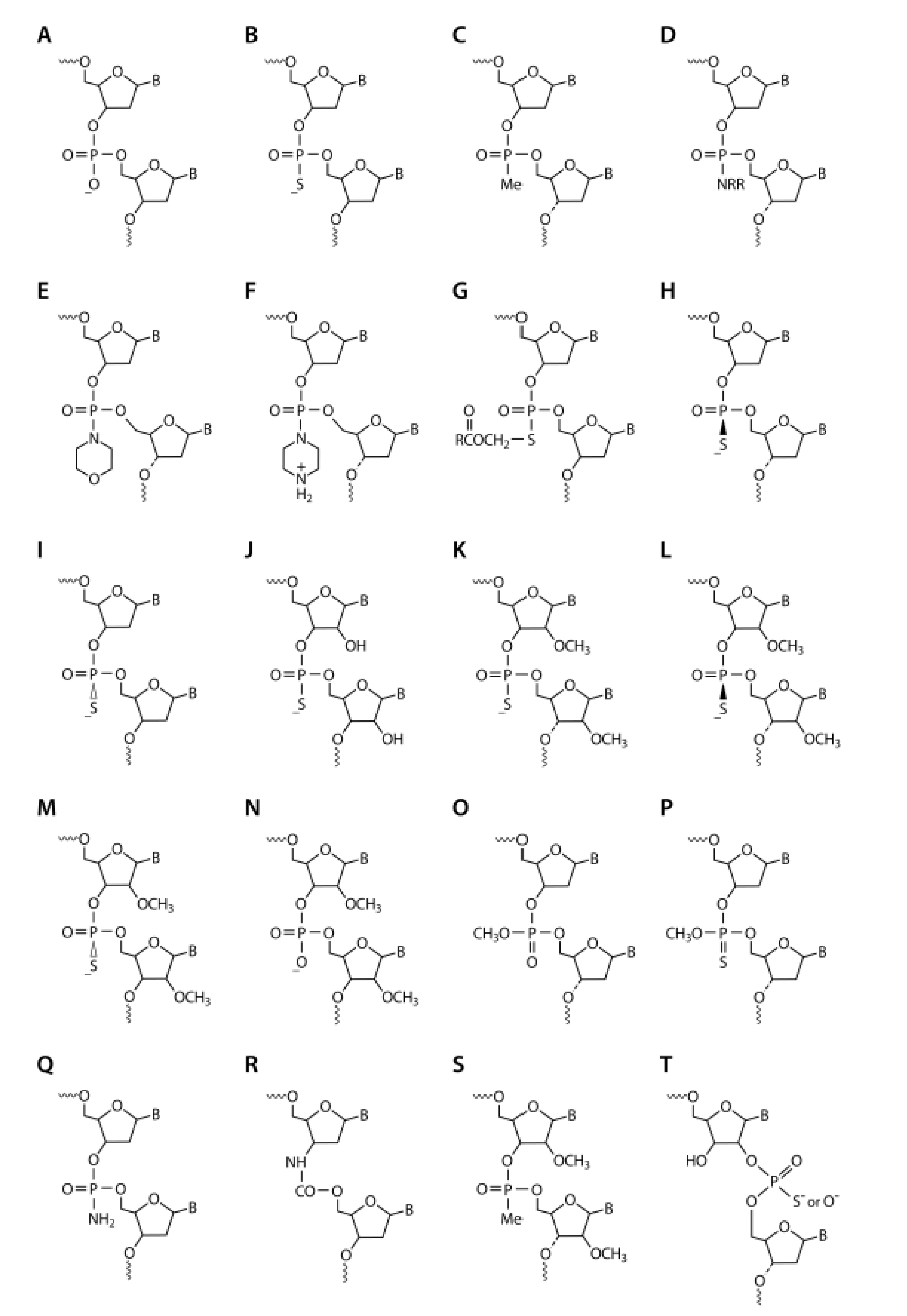
3. Stability against Nucleolytic Degradation
4. PS-ODNS—First Generation Antisense Therapeutics
5. Modified PS-Oligoribonucleotides (PS-ORNs)
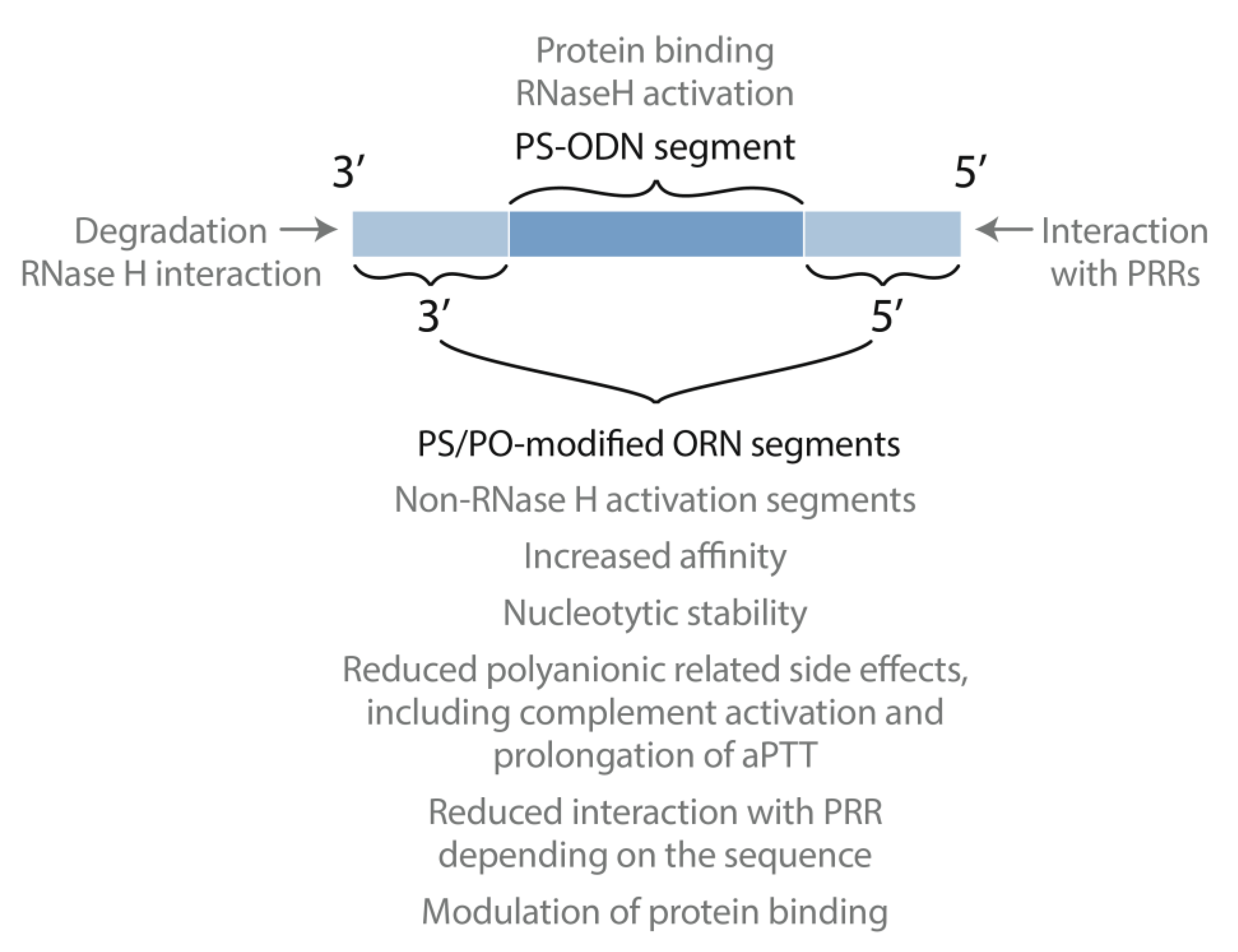
6. Putting the Pieces Together: Mixed Backbone Antisense
7. Paying the Tolls: The Role of Sequence and Modifications
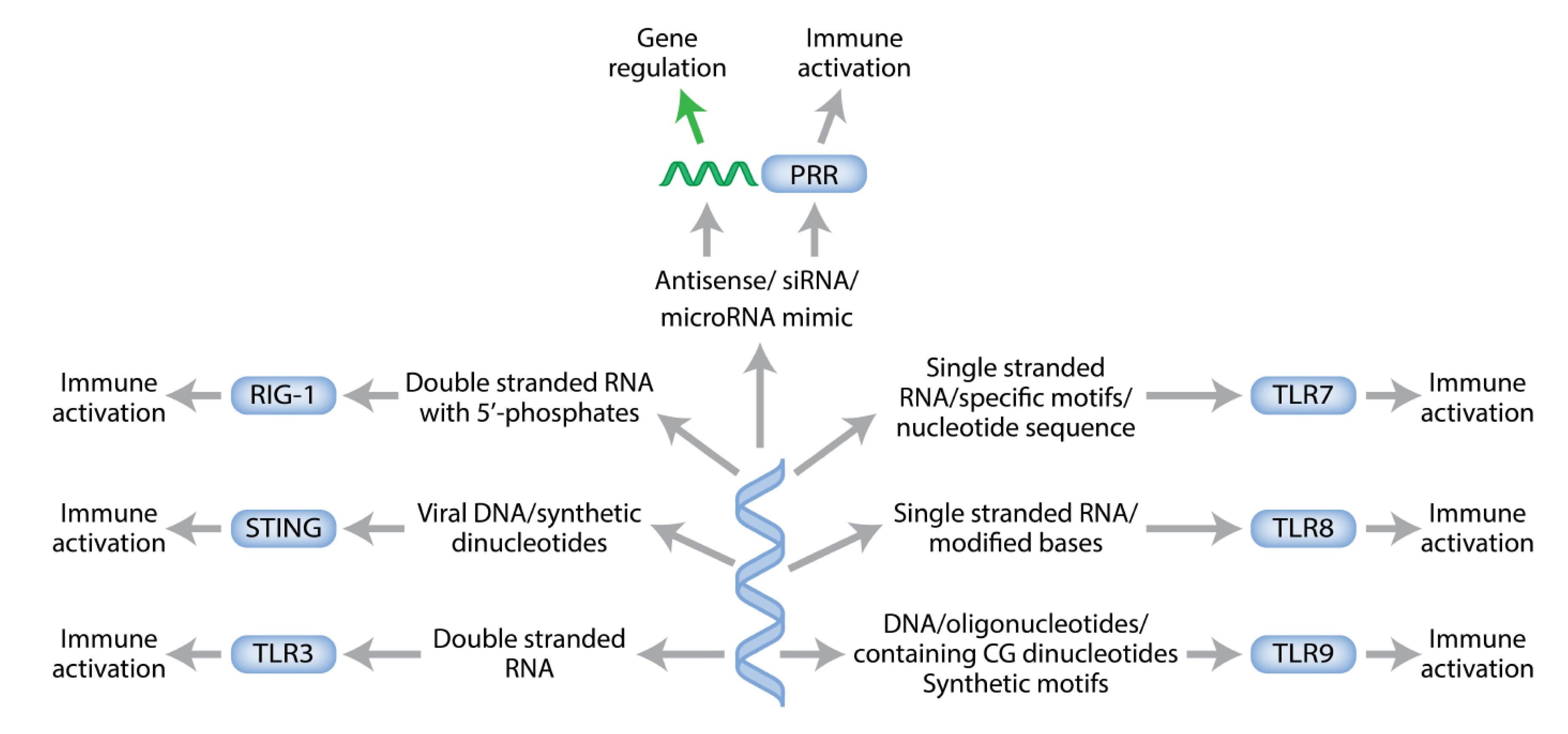
Immunotherapy with Synthetic Oligonucleotides
8. Connecting the Dots for New Antisense Chemistry
9. Distractions and Side Roads along the Way
10. Summary
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Agrawal, S.; Gait, M.J. (Eds.) Advances in Nucleic Acid Therapeutics; Royal Society of Chemistry: London, UK, 2019. [Google Scholar]
- Zamecnik, P.C. History of Antisense Oligonucleotides. In Antisense Therapeutics, 1st ed.; Agrawal, S., Walker, J.M., Eds.; Methods in Molecular Medicine; Humana Press: Totowa, NJ, USA, 1996. [Google Scholar]
- Agrawal, S.; Christodoulou, C.; Gait, M.J. Efficient methods for attaching non-radioactive labels to the 5’ ends of synthetic oligodeoxyribonucleotides. Nucleic Acids Res. 1986, 14, 6227–6245. [Google Scholar] [CrossRef]
- Brown, D.M. A brief history of oligonucleotide synthesis. Methods Mol. Biol. 1993, 20, 1–17. [Google Scholar] [CrossRef]
- Stephenson, M.L.; Zamecnik, P.C. Inhibition of Rous sarcoma virus replication and cell transformation by a specific oligodeoxynucleotide. Proc. Natl. Acad. Sci. USA 1978, 75, 280–284. [Google Scholar] [CrossRef]
- Stephenson, M.L.; Zamecnik, P.C. Inhibition of Rous sarcoma viral RNA translation by a specific oligodeoxyribonucleotide. Proc. Natl. Acad. Sci. USA 1978, 75, 285–288. [Google Scholar] [CrossRef]
- Zamecnik, P.C.; Goodchild, J.; Taguchi, Y.; Sarin, P.S. Inhibition of replication and expression of human T-cell lymphotropic virus type III in cultured cells by exogenous synthetic oligonucleotides complementary to viral RNA. Proc. Natl. Acad. Sci. USA 1986, 83, 4143–4146. [Google Scholar] [CrossRef]
- Matsukura, M.; Zon, G.; Shinozuka, K.; Robert-Guroff, M.; Shimada, T.; Stein, C.A.; Mitsuya, H.; Wong-Staal, F.; Cohen, J.S.; Broder, S. Regulation of viral expression of human immunodeficiency virus in vitro by an antisense phosphorothioate oligodeoxynucleotide against rev (art/trs) in chronically infected cells. Proc. Natl. Acad. Sci. USA 1989, 86, 4244–4248. [Google Scholar] [CrossRef]
- Eckstein, F. Nucleoside phosphorothioates. Annu. Rev. Biochem. 1985, 54, 367–402. [Google Scholar] [CrossRef]
- Miller, P.S.; McParland, K.B.; Jayaraman, K.; Ts’o, P.O. Biochemical and biological effects of nonionic nucleic acid methylphosphonates. Biochemistry 1981, 20, 1874–1880. [Google Scholar] [CrossRef]
- Agrawal, S.; Goodchild, J. Oligodeoxynucleoside methylphosphonates: Synthesis and enzymic degradation. Tetrahedron Lett. 1987, 28, 3539–3542. [Google Scholar] [CrossRef]
- Sarin, P.S.; Agrawal, S.; Civeira, M.P.; Goodchild, J.; Ikeuchi, T.; Zamecnik, P.C. Inhibition of acquired immunodeficiency syndrome virus by oligodeoxynucleoside methylphosphonates. Proc. Natl. Acad. Sci. USA 1988, 85, 7448–7451. [Google Scholar] [CrossRef]
- Agrawal, S.; Goodchild, J.; Civeira, M.P.; Thornton, A.H.; Sarin, P.S.; Zamecnik, P.C. Oligodeoxynucleoside phosphoramidates and phosphorothioates as inhibitors of human immunodeficiency virus. Proc. Natl. Acad. Sci. USA 1988, 85, 7079–7083. [Google Scholar] [CrossRef]
- Lisziewicz, J.; Sun, D.; Klotman, M.; Agrawal, S.; Zamecnik, P.; Gallo, R. Specific inhibition of human immunodeficiency virus type 1 replication by antisense oligonucleotides: An in vitro model for treatment. Proc. Natl. Acad. Sci. USA 1992, 89, 11209–11213. [Google Scholar] [CrossRef]
- Lisziewicz, J.; Sun, D.; Metelev, V.; Zamecnik, P.; Gallo, R.C.; Agrawal, S. Long-term treatment of human immunodeficiency virus-infected cells with antisense oligonucleotide phosphorothioates. Proc. Natl. Acad. Sci. USA 1993, 90, 3860–3864. [Google Scholar] [CrossRef]
- Leiter, J.M.; Agrawal, S.; Palese, P.; Zamecnik, P.C. Inhibition of influenza virus replication by phosphorothioate oligodeoxynucleotides. Proc. Natl. Acad. Sci. USA 1990, 87, 3430–3434. [Google Scholar] [CrossRef]
- Agrawal, S.; Mayrand, S.H.; Zamecnik, P.C.; Pederson, T. Site-specific excision from RNA by RNase H and mixed-phosphate-backbone oligodeoxynucleotides. Proc. Natl. Acad. Sci. USA 1990, 87, 1401–1405. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, S. Antisense oligonucleotides as antiviral agents. Trends Biotechnol. 1992, 10, 152–158. [Google Scholar] [CrossRef]
- Agrawal, S.; Temsamani, J.; Tang, J.Y. Pharmacokinetics, biodistribution, and stability of oligodeoxynucleotide phosphorothioates in mice. Proc. Natl. Acad. Sci. USA 1991, 88, 7595–7599. [Google Scholar] [CrossRef]
- Agrawal, S. Antisense oligonucleotides: Towards clinical trials. Trends Biotechnol. 1996, 14, 376–387. [Google Scholar] [CrossRef]
- Crooke, S.T.; Vickers, T.A.; Liang, X.H. Phosphorothioate modified oligonucleotide-protein interactions. Nucleic Acids Res. 2020, 48, 5235–5253. [Google Scholar] [CrossRef]
- Agrawal, S.; Zhang, X.; Cai, Q.; Kandimalla, E.R.; Manning, A.; Jiang, Z.; Marcel, T.; Zhang, R. Effect of aspirin on protein binding and tissue disposition of oligonucleotide phosphorothioate in rats. J. Drug Target 1998, 5, 303–312. [Google Scholar] [CrossRef]
- Temsamani, J.; Tang, J.Y.; Padmapriya, A.; Kubert, M.; Agrawal, S. Pharmacokinetics, biodistribution, and stability of capped oligodeoxynucleotide phosphorothioates in mice. Antisense Res. Dev. 1993, 3, 277–284. [Google Scholar] [CrossRef]
- Zhang, R.; Diasio, R.B.; Lu, Z.; Liu, T.; Jiang, Z.; Galbraith, W.M.; Agrawal, S. Pharmacokinetics and tissue distribution in rats of an oligodeoxynucleotide phosphorothioate (GEM 91) developed as a therapeutic agent for human immunodeficiency virus type-1. Biochem. Pharmacol. 1995, 49, 929–939. [Google Scholar] [CrossRef]
- Grindel, J.M.; Musick, T.J.; Jiang, Z.; Roskey, A.; Agrawal, S. Pharmacokinetics and metabolism of an oligodeoxynucleotide phosphorothioate (GEM91) in cynomolgus monkeys following intravenous infusion. Antisense Nucleic Acid Drug Dev. 1998, 8, 43–52. [Google Scholar] [CrossRef]
- Zhang, R.; Yan, J.; Shahinian, H.K.; Shahinian, H.; Amin, G.; Lu, Z.; Liu, T.; Saag, M.S.; Jiang, Z.; Temsamani, J.; et al. Pharmacokinetics of an anti-human immunodeficiency virus antisense oligodeoxynucleotide phosphorothioate (GEM 91) in HIV-infected subjects. Clin. Pharmacol. Ther. 1995, 58, 44–53. [Google Scholar] [CrossRef]
- Agrawal, S.; Zhao, Q.; Jiang, Z.; Oliver, C.; Giles, H.; Heath, J.; Serota, D. Toxicologic effects of an oligodeoxynucleotide phosphorothioate and its analogs following intravenous administration in rats. Antisense Nucleic Acid Drug Dev. 1997, 7, 575–584. [Google Scholar] [CrossRef]
- Agrawal, S.; Rustagi, P.K.; Shaw, D.R. Novel enzymatic and immunological responses to oligonucleotides. Toxicol. Lett. 1995, 82–83, 431–434. [Google Scholar] [CrossRef]
- Zhao, Q.; Temsamani, J.; Iadarola, P.L.; Jiang, Z.; Agrawal, S. Effect of different chemically modified oligodeoxynucleotides on immune stimulation. Biochem. Pharmacol. 1996, 51, 173–182. [Google Scholar] [CrossRef]
- Galbraith, W.M.; Hobson, W.C.; Giclas, P.C.; Schechter, P.J.; Agrawal, S. Complement activation and hemodynamic changes following intravenous administration of phosphorothioate oligonucleotides in the monkey. Antisense Res. Dev. 1994, 4, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Black, L.E.; Degeorge, J.J.; Cavagnaro, J.A.; Jordan, A.; Ahn, C.H. Regulatory considerations for evaluating the pharmacology and toxicology of antisense drugs. Antisense Res. Dev. 1993, 3, 399–404. [Google Scholar] [CrossRef]
- Padmapriya, A.A.; Tang, J.; Agrawal, S. Large-scale synthesis, purification, and analysis of oligodeoxynucleotide phosphorothioates. Antisense Res. Dev. 1994, 4, 185–199. [Google Scholar] [CrossRef]
- Agrawal, S.; Hybridon, Inc. FORM 8-K June 29, 2000. Available online: https://ir.iderapharma.com/node/8761/html (accessed on 14 April 2021).
- Agrawal, S.; Lisziewicz, J. Potential for HIV-1 treatment with antisense oligonucleotides. J. Biotech. Healthcare 1994, 1, 167–182. [Google Scholar]
- Metelev, V.; Lisziewicz, J.; Agrawal, S. Study of antisense oligonucleotide phosphorothioates containing segments of oligodeoxynucleotides and 2′-o- methyloligoribonucleotides. Bioorganic Med. Chem. Lett. 1994, 4, 2929–2934. [Google Scholar]
- Metelev, V.; Agrawal, S. Hybrid oligonucleotide phosphorothioates. 1992. [Google Scholar]
- Temsamani, J.; Agrawal, S.; Pederson, T. Biotinylated antisense methylphosphonate oligodeoxynucleotides. Inhibition of spliceosome assembly and affinity selection of U1 and U2 small nuclear RNPs. J. Biol. Chem. 1991, 266, 468–472. [Google Scholar] [CrossRef]
- Sierakowska, H.; Sambade, M.J.; Agrawal, S.; Kole, R. Repair of thalassemic human beta-globin mRNA in mammalian cells by antisense oligonucleotides. Proc. Natl. Acad. Sci. USA 1996, 93, 12840–12844. [Google Scholar] [CrossRef]
- Wilton, S.D.; Lloyd, F.; Carville, K.; Fletcher, S.; Honeyman, K.; Agrawal, S.; Kole, R. Specific removal of the nonsense mutation from the mdx dystrophin mRNA using antisense oligonucleotides. Neuromuscul. Disord. 1999, 9, 330–338. [Google Scholar] [CrossRef]
- Kandimalla, E.R.; Shaw, D.R.; Agrawal, S. Effects of phosphorothioate oligodeoxyribonucleotide and oligoribonucleotides on human complement and coagulation. Bioorganic Med. Chem. Lett. 1998, 8, 2103–2108. [Google Scholar] [CrossRef]
- Agrawal, S.; Jiang, Z.; Zhao, Q.; Shaw, D.; Cai, Q.; Roskey, A.; Channavajjala, L.; Saxinger, C.; Zhang, R. Mixed-backbone oligonucleotides as second generation antisense oligonucleotides: In vitro and in vivo studies. Proc. Natl. Acad. Sci. USA 1997, 94, 2620–2625. [Google Scholar] [CrossRef]
- Shen, L.X.; Kandimalla, E.R.; Agrawal, S. Impact of mixed-backbone oligonucleotides on target binding affinity and target cleaving specificity and selectivity by Escherichia coli RNase, H. Bioorganic Med. Chem. 1998, 6, 1695–1705. [Google Scholar] [CrossRef]
- Zhang, R.; Lu, Z.; Zhao, H.; Zhang, X.; Diasio, R.B.; Habus, I.; Jiang, Z.; Iyer, R.P.; Yu, D.; Agrawal, S. In vivo stability, disposition and metabolism of a “hybrid” oligonucleotide phosphorothioate in rats. Biochem. Pharmacol. 1995, 50, 545–556. [Google Scholar] [CrossRef]
- Agrawal, S.; Zhang, X.; Lu, Z.; Zhao, H.; Tamburin, J.M.; Yan, J.; Cai, H.; Diasio, R.B.; Habus, I.; Jiang, Z. Absorption, tissue distribution and in vivo stability in rats of a hybrid antisense oligonucleotide following oral administration. Biochem. Pharmacol. 1995, 50, 571–576. [Google Scholar] [CrossRef]
- Wang, H.; Cai, Q.; Zeng, X.; Yu, D.; Agrawal, S.; Zhang, R. Antitumor activity and pharmacokinetics of a mixed-backbone antisense oligonucleotide targeted to the RIalpha subunit of protein kinase A after oral administration. Proc. Natl. Acad. Sci. USA 1999, 96, 13989–13994. [Google Scholar] [CrossRef]
- Zhou, W.; Agrawal, S. Mixed-backbone oligonucleotides as second-generation antisense agents with reduced phosphorothioate-related side effects. Bioorganic Med. Chem. Lett. 1998, 8, 3269–3274. [Google Scholar] [CrossRef]
- Iyer, R.P.; Yu, D.; Devlin, T.; Ho, N.-H.; Johnson, S.; Agrawal, S. Synthesis, Biophysical Properties, and Stability Studies of Mixed Backbone Oligonucleotides Containing Novel Non-Ionic Linkages. Nucleosides Nucleotides 1997, 16, 1491–1495. [Google Scholar] [CrossRef]
- Iyer, R.P.; Yu, D.; Jiang, Z.; Agrawal, S. Synthesis, biophysical properties, and stability studies of mixed backbone oligonucleotides containing segments of methylphosphotriester internucleotidic linkages. Tetrahedron 1996, 52, 14419–14436. [Google Scholar] [CrossRef]
- Devlin, T.; Iyer, R.P.; Johnson, S.; Agrawal, S. Mixed backbone oligonucleotides containing internucleotidic primary phosphoramidate linkages. Bioorganic Med. Chem. Lett. 1996, 6, 2663–2668. [Google Scholar]
- Habus, I.; Temsamani, J.; Agrawal, S. Synthesis of di-, tri-, and tetrameric building blocks with novel carbamate internucleoside linkages and their incorporation into oligonucleotides. Bioorganic Med. Chem. Lett. 1994, 4, 1065–1070. [Google Scholar]
- Kandimalla, E.R.; Temsamani, J.; Agrawal, S. Synthesis and Properties of 2′-O-Methylribonucleotide Methylphosphonate Containing Chimeric Oligonucleotides. Nucleosides Nucleotides 1995, 14, 1031–1035. [Google Scholar] [CrossRef]
- Kandimalla, E.R.; Manning, A.; Zhao, Q.; Shaw, D.R.; Byrn, R.A.; Sasisekharan, V.; Agrawal, S. Mixed backbone antisense oligonucleotides: Design, biochemical and biological properties of oligonucleotides containing 2’-5’-ribo- and 3’-5’-deoxyribonucleotide segments. Nucleic Acids Res. 1997, 25, 370–378. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, S.; Hybridon, Inc. FORM 8-K, May 24, 2001. Available online: https://www.sec.gov/Archives/edgar/data/861838/000095013501501616/b39654hye8-k.txt (accessed on 14 April 2021).
- Geary, R.S.; Baker, B.F.; Moni, B.P. Antisense Technology: Liver Targeting and Beyond for Drug Discovery. In Advances in Nucleic Acid Therapeutics; Agrawal, S.G., Michael, J., Thurston, D., Eds.; Drug Discovery; Royal Society of Chemistry: London, UK, 2019; pp. 62–79. [Google Scholar]
- Seth, P.P.; Swayze, E.E. The Medicinal Chemistry of RNase H-activating Antisense Oligonucleotides. In Advances in Nucleic Acid Therapeutics; Agrawal, S., Gait, M.J., Eds.; Royal Society of Chemistry: London, UK, 2019; Volume 68, pp. 32–61. [Google Scholar]
- Zhao, Q.; Temsamani, J.; Zhou, R.Z.; Agrawal, S. Pattern and kinetics of cytokine production following administration of phosphorothioate oligonucleotides in mice. Antisense Nucleic Acid Drug Dev. 1997, 7, 495–502. [Google Scholar] [CrossRef]
- Agrawal, S.; Iyer, R.P. Perspectives in antisense therapeutics. Pharmacol. Ther. 1997, 76, 151–160. [Google Scholar] [CrossRef]
- Lewis, E.J.; Agrawal, S.; Bishop, J.; Chadwick, J.; Cristensen, N.D.; Cuthill, S.; Dunford, P.; Field, A.K.; Francis, J.; Gibson, V.; et al. Non-specific antiviral activity of antisense molecules targeted to the E1 region of human papillomavirus. Antivir. Res. 2000, 48, 187–196. [Google Scholar] [CrossRef]
- Agrawal, S.; Martin, R.R. Was induction of HIV-1 through TLR9? J. Immunol. 2003, 171, 1621, author reply 1621–1622. [Google Scholar] [CrossRef]
- De Clercq, E.; Eckstein, E.; Merigan, T.C. Interferon induction increased through chemical modification of a synthetic polyribonucleotide. Science 1969, 165, 1137–1139. [Google Scholar] [CrossRef]
- De Clercq, E.; Torrence, P.F.; Witkop, B. Interferon induction by synthetic polynucleotides: Importance of purine N-7 and strandwise rearrangement. Proc. Natl. Acad. Sci. USA 1974, 71, 182–186. [Google Scholar] [CrossRef]
- Tokunaga, T.; Yamamoto, H.; Shimada, S.; Abe, H.; Fukuda, T.; Fujisawa, Y.; Furutani, Y.; Yano, O.; Kataoka, T.; Sudo, T. Antitumor activity of deoxyribonucleic acid fraction from Mycobacterium bovis BCG. I. Isolation, physicochemical characterization, and antitumor activity. J. Natl. Cancer Inst. 1984, 72, 955–962. [Google Scholar] [PubMed]
- Messina, J.P.; Gilkeson, G.S.; Pisetsky, D.S. Stimulation of in vitro murine lymphocyte proliferation by bacterial DNA. J. Immunol. 1991, 147, 1759–1764. [Google Scholar] [PubMed]
- Krieg, A.M.; Yi, A.K.; Matson, S.; Waldschmidt, T.J.; Bishop, G.A.; Teasdale, R.; Koretzky, G.A.; Klinman, D.M. CpG motifs in bacterial DNA trigger direct B-cell activation. Nature 1995, 374, 546–549. [Google Scholar] [CrossRef]
- Saitoh, S.-I.; Miyake, K. Nucleic Acid Innate Immune Receptors. In Advances in Nucleic Acid Therapeutics; Agrawal, S.G., Michael, J., Eds.; Royal Society of Chemistry: London, UK, 2019; pp. 292–305. [Google Scholar]
- Zhao, Q.; Yu, D.; Agrawal, S. Immunostimulatory activity of CpG containing phosphorothioate oligodeoxynucleotide is modulated by modification of a single deoxynucleoside. Bioorganic Med. Chem. Lett. 2000, 10, 1051–1054. [Google Scholar] [CrossRef]
- Yu, D.; Kandimalla, E.R.; Zhao, Q.; Cong, Y.; Agrawal, S. Modulation of immunostimulatory activity of CpG oligonucleotides by site-specific deletion of nucleobases. Bioorganic Med. Chem. Lett. 2001, 11, 2263–2267. [Google Scholar] [CrossRef]
- Yu, D.; Kandimalla, E.R.; Zhao, Q.; Cong, Y.; Agrawal, S. Immunostimulatory activity of CpG oligonucleotides containing non-ionic methylphosphonate linkages. Bioorganic Med. Chem. 2001, 9, 2803–2808. [Google Scholar] [CrossRef]
- Yu, D.; Zhao, Q.; Kandimalla, E.R.; Agrawal, S. Accessible 5’-end of CpG-containing phosphorothioate oligodeoxynucleotides is essential for immunostimulatory activity. Bioorganic Med. Chem. Lett. 2000, 10, 2585–2588. [Google Scholar] [CrossRef]
- Yu, D.; Kandimalla, E.R.; Zhao, Q.; Cong, Y.; Agrawal, S. Immunostimulatory properties of phosphorothioate CpG DNA containing both 3’-5’- and 2’-5’-internucleotide linkages. Nucleic Acids Res. 2002, 30, 1613–1619. [Google Scholar] [CrossRef][Green Version]
- Kandimalla, E.R.; Bhagat, L.; Yu, D.; Cong, Y.; Tang, J.; Agrawal, S. Conjugation of ligands at the 5’-end of CpG DNA affects immunostimulatory activity. Bioconjug. Chem. 2002, 13, 966–974. [Google Scholar] [CrossRef]
- Putta, M.R.; Zhu, F.G.; Wang, D.; Bhagat, L.; Dai, M.; Kandimalla, E.R.; Agrawal, S. Peptide conjugation at the 5’-end of oligodeoxynucleotides abrogates toll-like receptor 9-mediated immune stimulatory activity. Bioconjug. Chem. 2010, 21, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Kandimalla, E.R.; Bhagat, L.; Tang, J.Y.; Cong, Y.; Tang, J.; Agrawal, S. ‘Immunomers’--novel 3’-3’-linked CpG oligodeoxyribonucleotides as potent immunomodulatory agents. Nucleic Acids Res. 2002, 30, 4460–4469. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Putta, M.R.; Bhagat, L.; Wang, D.; Zhu, F.G.; Kandimalla, E.R.; Agrawal, S. Immune-Stimulatory Dinucleotide at the 5’-End of Oligodeoxynucleotides Is Critical for TLR9-Mediated Immune Responses. ACS Med. Chem. Lett. 2013, 4, 302–305. [Google Scholar] [CrossRef][Green Version]
- Kandimalla, E.R.; Bhagat, L.; Li, Y.; Yu, D.; Wang, D.; Cong, Y.P.; Song, S.S.; Tang, J.X.; Sullivan, T.; Agrawal, S. Immunomodulatory oligonucleotides containing a cytosine-phosphate-2’-deoxy-7-deazaguanosine motif as potent toll-like receptor 9 agonists. Proc. Natl. Acad. Sci. USA 2005, 102, 6925–6930. [Google Scholar] [CrossRef] [PubMed]
- Putta, M.R.; Zhu, F.; Li, Y.; Bhagat, L.; Cong, Y.; Kandimalla, E.R.; Agrawal, S. Novel oligodeoxynucleotide agonists of TLR9 containing N3-Me-dC or N1-Me-dG modifications. Nucleic Acids Res. 2006, 34, 3231–3238. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Putta, M.R.; Bhagat, L.; Li, Y.; Zhu, F.; Wang, D.; Tang, J.X.; Kandimalla, E.R.; Agrawal, S. Agonists of Toll-like receptor 9 containing synthetic dinucleotide motifs. J. Med. Chem. 2007, 50, 6411–6418. [Google Scholar] [CrossRef]
- Lan, T.; Bhagat, L.; Wang, D.; Dai, M.; Kandimalla, E.R.; Agrawal, S. Synthetic oligoribonucleotides containing arabinonucleotides act as agonists of TLR7 and 8. Bioorganic Med. Chem. Lett. 2009, 19, 2044–2047. [Google Scholar] [CrossRef] [PubMed]
- Lan, T.; Putta, M.R.; Wang, D.; Dai, M.; Yu, D.; Kandimalla, E.R.; Agrawal, S. Synthetic oligoribonucleotides-containing secondary structures act as agonists of Toll-like receptors 7 and 8. Biochem. Biophys. Res. Commun. 2009, 386, 443–448. [Google Scholar] [CrossRef]
- Lan, T.; Dai, M.; Wang, D.; Zhu, F.G.; Kandimalla, E.R.; Agrawal, S. Toll-like receptor 7 selective synthetic oligoribonucleotide agonists: Synthesis and structure-activity relationship studies. J. Med. Chem. 2009, 52, 6871–6879. [Google Scholar] [CrossRef]
- Lan, T.; Wang, D.; Bhagat, L.; Philbin, V.J.; Yu, D.; Tang, J.X.; Putta, M.R.; Sullivan, T.; La Monica, N.; Kandimalla, E.R.; et al. Design of synthetic oligoribonucleotide-based agonists of Toll-like receptor 3 and their immune response profiles in vitro and in vivo. Org. Biomol. Chem. 2013, 11, 1049–1058. [Google Scholar] [CrossRef]
- Conforti, A.; Cipriani, B.; Peruzzi, D.; Dharmapuri, S.; Kandimalla, E.R.; Agrawal, S.; Mori, F.; Ciliberto, G.; La Monica, N.; Aurisicchio, L. A TLR9 agonist enhances therapeutic effects of telomerase genetic vaccine. Vaccine 2010, 28, 3522–3530. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Kandimalla, E.R.; Yu, D.; Tang, J.X.; Agrawal, S. Oral administration of second-generation immunomodulatory oligonucleotides induces mucosal Th1 immune responses and adjuvant activity. Vaccine 2005, 23, 2614–2622. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Kandimalla, E.R.; Yu, D.; Agrawal, S. Oligodeoxynucleotides containing synthetic immunostimulatory motifs augment potent Th1 immune responses to HBsAg in mice. Int. Immunopharmacol. 2005, 5, 981–991. [Google Scholar] [CrossRef]
- Idera Pharmaceuticals, I. Study of Combination Treatment With IMO-2125 and Ribavirin in Naïve Hepatitis C-infected, Genotype 1 Patients. Available online: https://clinicaltrials.gov/ct2/show/NCT00990938 (accessed on 6 April 2021).
- Agrawal, D.K.; Edwan, J.; Kandimalla, E.R.; Yu, D.; Bhagat, L.; Wang, D.; Agrawal, S. Novel immunomodulatory oligonucleotides prevent development of allergic airway inflammation and airway hyperresponsiveness in asthma. Int. Immunopharmacol. 2004, 4, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, S.; Kandimall, E.R. Synthetic Agonists of Toll-like Receptors and Therapeutic Applications. In Advances in Nucleic Acid Therapeutics; Agrawal, S.G., Michael, J., Eds.; Royal Society of Chemistry: London, UK, 2019; pp. 306–338. [Google Scholar]
- Damiano, V.; Caputo, R.; Bianco, R.; D’Armiento, F.P.; Leonardi, A.; De Placido, S.; Bianco, A.R.; Agrawal, S.; Ciardiello, F.; Tortora, G. Novel toll-like receptor 9 agonist induces epidermal growth factor receptor (EGFR) inhibition and synergistic antitumor activity with EGFR inhibitors. Clin. Cancer Res. 2006, 12, 577–583. [Google Scholar] [CrossRef]
- Damiano, V.; Garofalo, S.; Rosa, R.; Bianco, R.; Caputo, R.; Gelardi, T.; Merola, G.; Racioppi, L.; Garbi, C.; Kandimalla, E.R.; et al. A novel toll-like receptor 9 agonist cooperates with trastuzumab in trastuzumab-resistant breast tumors through multiple mechanisms of action. Clin. Cancer Res. 2009, 15, 6921–6930. [Google Scholar] [CrossRef]
- Wang, D.; Jiang, W.; Zhu, F.; Mao, X.; Agrawal, S. Modulation of the tumor microenvironment by intratumoral administration of IMO-2125, a novel TLR9 agonist, for cancer immunotherapy. Int. J. Oncol. 2018, 53, 1193–1203. [Google Scholar] [CrossRef]
- Haymaker, C.; Johnson, D.H.; Murthy, R.; Bentebibel, S.E.; Uemura, M.I.; Hudgens, C.W.; Safa, H.; James, M.; Andtbacka, R.H.; Johnson, D.B.; et al. Tilsotolimod with ipilimumab drives tumor responses in anti-PD-1 refractory melanoma. Cancer Discov. 2021. [Google Scholar] [CrossRef] [PubMed]
- Kandimalla, E.R.; Bhagat, L.; Wang, D.; Yu, D.; Sullivan, T.; La Monica, N.; Agrawal, S. Design, synthesis and biological evaluation of novel antagonist compounds of Toll-like receptors 7, 8 and 9. Nucleic Acids Res. 2013, 41, 3947–3961. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yu, D.; Wang, D.; Zhu, F.G.; Bhagat, L.; Dai, M.; Kandimalla, E.R.; Agrawal, S. Modifications incorporated in CpG motifs of oligodeoxynucleotides lead to antagonist activity of toll-like receptors 7 and 9. J. Med. Chem. 2009, 52, 5108–5114. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Bhagat, L.; Yu, D.; Zhu, F.G.; Tang, J.X.; Kandimalla, E.R.; Agrawal, S. Oligodeoxyribonucleotide-based antagonists for Toll-like receptors 7 and 9. J. Med. Chem. 2009, 52, 551–558. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Zhu, F.G.; Bhagat, L.; Yu, D.; Tang, J.X.; Kandimalla, E.R.; La Monica, N.; Agrawal, S. A Toll-like receptor 7, 8, and 9 antagonist inhibits Th1 and Th17 responses and inflammasome activation in a model of IL-23-induced psoriasis. J. Invest. Dermatol. 2013, 133, 1777–1784. [Google Scholar] [CrossRef]
- Zhu, F.G.; Jiang, W.; Bhagat, L.; Wang, D.; Yu, D.; Tang, J.X.; Kandimalla, E.R.; La Monica, N.; Agrawal, S. A novel antagonist of Toll-like receptors 7, 8 and 9 suppresses lupus disease-associated parameters in NZBW/F1 mice. Autoimmunity 2013, 46, 419–428. [Google Scholar] [CrossRef]
- Zhu, F.; Wang, D.; Jiang, W.; Bhagat, L.; Agrawal, S. Sa1757 Targeting Innate Immune Receptors to Treat Inflammatory Bowel Disease: Preclinical Activity of IMO-9200, an Antagonist of TLRS 7, 8, and 9 in Mouse Models of Colitis. Gastroenterology 2015, 148, S-324. [Google Scholar] [CrossRef]
- Bhagat, L.; Wang, D.; Jiang, W.; Agrawal, S. Abstract 2570: IMO-8400, a selective antagonist of TLRs 7, 8 and 9, inhibits MYD88 L265P mutation-driven signaling and cell survival: A potential novel approach for treatment of B-cell lymphomas harboring MYD88 L265P mutation. Cancer Res. 2014, 74, 2570. [Google Scholar]
- Idera Pharmaceuticals, I. Trial of IMO-3100 in Patients with Moderate to Severe Plaque Psoriasis. Available online: https://www.clinicaltrials.gov/ct2/show/NCT01622348 (accessed on 6 April 2021).
- Idera Pharmaceuticals, I. Extension Study of IMO-8400 in Patients with Waldenström’s Macroglobulinemia Who Completed Study 8400-8401. Available online: https://clinicaltrials.gov/ct2/show/NCT02363439 (accessed on 6 April 2021).
- Bhagat, L.; Putta, M.R.; Wang, D.; Yu, D.; Lan, T.; Jiang, W.; Sun, Z.; Wang, H.; Tang, J.X.; La Monica, N.; et al. Novel oligonucleotides containing two 3’-ends complementary to target mRNA show optimal gene-silencing activity. J. Med. Chem. 2011, 54, 3027–3036. [Google Scholar] [CrossRef] [PubMed]
- Iyer, R.P.; Yu, D.; Agrawal, S. Prodrugs of Oligonucleotides: The Acyloxyalkyl Esters of Oligodeoxyribonucleoside Phosphorothioates. Bioorganic Chem. 1995, 23, 1–21. [Google Scholar] [CrossRef]
- Tang, J.Y.; Temsamani, J.; Agrawal, S. Self-stabilized antisense oligodeoxynucleotide phosphorothioates: Properties and anti-HIV activity. Nucleic Acids Res. 1993, 21, 2729–2735. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Lu, Z.; Zhang, X.; Zhao, H.; Diasio, R.B.; Liu, T.; Jiang, Z.; Agrawal, S. In vivo stability and disposition of a self-stabilized oligodeoxynucleotide phosphorothioate in rats. Clin. Chem. 1995, 41, 836–843. [Google Scholar] [CrossRef] [PubMed]
- Kandimalla, E.R.; Manning, A.; Lathan, C.; Byrn, R.A.; Agrawal, S. Design, biochemical, biophysical and biological properties of cooperative antisense oligonucleotides. Nucleic Acids Res. 1995, 23, 3578–3584. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chaix, C.; Iyer, R.P.; Agrawal, S. 3′-3′-linked oligonucleotides: Synthesis and stability studies. Bioorganic Med. Chem. Lett. 1996, 6, 827–832. [Google Scholar]
- Summerton, J.; Weller, D. Morpholino antisense oligomers: Design, preparation, and properties. Antisense Nucleic Acid Drug Dev. 1997, 7, 187–195. [Google Scholar] [CrossRef]
- Tang, J.; Roskey, A.; Li, Y.; Agrawal, S. Enzymatic Synthesis of Stereoregular (All Rp) Oligonucleotide Phosphorothioate and Its Properties. Nucleosides Nucleotides 1995, 14, 985–990. [Google Scholar] [CrossRef]
- Iyer, R.P.; Guo, M.-J.; Yu, D.; Agrawal, S. Solid-phase stereoselective synthesis of oligonucleoside phosphorothioates: The nucleoside bicyclic oxazaphospholidines as novel synthons. Tetrahedron Lett. 1998, 39, 2491–2494. [Google Scholar] [CrossRef]
- Yu, D.; Kandimalla, E.R.; Roskey, A.; Zhao, Q.; Chen, L.; Chen, J.; Agrawal, S. Stereo-enriched phosphorothioate oligodeoxynucleotides: Synthesis, biophysical and biological properties. Bioorganic Med. Chem. 2000, 8, 275–284. [Google Scholar] [CrossRef]
- Agrawal, S. RNA Therapeutics Are Stepping Out of the Maze. Trends Mol. Med. 2020, 26, 1061–1064. [Google Scholar] [CrossRef]
- Kim, J.; Hu, C.; Moufawad El Achkar, C.; Black, L.E.; Douville, J.; Larson, A.; Pendergast, M.K.; Goldkind, S.F.; Lee, E.A.; Kuniholm, A.; et al. Patient-Customized Oligonucleotide Therapy for a Rare Genetic Disease. N. Engl. J. Med. 2019, 381, 1644–1652. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Agrawal, S. The Evolution of Antisense Oligonucleotide Chemistry—A Personal Journey. Biomedicines 2021, 9, 503. https://doi.org/10.3390/biomedicines9050503
Agrawal S. The Evolution of Antisense Oligonucleotide Chemistry—A Personal Journey. Biomedicines. 2021; 9(5):503. https://doi.org/10.3390/biomedicines9050503
Chicago/Turabian StyleAgrawal, Sudhir. 2021. "The Evolution of Antisense Oligonucleotide Chemistry—A Personal Journey" Biomedicines 9, no. 5: 503. https://doi.org/10.3390/biomedicines9050503
APA StyleAgrawal, S. (2021). The Evolution of Antisense Oligonucleotide Chemistry—A Personal Journey. Biomedicines, 9(5), 503. https://doi.org/10.3390/biomedicines9050503