
High Antiproliferative Activity of Hydroxythiopyridones over Hydroxypyridones and Their Organoruthenium Complexes

 ,
,  , , ,
, , ,  ,
,
Abstract
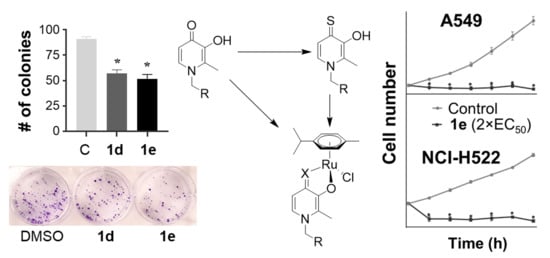
1. Introduction
2. Experimental Section
2.1. Materials and Methods
2.2. Syntheses
2.2.1. General Procedure for the Synthesis of 1-Substituted 2-Methyl-3-hydroxypyridin-4-(1H)-thiones (1d–1f)
2.2.2. 1-Benzyl-2-methyl-3-hydroxypyridin-4(1H)-thione, 1d
2.2.3. 1-Ethylbenzyl-2-methyl-3-hydroxypyridin-4(1H)-thione, 1e
2.2.4. 1-(p-Methylbenzyl)-2-methyl-3-hydroxypyridin-4(1H)-thione, 1f
2.3. General Procedure for the Synthesis of Ru(η6-p-Cymene) Complexes (2a–2f)
2.3.1. [Chlorido{3-oxo-1-benzyl-2-methylpyridin-4(1H)-onato-κ2O,O}(η6-p-cymene)ruthenium(II)], 2a
2.3.2. [Chlorido{3-oxo-1-ethylbenzyl-2-methylpyridin-4(1H)-onato-κ2O,O}(η6-p-cymene)ruthenium(II)], 2b
2.3.3. [Chlorido{3-oxo-1-(p-methylbenzyl)-2-methylpyridin-4(1H)-onato-κ2O,O}(η6-p-cymene)ruthenium(II)], 2c
2.3.4. [Chlorido{3-oxo-1-benzyl-2-methylpyridin-4(1H)-thionato-κ2O,S}(η6-p-cymene)ruthenium(II)], 2d
2.3.5. [Chlorido{3-oxo-1-ethylbenzyl-2-methylpyridin-4(1H)-thionato-κ2O,S}(η6-p-cymene)ruthenium(II)], 2e
2.3.6. [Chlorido{3-oxo-1-(p-methylbenzyl)-2-methylpyridin-4(1H)-thionato-κ2O,S}(η6-p-cymene)ruthenium(II)], 2f
2.4. Antiproliferative Activity Studies
2.4.1. Cell Maintenance
2.4.2. Drug Preparation
2.4.3. Sulforhodamine B (SRB) Cytotoxicity Assay
2.5. Clonogenic Assay
2.6. Cell Cycle Analysis
2.7. Apoptosis Assay
2.8. HDAC Inhibition
2.9. Western Blotting
2.10. Data Analysis
3. Results and Discussion
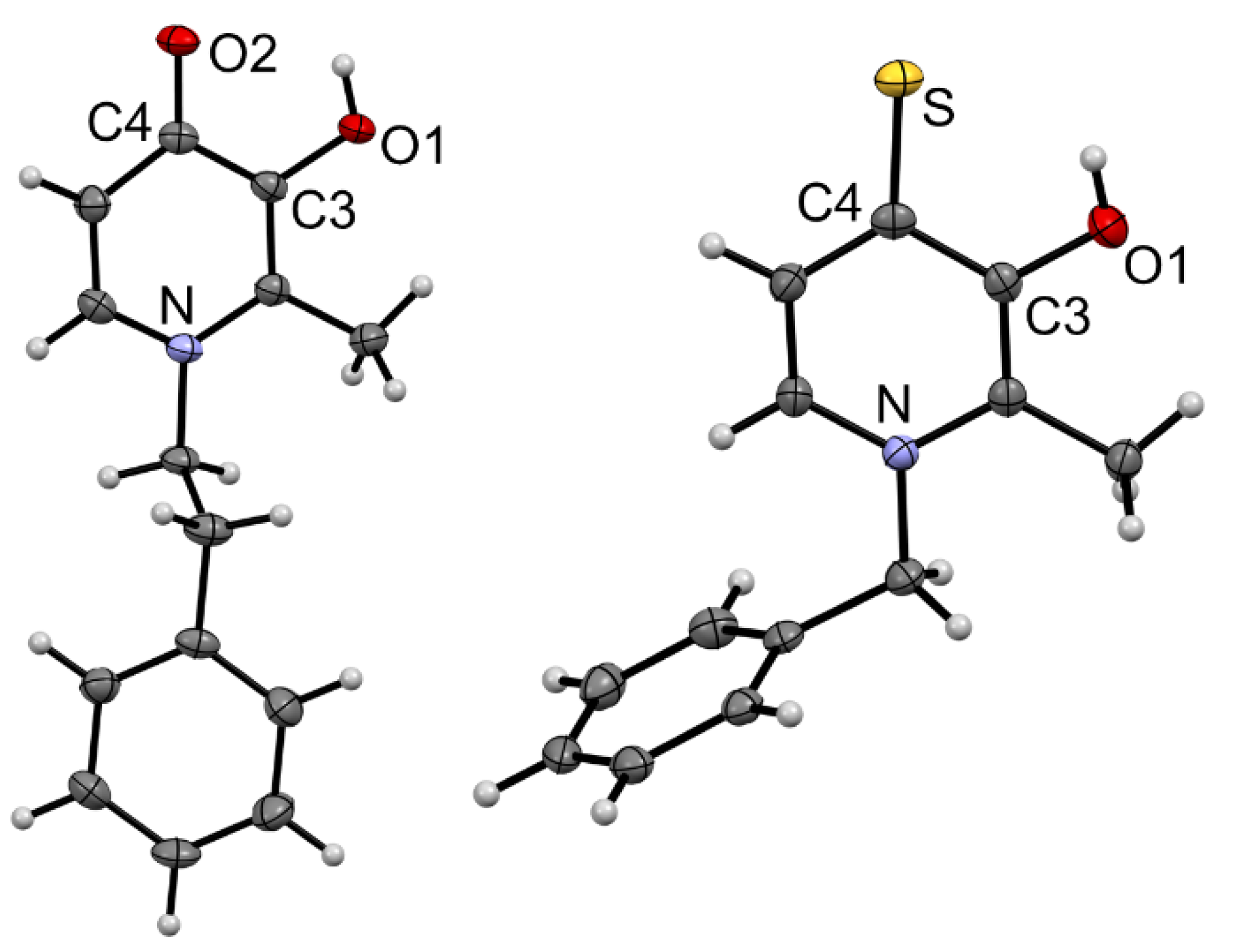
3.1. Synthesis and Characterization
3.2. Biological Properties
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Lewis, J.A.; Mongan, J.; McCammon, J.A.; Cohen, S.M. Evaluation and binding-mode prediction of thiopyrone-based inhibitors of anthrax lethal factor. ChemMedChem 2006, 1, 694–697. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.-L.; Miller, M.T.; Cao, Y.; Cohen, S.M. Synthesis of hydroxypyrone- and hydroxythiopyrone-based matrix metalloproteinase inhibitors: Developing a structure-activity relationship. Bioorg. Med. Chem. Lett. 2009, 19, 1970–1976. [Google Scholar] [CrossRef] [PubMed]
- Kandioller, W.; Kurzwernhart, A.; Hanif, M.; Meier, S.M.; Henke, H.; Keppler, B.K.; Hartinger, C.G. Pyrone derivatives and metals: From natural products to metal-based drugs. J. Organomet. Chem. 2011, 696, 999–1010. [Google Scholar] [CrossRef]
- Hanif, M.; Meier, S.M.; Adhireksan, Z.; Henke, H.; Martic, S.; Movassaghi, S.; Labib, M.; Kandioller, W.; Jamieson, S.M.F.; Hejl, M.; et al. Functionalization of Ruthenium(II)(η6-p-cymene)(3-hydroxy-2-pyridone) Complexes with (Thio)Morpholine: Synthesis and Bioanalytical Studies. ChemPlusChem 2017, 82, 841–847. [Google Scholar] [CrossRef]
- Feng, M.H.; van der Does, L.; Bantjes, A. Iron(III)-chelating resins. 3. Synthesis, iron(III)-chelating properties, and in vitro antibacterial activity of compounds containing 3-hydroxy-2-methyl-4(1H)-pyridinone ligands. J. Med. Chem. 1993, 36, 2822–2827. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, A.; de Oliveira, C.A.F.; Cheng, Y.; Jacobsen, J.A.; McCammon, J.A.; Cohen, S.M. Thioamide Hydroxypyrothiones Supersede Amide Hydroxypyrothiones in Potency against Anthrax Lethal Factor. J. Med. Chem. 2009, 52, 1063–1074. [Google Scholar] [CrossRef] [PubMed]
- Pierre, V.C.; Botta, M.; Aime, S.; Raymond, K.N. Tuning the Coordination Number of Hydroxypyridonate-Based Gadolinium Complexes: Implications for MRI Contrast Agents. J. Am. Chem. Soc. 2006, 128, 5344–5345. [Google Scholar] [CrossRef] [PubMed]
- Werner, E.J.; Avedano, S.; Botta, M.; Hay, B.P.; Moore, E.G.; Aime, S.; Raymond, K.N. Highly Soluble Tris-hydroxypyridonate Gd(III) Complexes with Increased Hydration Number, Fast Water Exchange, Slow Electronic Relaxation, and High Relaxivity. J. Am. Chem. Soc. 2007, 129, 1870–1871. [Google Scholar] [CrossRef]
- Jocher, C.J.; Botta, M.; Avedano, S.; Moore, E.G.; Xu, J.; Aime, S.; Raymond, K.N. Optimized Relaxivity and Stability of [Gd(H(2,2)-1,2-HOPO)(H2O)]- for Use as an MRI Contrast Agent. Inorg. Chem. 2007, 46, 4796–4798. [Google Scholar] [CrossRef]
- Jocher, C.J.; Moore, E.G.; Xu, J.; Avedano, S.; Botta, M.; Aime, S.; Raymond, K.N. 1,2-Hydroxypyridonates as Contrast Agents for Magnetic Resonance Imaging: TREN-1,2-HOPO. Inorg. Chem. 2007, 46, 9182–9191. [Google Scholar] [CrossRef]
- Werner, E.J.; Datta, A.; Jocher, C.J.; Raymond, K.N. High-relaxivity MRI contrast agents: Where coordination chemistry meets medical imaging. Angew. Chem. Int. Ed. 2008, 47, 8568–8580. [Google Scholar] [CrossRef] [PubMed]
- Datta, A.; Hooker, J.M.; Botta, M.; Francis, M.B.; Aime, S.; Raymond, K.N. High Relaxivity Gadolinium Hydroxypyridonate-Viral Capsid Conjugates: Nanosized MRI Contrast Agents. J. Am. Chem. Soc. 2008, 130, 2546–2552. [Google Scholar] [CrossRef]
- Moore, E.G.; Seitz, M.; Raymond, K.N. Use of YbIII-Centered Near-Infrared (NIR) Luminescence To Determine the Hydration State of a 3,2-HOPO-Based MRI Contrast Agent. Inorg. Chem. 2008, 47, 8571–8573. [Google Scholar] [CrossRef][Green Version]
- Datta, A.; Raymond, K.N. Gd-Hydroxypyridinone (HOPO)-Based High-Relaxivity Magnetic Resonance Imaging (MRI) Contrast Agents. Acc. Chem. Res. 2009, 42, 938–947. [Google Scholar] [CrossRef]
- Jacobsen, F.E.; Lewis, J.A.; Cohen, S.M. The design of inhibitors for medicinally relevant metalloproteins. ChemMedChem 2007, 2, 152–171. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-M.; Fan, X.; Yang, S.-M.; Scannevin, R.H.; Burke, S.L.; Rhodes, K.J.; Jackson, P.F. Syntheses and in vitro evaluation of arylsulfone-based MMP inhibitors with heterocycle-derived zinc-binding groups (ZBGs). Bioorg. Med. Chem. Lett. 2008, 18, 405–408. [Google Scholar] [CrossRef] [PubMed]
- Piotrowski, H.; Polborn, K.; Hilt, G.; Severin, K. A Self-Assembled Metallomacrocyclic Ionophore with High Affinity and Selectivity for Li+ and Na+. J. Am. Chem. Soc. 2001, 123, 2699–2700. [Google Scholar] [CrossRef] [PubMed]
- Grote, Z.; Lehaire, M.-L.; Scopelliti, R.; Severin, K. Selective Complexation of Li+ in Water at Neutral pH Using a Self-Assembled Ionophore. J. Am. Chem. Soc. 2003, 125, 13638–13639. [Google Scholar] [CrossRef] [PubMed]
- Grote, Z.; Scopelliti, R.; Severin, K. pH-Triggered Assembly of Organometallic Receptors for Lithium Ions. J. Am. Chem. Soc. 2004, 126, 16959–16972. [Google Scholar] [CrossRef]
- Jakusch, T.; Gajda-Schrantz, K.; Adachi, Y.; Sakurai, H.; Kiss, T.; Horvath, L. Solution equilibrium characterization of insulin-mimetic Zn(II) complexes. J. Inorg. Biochem. 2006, 100, 1521–1526. [Google Scholar] [CrossRef]
- Enyedy, E.A.; Lakatos, A.; Horvath, L.; Kiss, T. Interactions of insulin-mimetic zinc(II) complexes with cell constituents: Glutathione and ATP. J. Inorg. Biochem. 2008, 102, 1473–1485. [Google Scholar] [CrossRef] [PubMed]
- Jakusch, T.; Hollender, D.; Enyedy, E.A.; Gonzalez, C.S.; Montes-Bayon, M.; Sanz-Medel, A.; Pessoa, J.C.; Tomaz, I.; Kiss, T. Biospeciation of various antidiabetic VIVO compounds in serum. Dalton Trans. 2009, 13, 2428–2437. [Google Scholar] [CrossRef] [PubMed]
- Katoh, A.; Matsumura, Y.; Yoshikawa, Y.; Yasui, H.; Sakurai, H. Evaluation of insulin-mimetic activities of vanadyl and zinc(II) complexes from the viewpoint of heterocyclic bidentate ligands. J. Inorg. Biochem. 2009, 103, 567–574. [Google Scholar] [CrossRef]
- Santos, M.A. Recent developments on 3-hydroxy-4-pyridinones with respect to their clinical applications. Mono and combined ligand approaches. Coord. Chem. Rev. 2008, 252, 1213–1224. [Google Scholar] [CrossRef]
- Grazina, R.; Gano, L.; Sebestik, J.; Santos, M.A. New tripodal hydroxypyridinone based chelating agents for Fe(III), Al(III) and Ga(III): Synthesis, physico-chemical properties and bioevaluation. J. Inorg. Biochem. 2009, 103, 262–273. [Google Scholar] [CrossRef]
- Liu, Z.D.; Hider, R.C. Design of iron chelators with therapeutic application. Coord. Chem. Rev. 2002, 232, 151–171. [Google Scholar] [CrossRef]
- Liu, D.Y.; Liu, Z.D.; Hider, R.C. Oral iron chelators—development and application. Best Pract. Res. Clin. Haematol. 2002, 15, 369–384. [Google Scholar] [CrossRef]
- Khodaverdian, V.; Tapadar, S.; MacDonald, I.A.; Xu, Y.; Ho, P.-Y.; Bridges, A.; Rajpurohit, P.; Sanghani, B.A.; Fan, Y.; Thangaraju, M.; et al. Deferiprone: Pan-selective Histone Lysine Demethylase Inhibition Activity and Structure Activity Relationship Study. Sci. Rep. 2019, 9, 4802. [Google Scholar] [CrossRef]
- Hoyes, K.P.; Hider, R.C.; Porter, J.B. Cell Cycle Synchronization and Growth Inhibition by 3-Hydroxypyridin-4-one Iron Chelators in Leukemia Cell Lines. Cancer Res. 1992, 52, 4591–4599. [Google Scholar]
- Cooper, C.E.; Lynagh, G.R.; Hoyes, K.P.; Hider, R.C.; Cammack, R.; Porter, J.B. The Relationship of Intracellular Iron Chelation to the Inhibition and Regeneration of Human Ribonucleotide Reductase. J. Biol. Chem. 1996, 271, 20291–20299. [Google Scholar] [CrossRef]
- Maclean, K.H.; Cleveland, J.L.; Porter, J.B. Cellular zinc content is a major determinant of iron chelator–induced apoptosis of thymocytes. Blood 2001, 98, 3831–3839. [Google Scholar] [CrossRef] [PubMed]
- Yasumoto, E.; Nakano, K.; Nakayachi, T.; Morshed, S.R.M.; Hashimoto, K.; Kikuchi, H.; Nishikawa, H.; Kawase, M.; Sakagami, H. Cytotoxic Activity of Deferiprone, Maltol and Related Hydroxyketones against Human Tumor Cell Lines. Anticancer Res. 2004, 24, 755–762. [Google Scholar] [PubMed]
- Agrawal, A.; Johnson, S.L.; Jacobsen, J.A.; Miller, M.T.; Chen, L.-H.; Pellecchia, M.; Cohen, S.M. Chelator Fragment Libraries for Targeting Metalloproteinases. ChemMedChem 2010, 5, 195–199. [Google Scholar] [CrossRef] [PubMed]
- Hanif, M.; Schaaf, P.; Kandioller, W.; Hejl, M.; Jakupec, M.A.; Roller, A.; Keppler, B.K.; Hartinger, C.G. Influence of the Arene Ligand and the Leaving Group on the Anticancer Activity of (Thio)maltol Ruthenium(II)–(η6-Arene) Complexes. Aust. J. Chem. 2010, 63, 1521–1528. [Google Scholar] [CrossRef]
- Harringer, S.; Happl, B.; Ozenil, M.; Kast, C.; Hejl, M.; Wernitznig, D.; Legin, A.A.; Schweikert, A.; Gajic, N.; Roller, A.; et al. Synthesis, Modification, and Biological Evaluation of a Library of Novel Water-Soluble Thiopyridone-Based Organometallic Complexes and Their Unexpected (Biological) Behavior. Chem. Eur. J. 2020, 26, 5419–5433. [Google Scholar] [CrossRef]
- Patil, V.; Sodji, Q.H.; Kornacki, J.R.; Mrksich, M.; Oyelere, A.K. 3-Hydroxypyridin-2-thione as novel zinc binding group for selective histone deacetylase inhibition. J. Med. Chem. 2013, 56, 3492–3506. [Google Scholar] [CrossRef] [PubMed]
- Sodji, Q.H.; Patil, V.; Kornacki, J.R.; Mrksich, M.; Oyelere, A.K. Synthesis and structure-activity relationship of 3-hydroxypyridine-2-thione-based histone deacetylase inhibitors. J. Med. Chem. 2013, 56, 9969–9981. [Google Scholar] [CrossRef]
- Kandioller, W.; Hartinger, C.G.; Nazarov, A.A.; Bartel, C.; Skocic, M.; Jakupec, M.A.; Arion, V.B.; Keppler, B.K. Maltol-Derived Ruthenium-Cymene Complexes with Tumor Inhibiting Properties: The Impact of Ligand-Metal Bond Stability on Anticancer Activity In Vitro. Chem. Eur. J. 2009, 15, 12283–12291. [Google Scholar] [CrossRef]
- Kandioller, W.; Hartinger, C.G.; Nazarov, A.A.; Kuznetsov, M.L.; John, R.O.; Bartel, C.; Jakupec, M.A.; Arion, V.B.; Keppler, B.K. From Pyrone to Thiopyrone Ligands-Rendering Maltol-Derived Ruthenium(II)-Arene Complexes That Are Anticancer Active in Vitro. Organometallics 2009, 28, 4249–4251. [Google Scholar] [CrossRef]
- Hanif, M.; Henke, H.; Meier, S.M.; Martic, S.; Labib, M.; Kandioller, W.; Jakupec, M.A.; Arion, V.B.; Kraatz, H.-B.; Keppler, B.K.; et al. Is the Reactivity of M(II)-Arene Complexes of 3-Hydroxy-2(1H)-pyridones to Biomolecules the Anticancer Activity Determining Parameter? Inorg. Chem. 2010, 49, 7953–7963. [Google Scholar] [CrossRef]
- Parveen, S.; Hanif, M.; Movassaghi, S.; Sullivan, M.P.; Kubanik, M.; Shaheen, M.A.; Söhnel, T.; Jamieson, S.M.F.; Hartinger, C.G. Cationic Ru(η6-p-cymene) Complexes of 3-Hydroxy-4-pyr(id)ones—Lipophilic Triphenylphosphine as Co-Ligand Is Key to Highly Stable and Cytotoxic Anticancer Agents. Eur. J. Inorg. Chem. 2017, 2017, 1721–1727. [Google Scholar] [CrossRef]
- Mendoza-Ferri, M.G.; Hartinger, C.G.; Mendoza, M.A.; Groessl, M.; Egger, A.E.; Eichinger, R.E.; Mangrum, J.B.; Farrell, N.P.; Maruszak, M.; Bednarski, P.J.; et al. Transferring the concept of multinuclearity to ruthenium complexes for improvement of anticancer activity. J. Med. Chem. 2009, 52, 916–925. [Google Scholar] [CrossRef] [PubMed]
- Parveen, S.; Hanif, M.; Leung, E.; Tong, K.K.H.; Yang, A.; Astin, J.; De Zoysa, G.H.; Steel, T.R.; Goodman, D.; Movassaghi, S.; et al. Anticancer organorhodium and -iridium complexes with low toxicity in vivo but high potency in vitro: DNA damage, reactive oxygen species formation, and haemolytic activity. Chem. Commun. 2019, 55, 12016–12019. [Google Scholar] [CrossRef] [PubMed]
- Hajar, S.; Afshin, F.; Simone, B.; Giuseppe, C.; Frauke, C.; Zeger, D.; Sandra, G.; Stefania, B.; Giulia, C.; Alessandro, G.; et al. Synthesis, Molecular Modelling and Biological Studies of 3-hydroxypyrane-4-one and 3-hydroxy-pyridine-4-one Derivatives as HIV-1 Integrase Inhibitors. Med. Chem. 2019, 15, 755–770. [Google Scholar] [CrossRef]
- Bennett, M.A.; Smith, A.K. Arene ruthenium(II) complexes formed by dehydrogenation by cyclohexadienes with ruthenium(III) trichloride. J. Chem. Soc. Dalton Trans. 1974, 233–241. [Google Scholar] [CrossRef]
- SAINT. Area Detector Integration Software; Siemens Analytical Instruments Inc.: Madison, WI, USA, 1995. [Google Scholar]
- Sheldrick, G.M. SADABS, Program for Semi-Empirical Absorption Correction; University of Göttingen: Göttingen, Germany, 1997. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A Found. Crystallogr. 2008, A64, 112–122. [Google Scholar] [CrossRef]
- Yadav, B.; Taurin, S.; Rosengren, R.J.; Schumacher, M.; Diederich, M.; Somers-Edgar, T.J.; Larsen, L. Synthesis and cytotoxic potential of heterocyclic cyclohexanone analogues of curcumin. Bioorg. Med. Chem. 2010, 18, 6701–6707. [Google Scholar] [CrossRef]
- Badisa, R.B.; Darling-Reed, S.F.; Joseph, P.; Cooperwood, J.S.; Latinwo, L.M.; Goodman, C.B. Selective cytotoxic activities of two novel synthetic drugs on human breast carcinoma MCF-7 cells. Anticancer Res. 2009, 29, 2993–2996. [Google Scholar]
- Tripathi, S.K.; Rengasamy, K.R.R.; Biswal, B.K. Plumbagin engenders apoptosis in lung cancer cells via caspase-9 activation and targeting mitochondrial-mediated ROS induction. Arch. Pharm. Res. 2020, 43, 242–256. [Google Scholar] [CrossRef]
- Shrestha, N.; Nimick, M.; Dass, P.; Rosengren, R.J.; Ashton, J.C. Mechanisms of suppression of cell growth by dual inhibition of ALK and MEK in ALK-positive non-small cell lung cancer. Sci. Rep. 2019, 9, 18842. [Google Scholar] [CrossRef]
- Chen, S.; Nimick, M.; Cridge, A.G.; Hawkins, B.C.; Rosengren, R.J. Anticancer potential of novel curcumin analogs towards castrate-resistant prostate cancer. Int. J. Oncol. 2018, 52, 579–588. [Google Scholar] [CrossRef] [PubMed]
- Henke, H.; Kandioller, W.; Hanif, M.; Keppler, B.K.; Hartinger, C.G. Organometallic Ru and Os compounds of 2- and 4-Pyridones as Potential Anticancer Agents. Chem. Biodivers. 2012, 9, 1718–1727. [Google Scholar] [CrossRef] [PubMed]
- Harringer, S.; Wernitznig, D.; Gajic, N.; Diridl, A.; Wenisch, D.; Hejl, M.; Jakupec, M.A.; Theiner, S.; Koellensperger, G.; Kandioller, W.; et al. Introducing N-, P-, and S-donor leaving groups: An investigation of the chemical and biological properties of ruthenium, rhodium and iridium thiopyridone piano stool complexes. Dalton Trans. 2020, 49, 15693–15711. [Google Scholar] [CrossRef] [PubMed]
- Rotow, J.; Bivona, T.G. Understanding and targeting resistance mechanisms in NSCLC. Nat. Rev. Cancer 2017, 17, 637–658. [Google Scholar] [CrossRef]
- Phi, L.T.H.; Sari, I.N.; Yang, Y.G.; Lee, S.H.; Jun, N.; Kim, K.S.; Lee, Y.K.; Kwon, H.Y. Cancer Stem Cells (CSCs) in Drug Resistance and their Therapeutic Implications in Cancer Treatment. Stem Cells Int. 2018, 2018, 5416923. [Google Scholar] [CrossRef]
- Parvathaneni, V.; Kulkarni, N.S.; Shukla, S.K.; Farrales, P.T.; Kunda, N.K.; Muth, A.; Gupta, V. Systematic Development and Optimization of Inhalable Pirfenidone Liposomes for Non-Small Cell Lung Cancer Treatment. Pharmaceutics 2020, 12, 206. [Google Scholar] [CrossRef]
- Lee, W.Y.; Chen, P.C.; Wu, W.S.; Wu, H.C.; Lan, C.H.; Huang, Y.H.; Cheng, C.H.; Chen, K.C.; Lin, C.W. Panobinostat sensitizes KRAS-mutant non-small-cell lung cancer to gefitinib by targeting TAZ. Int. J. Cancer 2017, 141, 1921–1931. [Google Scholar] [CrossRef]
- Rana, Z.; Diermeier, S.; Hanif, M.; Rosengren, R.J. Understanding Failure and Improving Treatment Using HDAC Inhibitors for Prostate Cancer. Biomedicines 2020, 8, 22. [Google Scholar] [CrossRef]
- Hanif, M.; Arshad, J.; Astin, J.W.; Rana, Z.; Zafar, A.; Movassaghi, S.; Leung, E.; Patel, K.; Sohnel, T.; Reynisson, J.; et al. A Multitargeted Approach: Organorhodium Anticancer Agent Based on Vorinostat as a Potent Histone Deacetylase Inhibitor. Angew. Chem. Int. Ed. Engl. 2020, 59, 14609–14614. [Google Scholar] [CrossRef]
- Li, Y.; Fan, J.; Ju, D. Neurotoxicity concern about the brain targeting delivery systems. In Brain Targeted Drug Delivery System; Elsevier: Amsterdam, The Netherlands, 2019; pp. 377–408. [Google Scholar] [CrossRef]
- Shapiro, G.I.; Harper, J.W. Anticancer drug targets: Cell cycle and checkpoint control. J. Clin. Investig. 1999, 104, 1645–1653. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50/µM | |||
|---|---|---|---|---|
| HCT116 | NCI-H460 | SW480 | SiHa | |
| 1a | 11 ± 2 | 10 ± 1 | 13 ± 0.1 | 25 ± 1 |
| 1b | 6.3 ± 5.3 | 7.0 ± 2.1 | 9.8 ± 1.1 | 24 ± 8 |
| 1c | 5.4 ± 0.02 | 4.2 ± 0.3 | 5.3 ± 1.0 | 14 ± 0.1 |
| 1d | 0.16 ± 0.04 | 0.42 ± 0.07 | 0.28 ± 0.04 | 0.74 ± 0.06 |
| 1e | 0.06 ± 0.01 | 0.18 ± 0.05 | 0.04 ± 0.03 | 0.40 ± 0.02 |
| 1f | 0.17 ± 0.08 | 1.5 ± 2.5 | 3.6 ± 6.2 | 0.50 ± 0.02 |
| 2a | 16 ± 2 | 13 ± 1 | 14 ± 0.3 | 25 ± 1 |
| 2b | 11 ± 1 | 9.5 ± 3.4 | 11 ± 4 | 23 ± 6 |
| 2c | 13 ± 0.4 | 13 ± 1 | 16 ± 0.4 | 34 ± 0.1 |
| 2d | 0.35 ± 0.04 | 0.94 ± 0.06 | 0.50 ± 0.22 | 1.6 ± 0.1 |
| 2e | 0.48 ± 0.02 | 1.3 ± 0.1 | 1.1 ± 0.1 | 1.4 ± 0.1 |
| 2f | 0.35 ± 0.03 | 0.90 ± 0.13 | 0.79 ± 0.13 | 1.5 ± 0.0 |
| SAHA | 0.46 ± 0.09 | 0.57 ± 0.01 | 1.3 ± 0.1 | 1.6 ± 0.1 |
| Compound | EC50/µM | ||||
|---|---|---|---|---|---|
| A549 | NCI-H522 | MDA-MB-231 | MDA-MB-468 | PC3 | |
| 1d | 0.36 ± 0.01 | 0.28 ± 0.01 | 2.79 ± 0.14 | 1.70 ± 0.19 | 0.33 ± 0.04 |
| 1e | 0.32 ± 0.01 | 0.23 ± 0.02 | 2.64 ± 0.11 | 3.33 ± 0.63 | 1.44 ± 0.21 |
| 2d | 1.73 ± 0.02 | 6.67 ± 0.30 | 7.24 ± 0.31 | 5.46 ± 0.41 | 7.93 ± 0.25 |
| 2e | 1.66 ± 0.06 | 7.42 ± 0.19 | 7.28 ± 0.51 | 8.70 ± 0.51 | 11.11 ± 0.89 |
| SAHA | 1.01 ± 0.05 | 0.35 ± 0.02 | 2.01 ± 0.11 | 5.56 ± 0.64 | 1.13 ± 0.19 |
| Compound | HDAC8 Activity/% |
|---|---|
| 1a | 107 (110,104) |
| 1b | 108 (107,108) |
| 2a | 59 (58,59) |
| 2b | 94 (96,92) |
| 1d | 52 (53,50) |
| 1e | 30 (32,28) |
| 2d | 23 (23,23) |
| 2e | 35 (35,34) |
| SAHA, IC50 = 0.84 ± 0.02 μM [61] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shakil, M.S.; Parveen, S.; Rana, Z.; Walsh, F.; Movassaghi, S.; Söhnel, T.; Azam, M.; Shaheen, M.A.; Jamieson, S.M.F.; Hanif, M.; et al. High Antiproliferative Activity of Hydroxythiopyridones over Hydroxypyridones and Their Organoruthenium Complexes. Biomedicines 2021, 9, 123. https://doi.org/10.3390/biomedicines9020123
Shakil MS, Parveen S, Rana Z, Walsh F, Movassaghi S, Söhnel T, Azam M, Shaheen MA, Jamieson SMF, Hanif M, et al. High Antiproliferative Activity of Hydroxythiopyridones over Hydroxypyridones and Their Organoruthenium Complexes. Biomedicines. 2021; 9(2):123. https://doi.org/10.3390/biomedicines9020123
Chicago/Turabian StyleShakil, Md. Salman, Shahida Parveen, Zohaib Rana, Fearghal Walsh, Sanam Movassaghi, Tilo Söhnel, Mayur Azam, Muhammad Ashraf Shaheen, Stephen M. F. Jamieson, Muhammad Hanif, and et al. 2021. "High Antiproliferative Activity of Hydroxythiopyridones over Hydroxypyridones and Their Organoruthenium Complexes" Biomedicines 9, no. 2: 123. https://doi.org/10.3390/biomedicines9020123
APA StyleShakil, M. S., Parveen, S., Rana, Z., Walsh, F., Movassaghi, S., Söhnel, T., Azam, M., Shaheen, M. A., Jamieson, S. M. F., Hanif, M., Rosengren, R. J., & Hartinger, C. G. (2021). High Antiproliferative Activity of Hydroxythiopyridones over Hydroxypyridones and Their Organoruthenium Complexes. Biomedicines, 9(2), 123. https://doi.org/10.3390/biomedicines9020123