
Alteration in the Cerebrospinal Fluid Lipidome in Parkinson’s Disease: A Post-Mortem Pilot Study

 , ,
, , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
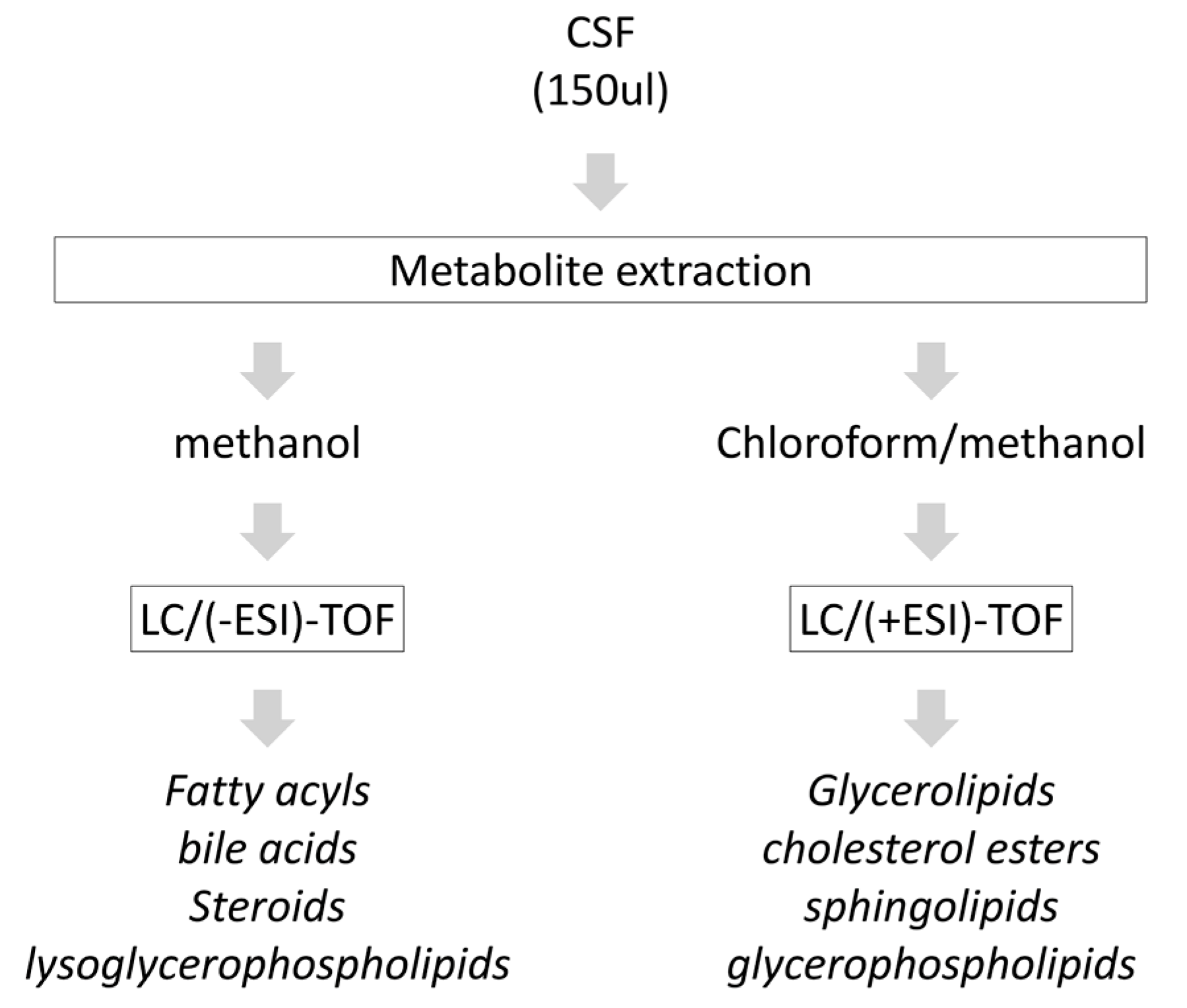
2.2. Metabolite Extraction from CSF
2.3. Chromatography and Mass-Spectrometry
2.4. Data Processing and Normalization
2.5. Data Analysis
3. Results
3.1. Categorization of the Detected CSF Lipidome
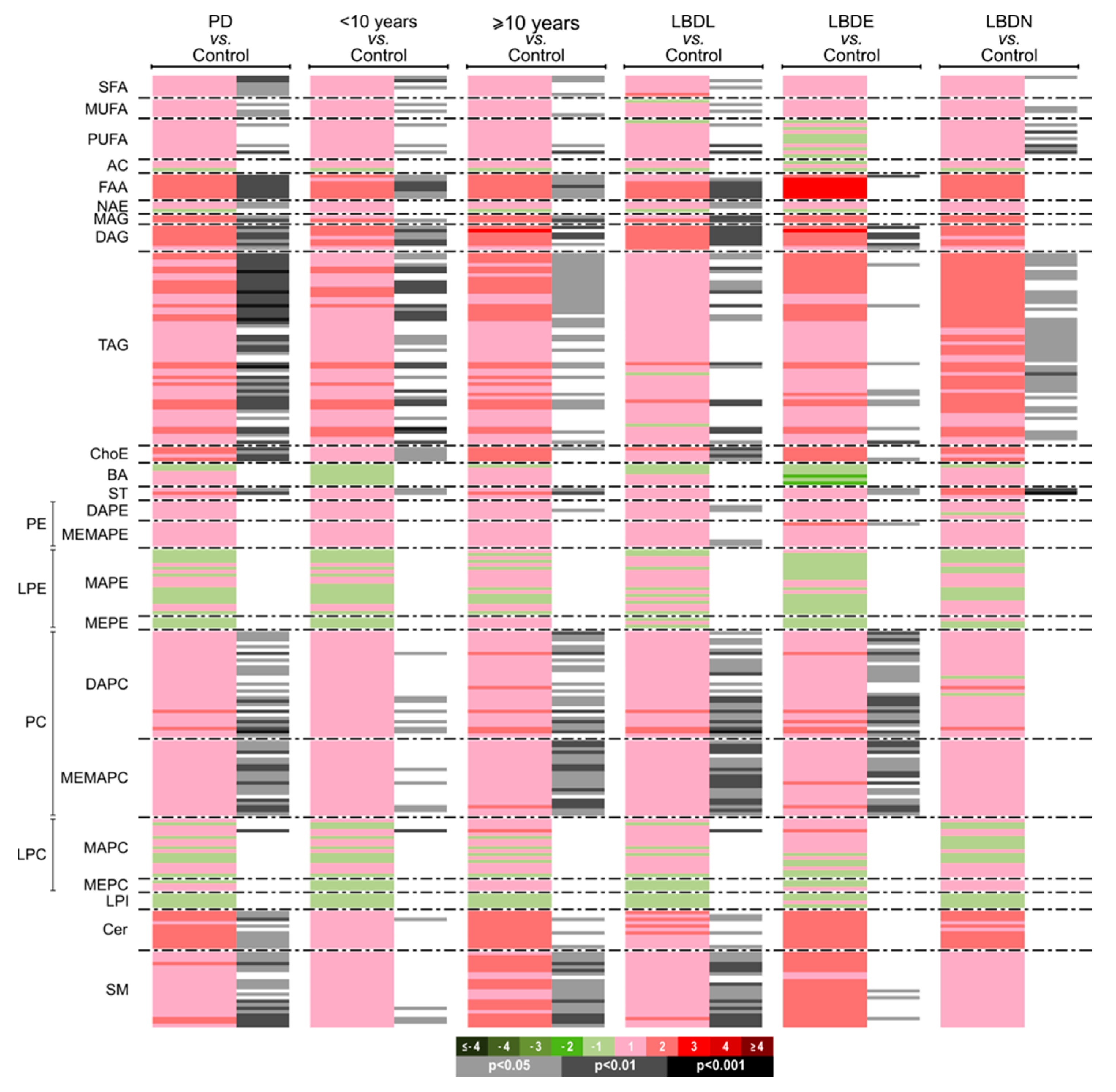
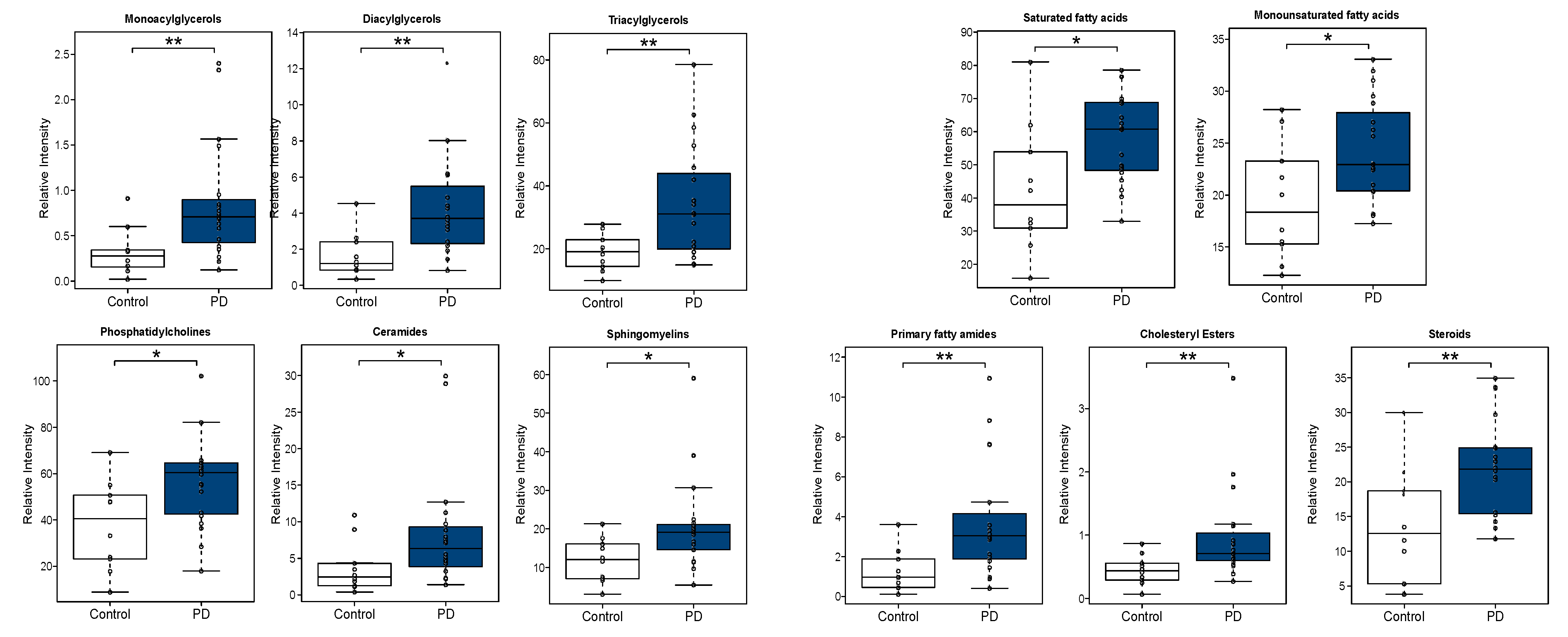
3.2. CSF Lipidomic Profiling in Parkinson’s Disease
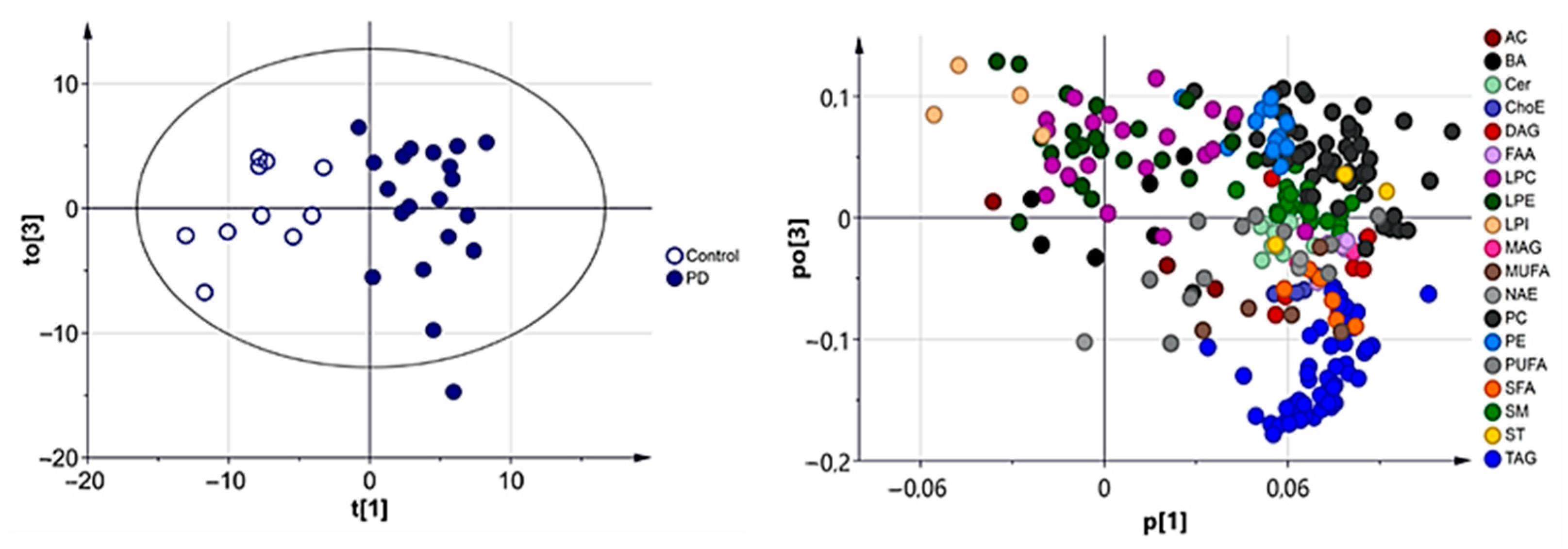
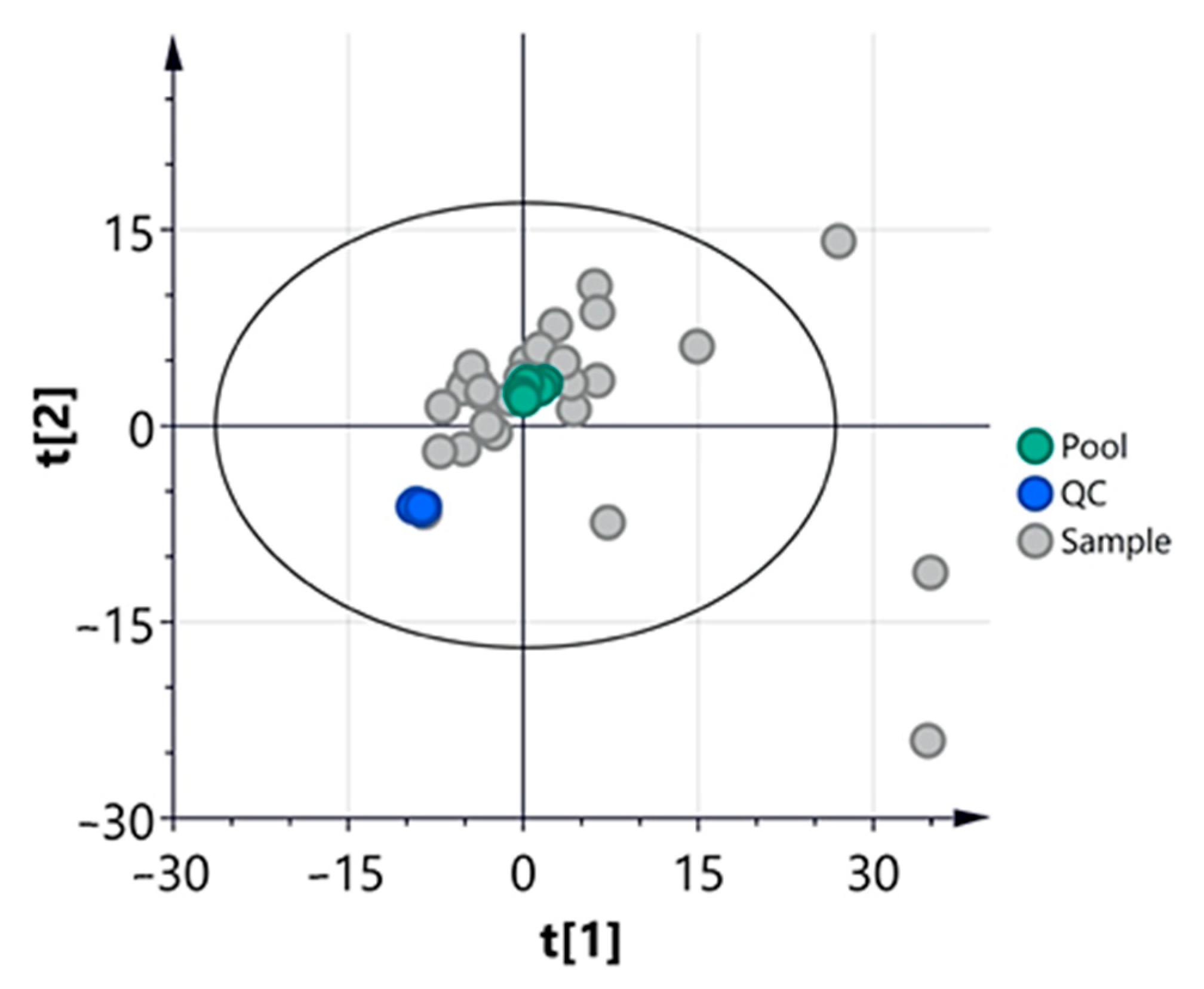
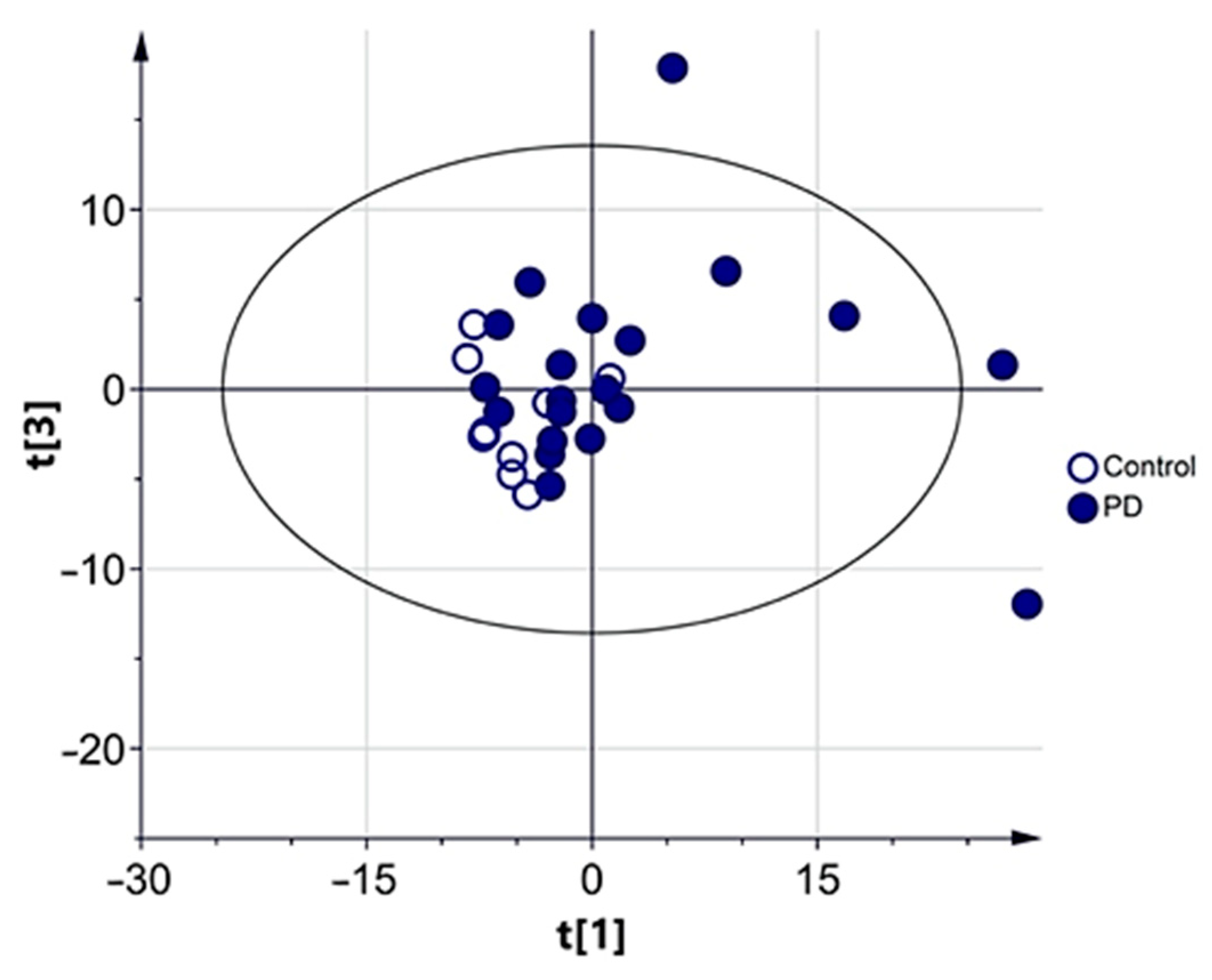
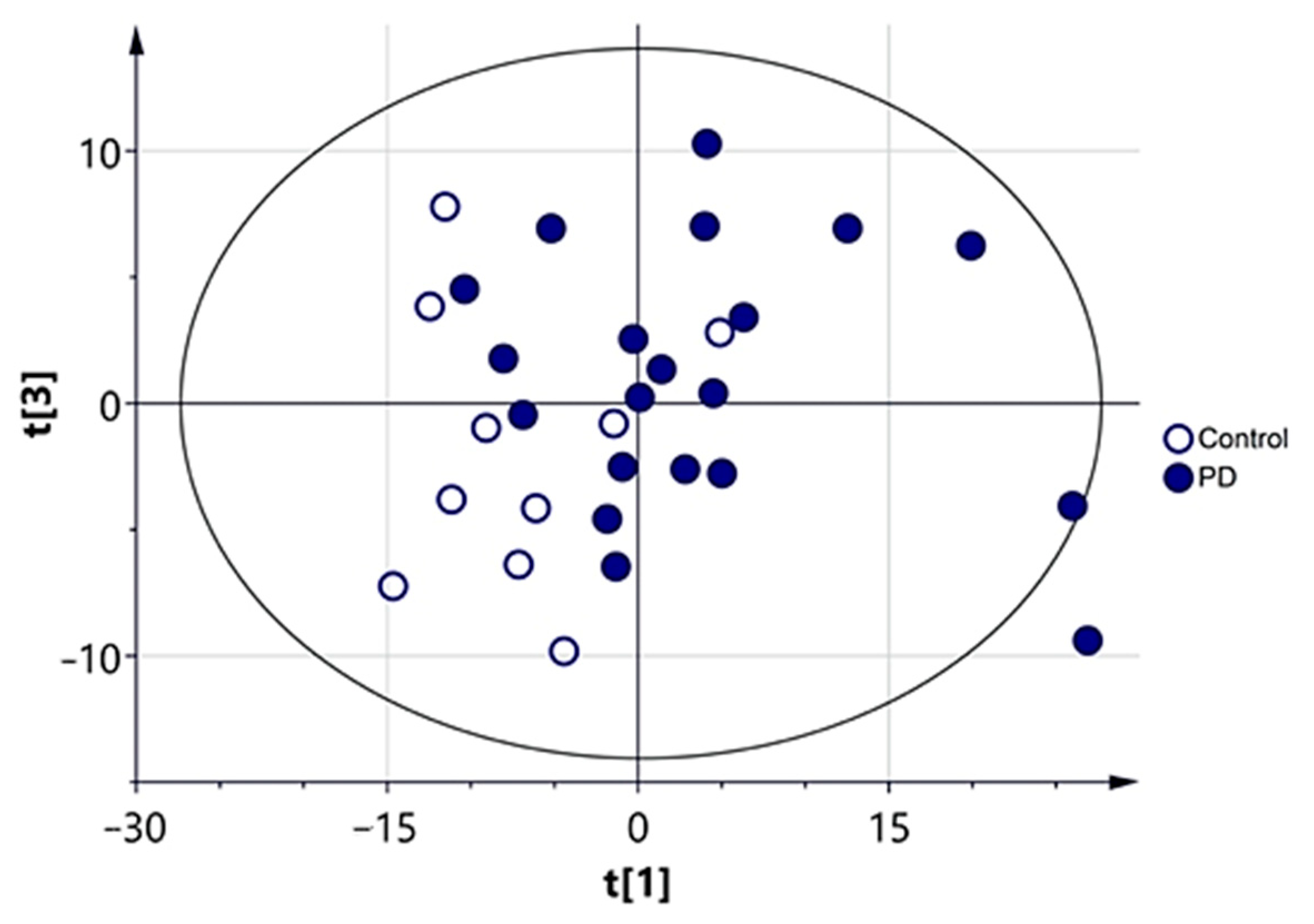
3.2.1. Multivariate Analysis
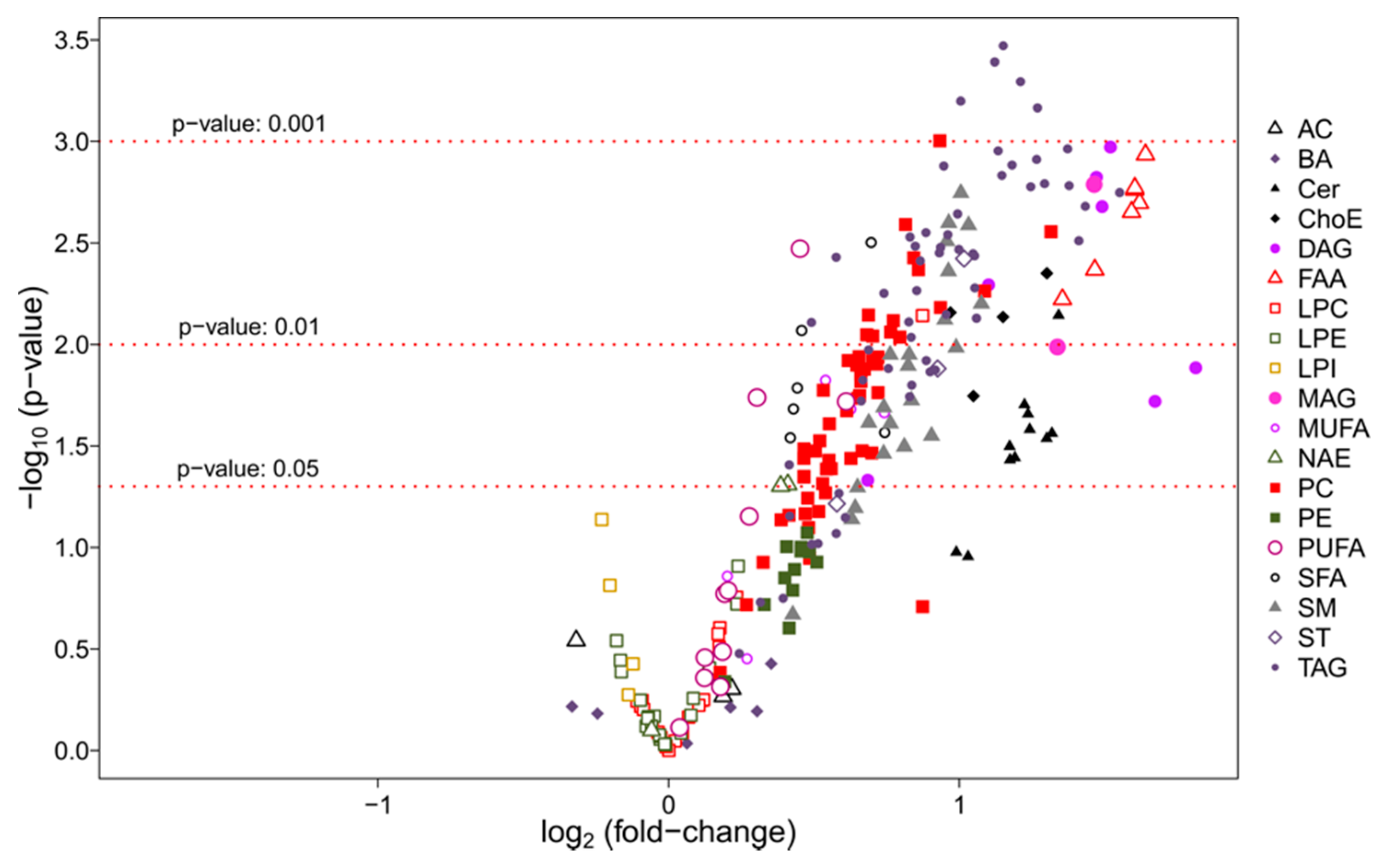
3.2.2. Univariate Analysis
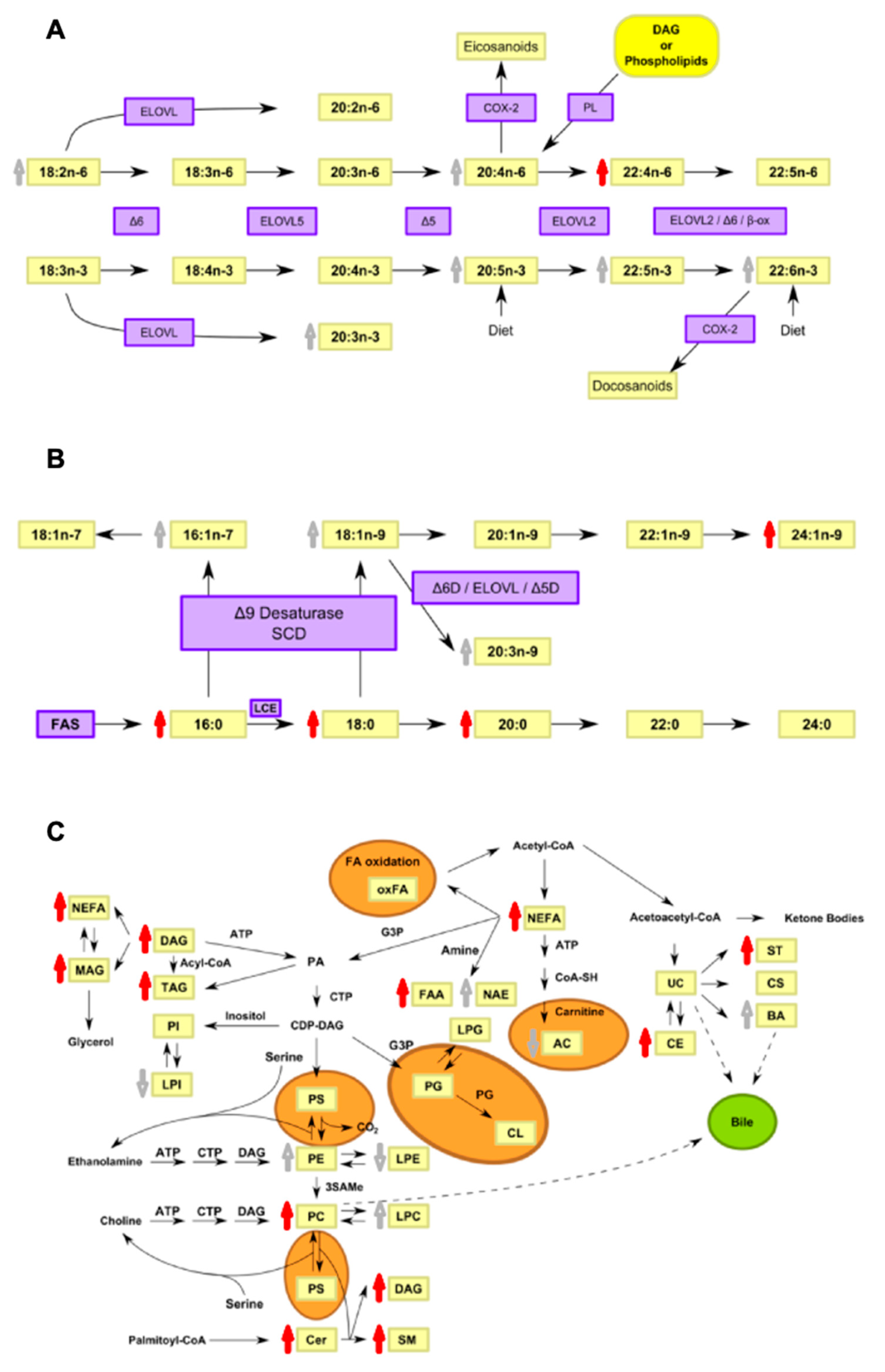
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IS | IS Stock Solution (µg/mL) | IS Stock Solution | IS Intermediate Solution in CHCl3:MeOH (2:1) (µg/mL) | IS Working Solution in MeOH (µg/mL) |
|---|---|---|---|---|
| 13:0 Lyso PC | 10,000 | CHCl3 | 10 | 0.1 |
| Dehydrocholic acid | 5000 | CHCl3:MeOH (1:1) | 30 | 0.3 |
| Nonadecanoic acid | 10,000 | CHCl3 | 500 | 5.0 |
| Tryptophan-(indole-d5) | 5000 | 0.05% Formic acid in water | 200 | 2.0 |
| IS | IS Stock Solution (µg/mL) | IS Stock Solution | Working IS Solution CHCl3:MeOH (2:1) (µg/mL) |
|---|---|---|---|
| SM (d18:1/6:0) | 5000 | CHCl3 | 5 |
| PE (17:0/17:0) | 10,000 | CHCl3:MeOH:H2O | 50 |
| PC (19:0/19:0) | 10,000 | CHCl3 | 10 |
| TG (13:0/13:0/13:0) | 10,000 | CHCl3 | 5 |
| TG (17:0/17:0/17:0) | 10,000 | CHCl3 | 5 |
| Cer(d18:1/17:0) | 10,000 | CHCl3 | 10 |
| ChoE(12:0) | 10,000 | CHCl3 | 250 |




References
- Shevchenko, A.; Simons, K. Lipidomics: Coming to Grips with Lipid Diversity. Nat. Rev. Mol. Cell Biol. 2010, 11, 593–598. [Google Scholar] [CrossRef]
- Brugger, B. Lipidomics: Analysis of the Lipid Composition of Cells and Subcellular Organelles by Electrospray Ionization Mass Spectrometry. Annu. Rev. Biochem. 2014, 83, 79–98. [Google Scholar] [CrossRef]
- Piomelli, D.; Astarita, G.; Rapaka, R. A Neuroscientist’s Guide to Lipidomics. Nat. Rev. Neurosci. 2007, 8, 743–754. [Google Scholar] [CrossRef]
- Gross, R.W.; Han, X. Lipidomics at the Interface of Structure and Function in Systems Biology. Chem. Biol. 2011, 18, 284–291. [Google Scholar] [CrossRef]
- Lauwers, E.; Goodchild, R.; Verstreken, P. Membrane Lipids in Presynaptic Function and Disease. Neuron 2016, 90, 11–25. [Google Scholar] [CrossRef]
- Klemann, C.; Martens, G.J.M.; Sharma, M.; Martens, M.B.; Isacson, O.; Gasser, T.; Visser, J.E.; Poelmans, G. Integrated Molecular Landscape of Parkinson’s Disease. NPJ Parkinsons Dis. 2017, 3, 14. [Google Scholar] [CrossRef]
- Sidransky, E.; Lopez, G. The Link between the GBA Gene and Parkinsonism. Lancet Neurol. 2012, 11, 986–998. [Google Scholar] [CrossRef]
- Do, C.B.; Tung, J.Y.; Dorfman, E.; Kiefer, A.K.; Drabant, E.M.; Francke, U.; Mountain, J.L.; Goldman, S.M.; Tanner, C.M.; Langston, J.W.; et al. Web-Based Genome-Wide Association Study Identifies Two Novel Loci and a Substantial Genetic Component for Parkinson’s Disease. PLoS Genet. 2011, 7, e1002141. [Google Scholar] [CrossRef] [PubMed]
- Pankratz, N.; Wilk, J.B.; Latourelle, J.C.; DeStefano, A.L.; Halter, C.; Pugh, E.W.; Doheny, K.F.; Gusella, J.F.; Nichols, W.C.; Foroud, T.; et al. Genomewide Association Study for Susceptibility Genes Contributing to Familial Parkinson Disease. Hum. Genet 2009, 124, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Robak, L.A.; Jansen, I.E.; van Rooij, J.; Uitterlinden, A.G.; Kraaij, R.; Jankovic, J.; Heutink, P.; Shulman, J.M. Excessive Burden of Lysosomal Storage Disorder Gene Variants in Parkinson’s Disease. Brain 2017, 140, 3191–3203. [Google Scholar] [CrossRef]
- Fanning, S.; Selkoe, D.; Dettmer, U. Vesicle Trafficking and Lipid Metabolism in Synucleinopathy. Acta Neuropathol. 2021, 141, 491–510. [Google Scholar] [CrossRef] [PubMed]
- Galvagnion, C. The Role of Lipids Interacting with Alpha-Synuclein in the Pathogenesis of Parkinson’s Disease. J. Parkinsons Dis. 2017, 7, 433–450. [Google Scholar] [CrossRef] [PubMed]
- Shahmoradian, S.H.; Lewis, A.J.; Genoud, C.; Hench, J.; Moors, T.E.; Navarro, P.P.; Castano-Diez, D.; Schweighauser, G.; Graff-Meyer, A.; Goldie, K.N.; et al. Lewy Pathology in Parkinson’s Disease Consists of Crowded Organelles and Lipid Membranes. Nat. Neurosci. 2019, 22, 1099–1109. [Google Scholar] [CrossRef]
- Seyer, A.; Boudah, S.; Broudin, S.; Junot, C.; Colsch, B. Annotation of the Human Cerebrospinal Fluid Lipidome Using High Resolution Mass Spectrometry and a Dedicated Data Processing Workflow. Metabolomics 2016, 12, 91. [Google Scholar] [CrossRef]
- Naudi, A.; Cabre, R.; Jove, M.; Ayala, V.; Gonzalo, H.; Portero-Otin, M.; Ferrer, I.; Pamplona, R. Lipidomics of Human Brain Aging and Alzheimer’s Disease Pathology. Int. Rev. Neurobiol. 2015, 122, 133–189. [Google Scholar] [PubMed]
- Liebisch, G.; Ahrends, R.; Makoto, A.; Masanori, A.; Bowden, J.A.; Ejsing, C.S.; Griffiths, W.J.; Holcapek, M.; Köfeler, H.; Mitchell, T.W. Lipidomics Needs More Standardization. Nat. Metab. 2019, 1, 745–747. [Google Scholar]
- Proitsi, P.; Kim, M.; Whiley, L.; Simmons, A.; Sattlecker, M.; Velayudhan, L.; Lupton, M.K.; Soininen, H.; Kloszewska, I.; Mecocci, P.; et al. Association of Blood Lipids with Alzheimer’s Disease: A Comprehensive Lipidomics Analysis. Alzheimers Dement 2017, 13, 140–151. [Google Scholar] [CrossRef]
- Wong, M.W.; Braidy, N.; Poljak, A.; Pickford, R.; Thambisetty, M.; Sachdev, P.S. Dysregulation of Lipids in Alzheimer’s Disease and Their Role as Potential Biomarkers. Alzheimers Dement 2017, 13, 810–827. [Google Scholar] [CrossRef] [PubMed]
- Gonzalo, H.; Brieva, L.; Tatzber, F.; Jove, M.; Cacabelos, D.; Cassanye, A.; Lanau-Angulo, L.; Boada, J.; Serrano, J.C.; Gonzalez, C.; et al. Lipidome Analysis in Multiple Sclerosis Reveals Protein Lipoxidative Damage as a Potential Pathogenic Mechanism. J. Neurochem. 2012, 123, 622–634. [Google Scholar] [CrossRef]
- Pieragostino, D.; Cicalini, I.; Lanuti, P.; Ercolino, E.; di Ioia, M.; Zucchelli, M.; Zappacosta, R.; Miscia, S.; Marchisio, M.; Sacchetta, P.; et al. Enhanced Release of Acid Sphingomyelinase-Enriched Exosomes Generates a Lipidomics Signature in CSF of Multiple Sclerosis Patients. Sci. Rep. 2018, 8, 3071. [Google Scholar] [CrossRef]
- Blasco, H.; Veyrat-Durebex, C.; Bocca, C.; Patin, F.; Vourc’h, P.; Kouassi Nzoughet, J.; Lenaers, G.; Andres, C.R.; Simard, G.; Corcia, P.; et al. Lipidomics Reveals Cerebrospinal-Fluid Signatures of ALS. Sci. Rep. 2017, 7, 17652. [Google Scholar] [CrossRef]
- Millan, L.; Fernandez-Irigoyen, J.; Santamaria, E.; Mayo, R. Mass Spectrometry Applied to Human Cerebrospinal Fluid Lipidome. Methods Mol. Biol. 2019, 2044, 353–361. [Google Scholar]
- Barr, J.; Vazquez-Chantada, M.; Alonso, C.; Perez-Cormenzana, M.; Mayo, R.; Galan, A.; Caballeria, J.; Martin-Duce, A.; Tran, A.; Wagner, C.; et al. Liquid Chromatography-Mass Spectrometry-Based Parallel Metabolic Profiling of Human and Mouse Model Serum Reveals Putative Biomarkers Associated with the Progression of Nonalcoholic Fatty Liver Disease. J. Proteome Res. 2010, 9, 4501–4512. [Google Scholar] [CrossRef]
- Fonteh, A.N.; Harrington, R.J.; Huhmer, A.F.; Biringer, R.G.; Riggins, J.N.; Harrington, M.G. Identification of Disease Markers in Human Cerebrospinal Fluid Using Lipidomic and Proteomic Methods. Dis. Markers 2006, 22, 39–64. [Google Scholar] [CrossRef] [PubMed]
- Fanning, S.; Selkoe, D.; Dettmer, U. Parkinson’s Disease: Proteinopathy or Lipidopathy? NPJ Parkinsons Dis. 2020, 6, 3. [Google Scholar] [CrossRef]
- Xicoy, H.; Wieringa, B.; Martens, G.J.M. The Role of Lipids in Parkinson’s Disease. Cells 2019, 8, 27. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Blood-Brain Barrier Breakdown in Alzheimer Disease and Other Neurodegenerative Disorders. Nat. Rev. Neurol. 2018, 14, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Kamel, F.; Goldman, S.M.; Umbach, D.M.; Chen, H.; Richardson, G.; Barber, M.R.; Meng, C.; Marras, C.; Korell, M.; Kasten, M.; et al. Dietary Fat Intake, Pesticide Use, and Parkinson’s Disease. Parkinsonism Relat. Disord. 2014, 20, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Fabelo, N.; Martin, V.; Santpere, G.; Marin, R.; Torrent, L.; Ferrer, I.; Diaz, M. Severe Alterations in Lipid Composition of Frontal Cortex Lipid Rafts from Parkinson’s Disease and Incidental Parkinson’s Disease. Mol. Med. 2011, 17, 1107–1118. [Google Scholar] [CrossRef] [PubMed]
- Mori, A.; Hatano, T.; Inoshita, T.; Shiba-Fukushima, K.; Koinuma, T.; Meng, H.; Kubo, S.I.; Spratt, S.; Cui, C.; Yamashita, C.; et al. Parkinson’s Disease-Associated iPLA2-VIA/PLA2G6 Regulates Neuronal Functions and Alpha-Synuclein Stability Through Membrane Remodeling. Proc. Natl. Acad. Sci. USA 2019, 116, 20689–20699. [Google Scholar] [CrossRef]
- Imberdis, T.; Negri, J.; Ramalingam, N.; Terry-Kantor, E.; Ho, G.P.H.; Fanning, S.; Stirtz, G.; Kim, T.E.; Levy, O.A.; Young-Pearse, T.L.; et al. Cell Models of Lipid-Rich Alpha-Synuclein Aggregation Validate Known Modifiers of Alpha-Synuclein Biology and Identify Stearoyl-CoA Desaturase. Proc. Natl. Acad. Sci. USA 2019, 116, 20760–20769. [Google Scholar] [CrossRef] [PubMed]
- Nuber, S.; Nam, A.Y.; Rajsombath, M.M.; Cirka, H.; Hronowski, X.; Wang, J.; Hodgetts, K.; Kalinichenko, L.S.; Muller, C.P.; Lambrecht, V.; et al. A Stearoyl-Coenzyme A Desaturase Inhibitor Prevents Multiple Parkinson Disease Phenotypes in alpha-Synuclein Mice. Ann. Neurol. 2021, 89, 74–90. [Google Scholar] [CrossRef] [PubMed]
- Julien, C.; Berthiaume, L.; Hadj-Tahar, A.; Rajput, A.H.; Bedard, P.J.; Di Paolo, T.; Julien, P.; Calon, F. Postmortem Brain Fatty Acid Profile of Levodopa-Treated Parkinson Disease Patients and Parkinsonian Monkeys. Neurochem. Int. 2006, 48, 404–414. [Google Scholar] [CrossRef] [PubMed]
- Schmid, S.P.; Schleicher, E.D.; Cegan, A.; Deuschle, C.; Baur, S.; Hauser, A.K.; Synofzik, M.; Srulijes, K.; Brockmann, K.; Berg, D.; et al. Cerebrospinal Fluid Fatty Acids in Glucocerebrosidase-Associated Parkinson’s Disease. Mov. Disord. 2012, 27, 288–292. [Google Scholar] [CrossRef]
- Abbott, S.K.; Jenner, A.M.; Spiro, A.S.; Batterham, M.; Halliday, G.M.; Garner, B. Fatty Acid Composition of the Anterior Cingulate Cortex Indicates a High Susceptibility to Lipid Peroxidation in Parkinson’s Disease. J. Parkinsons Dis. 2015, 5, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Ben Gedalya, T.; Loeb, V.; Israeli, E.; Altschuler, Y.; Selkoe, D.J.; Sharon, R. Alpha-Synuclein and Polyunsaturated Fatty Acids Promote Clathrin-Mediated Endocytosis and Synaptic Vesicle Recycling. Traffic 2009, 10, 218–234. [Google Scholar] [CrossRef] [PubMed]
- Sharon, R.; Bar-Joseph, I.; Frosch, M.P.; Walsh, D.M.; Hamilton, J.A.; Selkoe, D.J. The Formation of Highly Soluble Oligomers of Alpha-Synuclein is Regulated by Fatty Acids and Enhanced in Parkinson’s Disease. Neuron 2003, 37, 583–595. [Google Scholar] [CrossRef]
- Perrin, R.J.; Woods, W.S.; Clayton, D.F.; George, J.M. Exposure to Long Chain Polyunsaturated Fatty Acids Triggers Rapid Multimerization of Synucleins. J. Biol. Chem. 2001, 276, 41958–41962. [Google Scholar] [CrossRef] [PubMed]
- Tu-Sekine, B.; Goldschmidt, H.; Raben, D.M. Diacylglycerol, Phosphatidic Acid, and Their Metabolic Enzymes in Synaptic Vesicle Recycling. Adv. Biol. Regul. 2015, 57, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Almena, M.; Merida, I. Shaping Up the Membrane: Diacylglycerol Coordinates Spatial Orientation of Signaling. Trends Biochem. Sci. 2011, 36, 593–603. [Google Scholar] [CrossRef]
- Ahmadian, M.; Duncan, R.E.; Jaworski, K.; Sarkadi-Nagy, E.; Sul, H.S. Triacylglycerol Metabolism in Adipose Tissue. Future Lipidol. 2007, 2, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, X.; Wang, L.; Yang, C. High Performance Liquid Chromatography-Mass Spectrometry (LC-MS) Based Quantitative Lipidomics Study of Ganglioside-NANA-3 Plasma to Establish Its Association with Parkinson’s Disease Patients. Med. Sci. Monit. 2017, 23, 5345–5353. [Google Scholar] [CrossRef] [PubMed]
- Chan, R.B.; Perotte, A.J.; Zhou, B.; Liong, C.; Shorr, E.J.; Marder, K.S.; Kang, U.J.; Waters, C.H.; Levy, O.A.; Xu, Y.; et al. Elevated GM3 Plasma Concentration in Idiopathic Parkinson’s Disease: A Lipidomic Analysis. PLoS ONE 2017, 12, e0172348. [Google Scholar] [CrossRef]
- Vikdahl, M.; Backman, L.; Johansson, I.; Forsgren, L.; Haglin, L. Cardiovascular Risk Factors and the Risk of Parkinson’s Disease. Eur. J. Clin. Nutr. 2015, 69, 729–733. [Google Scholar] [CrossRef]
- He, Q.; Wang, M.; Petucci, C.; Gardell, S.J.; Han, X. Rotenone Induces Reductive Stress and Triacylglycerol Deposition in C2C12 Cells. Int. J. Biochem. Cell Biol. 2013, 45, 2749–2755. [Google Scholar] [CrossRef]
- Sere, Y.Y.; Regnacq, M.; Colas, J.; Berges, T. A Saccharomyces Cerevisiae Strain Unable to Store Neutral Lipids is Tolerant to Oxidative Stress Induced by Alpha-Synuclein. Free. Radic. Biol. Med. 2010, 49, 1755–1764. [Google Scholar] [CrossRef]
- Spann, N.J.; Glass, C.K. Sterols and Oxysterols in Immune Cell Function. Nat. Immunol. 2013, 14, 893–900. [Google Scholar] [CrossRef]
- Hannich, J.T.; Umebayashi, K.; Riezman, H. Distribution and Functions of Sterols and Sphingolipids. Cold Spring Harb. Perspect. Biol. 2011, 3, a004762. [Google Scholar] [CrossRef]
- Cheng, D.; Jenner, A.M.; Shui, G.; Cheong, W.F.; Mitchell, T.W.; Nealon, J.R.; Kim, W.S.; McCann, H.; Wenk, M.R.; Halliday, G.M.; et al. Lipid Pathway Alterations in Parkinson’s Disease Primary Visual Cortex. PLoS ONE 2011, 6, e17299. [Google Scholar] [CrossRef] [PubMed]
- Musanti, R.; Parati, E.; Lamperti, E.; Ghiselli, G. Decreased Cholesterol Biosynthesis in Fibroblasts from Patients with Parkinson Disease. Biochem. Med. Metab. Biol. 1993, 49, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Magalhaes, J.; Gegg, M.E.; Migdalska-Richards, A.; Doherty, M.K.; Whitfield, P.D.; Schapira, A.H. Autophagic Lysosome Reformation Dysfunction in Glucocerebrosidase Deficient Cells: Relevance to Parkinson Disease. Hum. Mol. Genet. 2016, 25, 3432–3445. [Google Scholar] [CrossRef]
- Zhang, S.; Glukhova, S.A.; Caldwell, K.A.; Caldwell, G.A. NCEH-1 Modulates Cholesterol Metabolism and Protects Against Alpha-Synuclein Toxicity in a C. Elegans Model of Parkinson’s Disease. Hum. Mol. Genet. 2017, 26, 3823–3836. [Google Scholar] [CrossRef]
- Treede, I.; Braun, A.; Sparla, R.; Kuhnel, M.; Giese, T.; Turner, J.R.; Anes, E.; Kulaksiz, H.; Fullekrug, J.; Stremmel, W.; et al. Anti-Inflammatory Effects of Phosphatidylcholine. J. Biol. Chem. 2007, 282, 27155–27164. [Google Scholar] [CrossRef]
- Lagace, T.A. Phosphatidylcholine: Greasing the Cholesterol Transport Machinery. Lipid Insights 2015, 8, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Marcucci, H.; Paoletti, L.; Jackowski, S.; Banchio, C. Phosphatidylcholine Biosynthesis during Neuronal Differentiation and Its Role in Cell Fate Determination. J. Biol. Chem. 2010, 285, 25382–25393. [Google Scholar] [CrossRef]
- Wood, P.L.; Tippireddy, S.; Feriante, J.; Woltjer, R.L. Augmented Frontal Cortex Diacylglycerol Levels in Parkinson’s Disease and Lewy Body Disease. PLoS ONE 2018, 13, e0191815. [Google Scholar] [CrossRef] [PubMed]
- Seyfried, T.N.; Choi, H.; Chevalier, A.; Hogan, D.; Akgoc, Z.; Schneider, J.S. Sex-Related Abnormalities in Substantia Nigra Lipids in Parkinson’s Disease. ASN Neuro 2018, 10, 1759091418781889. [Google Scholar] [CrossRef] [PubMed]
- Farmer, K.; Smith, C.A.; Hayley, S.; Smith, J. Major Alterations of Phosphatidylcholine and Lysophosphotidylcholine Lipids in the Substantia Nigra Using an Early Stage Model of Parkinson’s Disease. Int. J. Mol. Sci. 2015, 16, 18865–18877. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Wang, J.; Li, M.; Liu, Q.; Wei, D.; Yang, M.; Kong, L. (1)H NMR-Based Metabolomics Study on a Goldfish Model of Parkinson’s Disease Induced by 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine (MPTP). Chem. Biol. Interact. 2014, 223, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Stockl, M.; Fischer, P.; Wanker, E.; Herrmann, A. Alpha-Synuclein Selectively Binds to Anionic Phospholipids Embedded in Liquid-Disordered Domains. J. Mol. Biol. 2008, 375, 1394–1404. [Google Scholar] [CrossRef]
- Jiang, Z.; de Messieres, M.; Lee, J.C. Membrane Remodeling by Alpha-Synuclein and Effects on Amyloid Formation. J. Am. Chem. Soc. 2013, 135, 15970–15973. [Google Scholar] [CrossRef] [PubMed]
- Di Pasquale, E.; Fantini, J.; Chahinian, H.; Maresca, M.; Taieb, N.; Yahi, N. Altered Ion Channel Formation by the Parkinson’s-Disease-Linked E46K Mutant of Alpha-Synuclein is Corrected by GM3 but not by GM1 Gangliosides. J. Mol. Biol. 2010, 397, 202–218. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, E.I.; Jiang, Z.; Strub, M.P.; Lee, J.C. Effects of Phosphatidylcholine Membrane Fluidity on the Conformation and Aggregation of N-Terminally Acetylated Alpha-Synuclein. J. Biol. Chem. 2018, 293, 11195–11205. [Google Scholar] [CrossRef]
- Hirsch, E.C.; Standaert, D.G. Ten Unsolved Questions About Neuroinflammation in Parkinson’s Disease. Mov. Disord. 2021, 36, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Jin, H.K.; Bae, J.S. Sphingolipids in Neuroinflammation: A Potential Target for Diagnosis and Therapy. BMB Rep. 2020, 53, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Hannun, Y.A.; Obeid, L.M. Sphingolipids and Their Metabolism in Physiology and Disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 175–191. [Google Scholar] [CrossRef]
- Assi, E.; Cazzato, D.; De Palma, C.; Perrotta, C.; Clementi, E.; Cervia, D. Sphingolipids and Brain Resident Macrophages in Neuroinflammation: An Emerging Aspect of Nervous System Pathology. Clin. Dev. Immunol. 2013, 2013, 309302. [Google Scholar] [CrossRef]
- Young, M.M.; Kester, M.; Wang, H.G. Sphingolipids: Regulators of Crosstalk between Apoptosis and Autophagy. J. Lipid Res. 2013, 54, 5–19. [Google Scholar] [CrossRef]
- de Wit, N.M.; den Hoedt, S.; Martinez-Martinez, P.; Rozemuller, A.J.; Mulder, M.T.; de Vries, H.E. Astrocytic Ceramide as Possible Indicator of Neuroinflammation. J. Neuroinflammation 2019, 16, 48. [Google Scholar] [CrossRef]
- Mielke, M.M.; Maetzler, W.; Haughey, N.J.; Bandaru, V.V.; Savica, R.; Deuschle, C.; Gasser, T.; Hauser, A.K.; Graber-Sultan, S.; Schleicher, E.; et al. Plasma Ceramide and Glucosylceramide Metabolism is Altered in Sporadic Parkinson’s Disease and Associated with Cognitive Impairment: A Pilot Study. PLoS ONE 2013, 8, e73094. [Google Scholar] [CrossRef]
- Atashrazm, F.; Hammond, D.; Perera, G.; Dobson-Stone, C.; Mueller, N.; Pickford, R.; Kim, W.S.; Kwok, J.B.; Lewis, S.J.G.; Halliday, G.M.; et al. Reduced Glucocerebrosidase Activity in Monocytes from Patients with Parkinson’s Disease. Sci. Rep. 2018, 8, 15446. [Google Scholar] [CrossRef] [PubMed]
- Lupescu, A.; Jilani, K.; Zbidah, M.; Lang, F. Induction of Apoptotic Erythrocyte Death by Rotenone. Toxicology 2012, 300, 132–137. [Google Scholar] [CrossRef] [PubMed]
- Lin, G.; Lee, P.T.; Chen, K.; Mao, D.; Tan, K.L.; Zuo, Z.; Lin, W.W.; Wang, L.; Bellen, H.J. Phospholipase PLA2G6, a Parkinsonism-Associated Gene, Affects Vps26 and Vps35, Retromer Function, and Ceramide Levels, Similar to Alpha-Synuclein Gain. Cell Metab. 2018, 28, 605–618.e6. [Google Scholar] [CrossRef] [PubMed]
- Ferrazza, R.; Cogo, S.; Melrose, H.; Bubacco, L.; Greggio, E.; Guella, G.; Civiero, L.; Plotegher, N. LRRK2 Deficiency Impacts Ceramide Metabolism in Brain. Biochem. Biophys. Res. Commun. 2016, 478, 1141–1146. [Google Scholar] [CrossRef] [PubMed]
- Torres-Odio, S.; Key, J.; Hoepken, H.H.; Canet-Pons, J.; Valek, L.; Roller, B.; Walter, M.; Morales-Gordo, B.; Meierhofer, D.; Harter, P.N.; et al. Progression of Pathology in PINK1-Deficient Mouse Brain from Splicing via Ubiquitination, ER Stress, and Mitophagy Changes to Neuroinflammation. J. Neuroinflamm. 2017, 14, 154. [Google Scholar] [CrossRef] [PubMed]
- Satoi, H.; Tomimoto, H.; Ohtani, R.; Kitano, T.; Kondo, T.; Watanabe, M.; Oka, N.; Akiguchi, I.; Furuya, S.; Hirabayashi, Y.; et al. Astroglial Expression of Ceramide in Alzheimer’s Disease Brains: A Role during Neuronal Apoptosis. Neuroscience 2005, 130, 657–666. [Google Scholar] [CrossRef] [PubMed]
- Nixon, G.F. Sphingolipids in Inflammation: Pathological Implications and Potential Therapeutic Targets. Br. J. Pharmacol. 2009, 158, 982–993. [Google Scholar] [CrossRef] [PubMed]
- Norris, G.H.; Blesso, C.N. Dietary and Endogenous Sphingolipid Metabolism in Chronic Inflammation. Nutrients 2017, 9, 1180. [Google Scholar] [CrossRef]
- Kiraz, Y.; Adan, A.; Kartal Yandim, M.; Baran, Y. Major Apoptotic Mechanisms and Genes Involved in Apoptosis. Tumor Biol. 2016, 37, 8471–8486. [Google Scholar] [CrossRef]
- Tommasino, C.; Marconi, M.; Ciarlo, L.; Matarrese, P.; Malorni, W. Autophagic Fux and Autophagosome Morphogenesis Require the Participation of Sphingolipids. Apoptosis 2015, 20, 645–657. [Google Scholar] [CrossRef] [PubMed]
- den Jager, W.A. Sphingomyelin in Lewy Inclusion Bodies in Parkinson’s Disease. Arch. Neurol. 1969, 21, 615–619. [Google Scholar] [CrossRef] [PubMed]
- Foo, J.N.; Liany, H.; Bei, J.X.; Yu, X.Q.; Liu, J.; Au, W.L.; Prakash, K.M.; Tan, L.C.; Tan, E.K. A Rare Lysosomal Enzyme Gene SMPD1 Variant (p.R591C) Associates with Parkinson’s Disease. Neurobiol. Aging 2013, 34, 2890.e13–2890.e15. [Google Scholar] [CrossRef] [PubMed]
- Mao, C.Y.; Yang, J.; Wang, H.; Zhang, S.Y.; Yang, Z.H.; Luo, H.Y.; Li, F.; Shi, M.; Liu, Y.T.; Zhuang, Z.P.; et al. SMPD1 Variants in Chinese Han Patients with Sporadic Parkinson’s Disease. Parkinsonism Relat. Disord. 2017, 34, 59–61. [Google Scholar] [CrossRef] [PubMed]
- Mattace Raso, G.; Russo, R.; Calignano, A.; Meli, R. Palmitoylethanolamide in CNS Health and Disease. Pharmacol. Res. 2014, 86, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Skaper, S.D.; Facci, L.; Barbierato, M.; Zusso, M.; Bruschetta, G.; Impellizzeri, D.; Cuzzocrea, S.; Giusti, P. N-Palmitoylethanolamine and Neuroinflammation: A Novel Therapeutic Strategy of Resolution. Mol. Neurobiol. 2015, 52, 1034–1042. [Google Scholar] [CrossRef]
- Esposito, E.; Impellizzeri, D.; Mazzon, E.; Paterniti, I.; Cuzzocrea, S. Neuroprotective Activities of Palmitoylethanolamide in an Animal Model of Parkinson’s Disease. PLoS ONE 2012, 7, e41880. [Google Scholar] [CrossRef]
- Galan-Rodriguez, B.; Suarez, J.; Gonzalez-Aparicio, R.; Bermudez-Silva, F.J.; Maldonado, R.; Robledo, P.; Rodriguez de Fonseca, F.; Fernandez-Espejo, E. Oleoylethanolamide Exerts Partial and Dose-Dependent Neuroprotection of Substantia Nigra Dopamine Neurons. Neuropharmacology 2009, 56, 653–664. [Google Scholar] [CrossRef]
- Farrell, E.K.; Chen, Y.; Barazanji, M.; Jeffries, K.A.; Cameroamortegui, F.; Merkler, D.J. Primary Fatty Acid Amide Metabolism: Conversion of Fatty Acids and an Ethanolamine in N18TG2 and SCP Cells. J. Lipid Res. 2012, 53, 247–256. [Google Scholar] [CrossRef]
- Varvel, S.A.; Cravatt, B.F.; Engram, A.E.; Lichtman, A.H. Fatty Acid Amide Hydrolase (-/-) Mice Exhibit an Increased Sensitivity to the Disruptive Effects of Anandamide or Oleamide in a Working Memory Water Maze Task. J. Pharmacol. Exp. Ther. 2006, 317, 251–257. [Google Scholar] [CrossRef]
- Huitron-Resendiz, S.; Gombart, L.; Cravatt, B.F.; Henriksen, S.J. Effect of Oleamide on Sleep and Its Relationship to Blood Pressure, Body Temperature, and Locomotor Activity in Rats. Exp. Neurol. 2001, 172, 235–243. [Google Scholar] [CrossRef]
- Farrell, E.K.; Merkler, D.J. Biosynthesis, Degradation and Pharmacological Importance of the Fatty Acid Amides. Drug Discov. Today 2008, 13, 558–568. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Snowden, S.; Suvitaival, T.; Ali, A.; Merkler, D.J.; Ahmad, T.; Westwood, S.; Baird, A.; Proitsi, P.; Nevado-Holgado, A.; et al. Primary Fatty Amides in Plasma Associated with Brain Amyloid Burden, Hippocampal Volume, and Memory in the European Medical Information Framework for Alzheimer’s Disease Biomarker Discovery Cohort. Alzheimers Dement 2019, 15, 817–827. [Google Scholar] [CrossRef] [PubMed]
- Ren, S.Y.; Wang, Z.Z.; Zhang, Y.; Chen, N.H. Potential Application of Endocannabinoid System Agents in Neuropsychiatric and Neurodegenerative Diseases-Focusing on FAAH/MAGL Inhibitors. Acta Pharmacol. Sin. 2020, 41, 1263–1271. [Google Scholar] [CrossRef]
- Alecu, I.; Bennett, S.A.L. Dysregulated Lipid Metabolism and Its Role in Alpha-Synucleinopathy in Parkinson’s Disease. Front. Neurosci. 2019, 13, 328. [Google Scholar] [CrossRef] [PubMed]
- Belarbi, K.; Cuvelier, E.; Bonte, M.A.; Desplanque, M.; Gressier, B.; Devos, D.; Chartier-Harlin, M.C. Glycosphingolipids and Neuroinflammation in Parkinson’s Disease. Mol. Neurodegener. 2020, 15, 59. [Google Scholar] [CrossRef]
- Stoessel, D.; Schulte, C.; Teixeira Dos Santos, M.C.; Scheller, D.; Rebollo-Mesa, I.; Deuschle, C.; Walther, D.; Schauer, N.; Berg, D.; Nogueira da Costa, A.; et al. Promising Metabolite Profiles in the Plasma and CSF of Early Clinical Parkinson’s Disease. Front. Aging Neurosci. 2018, 10, 51. [Google Scholar] [CrossRef]
- Kosicek, M.; Zetterberg, H.; Andreasen, N.; Peter-Katalinic, J.; Hecimovic, S. Elevated Cerebrospinal Fluid Sphingomyelin Levels in Prodromal Alzheimer’s Disease. Neurosci. Lett. 2012, 516, 302–305. [Google Scholar] [CrossRef]
- Wood, P.L.; Barnette, B.L.; Kaye, J.A.; Quinn, J.F.; Woltjer, R.L. Non-Targeted Lipidomics of CSF and Frontal Cortex Grey and White Matter in Control, Mild Cognitive Impairment, and Alzheimer’s Disease Subjects. Acta Neuropsychiatr. 2015, 27, 270–278. [Google Scholar] [CrossRef]






| Platform 1 | Platform 2 | |
|---|---|---|
| Column type | UPLC BEH C18, 1.0 × 100 mm, 1.7 μm | UPLC BEH C18, 2.1 × 100 mm, 1.7 μm |
| Flow rate | 0.140 mL/min | 0.400 mL/min |
| Solvent A | 0.05% Formic Acid in water | Water:Acetronitrile (2:3) + 10 mM Ammonium Formate |
| Solvent B | 0.05% Formic Acid in acetonitrile | Acetonitrile:Isopropanol (1:9) + 10 mM Ammonium Formate |
| (%B), time | 0%, 0 min | 40%, 0 min |
| (%B), time | 50%, 2 min | 100%, 10 min |
| (%B), time | 100%, 13 min | 40%, 15 min |
| (%B), time | 0%, 18 min | 40%, 17 min |
| Column temperature | 40 °C | 60 °C |
| Injection volume | 2 μL | 3 μL |
| Autosampler temperature | 10 °C | 10 °C |
| Source temperature | 120 °C | 120 °C |
| Nebulisation N2 flow | 600 L/hour | 1000 L/hour |
| Nebulisation N2 temperature | 350 °C | 500 °C |
| Cone N2 flow | 30 L/hour | 30 L/hour |
| Ionization | ESI −ve | ESI +ve |
| Capillary voltage | 2.8 kV | 3.2 kV |
| Cone voltage | 50 V | 30 V |
| Type of data | Centroid | Centroid |
| Scan time | 0.2 s | 0.2 s |
| Acquisition range | 50–1000 Da | 50–1200 Da |
| SAMPLE.ID | Age | Sex | Onset | Duration | NPD |
|---|---|---|---|---|---|
| PD354 | 88 | F | 77 | 11 | LBDE |
| PD423 | 66 | F | 53 | 13 | LBDE |
| PD436 | 90 | M | 82 | 8 | LBDE |
| PD520 | 80 | M | 56 | 24 | LBDE |
| PD530 | 85 | M | 77 | 8 | LBDE |
| PD357 | 71 | M | 37 | 34 | LBDN |
| PD450 | 66 | M | 47 | 19 | LBDN |
| PD495 | 88 | F | 78 | 10 | LBDN |
| PD501 | 89 | F | 82 | 7 | LBDN |
| PD537 | 84 | M | 74 | 9 | LBDN |
| PD550 | 83 | F | 77 | 7 | LBDN |
| PD562 | 79 | M | 72 | 7 | LBDN |
| PD636 | 84 | M | 65 | 20 | LBDN |
| PD295 | 83 | M | 67 | 16 | LBDL |
| PD340 | 67 | M | 53 | 14 | LBDL |
| PD356 | 86 | F | 75 | 9 | LBDL |
| PD541 | 72 | M | 66 | 6 | LBDL |
| PD546 | 84 | F | 71 | 13 | LBDL |
| PD579 | 76 | M | 55 | 21 | LBDL |
| PD591 | 77 | M | 68 | 9 | LBDL |
| C022 | 65 | M | aging-related changes | ||
| C023 | 78 | F | aging-related changes | ||
| C030 | 77 | M | aging-related changes | ||
| C008 | 93 | F | aging-related changes | ||
| C015 | 82 | M | possible ischaemia | ||
| C026 | 78 | F | minimal leukostasis | ||
| C032 | 88 | M | aging-related changes | ||
| C054 | 66 | M | mild aging-related changes | ||
| C064 | 63 | F | microvascular pathology | ||
| C076 | 87 | M | aging-related changes |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernández-Irigoyen, J.; Cartas-Cejudo, P.; Iruarrizaga-Lejarreta, M.; Santamaría, E. Alteration in the Cerebrospinal Fluid Lipidome in Parkinson’s Disease: A Post-Mortem Pilot Study. Biomedicines 2021, 9, 491. https://doi.org/10.3390/biomedicines9050491
Fernández-Irigoyen J, Cartas-Cejudo P, Iruarrizaga-Lejarreta M, Santamaría E. Alteration in the Cerebrospinal Fluid Lipidome in Parkinson’s Disease: A Post-Mortem Pilot Study. Biomedicines. 2021; 9(5):491. https://doi.org/10.3390/biomedicines9050491
Chicago/Turabian StyleFernández-Irigoyen, Joaquín, Paz Cartas-Cejudo, Marta Iruarrizaga-Lejarreta, and Enrique Santamaría. 2021. "Alteration in the Cerebrospinal Fluid Lipidome in Parkinson’s Disease: A Post-Mortem Pilot Study" Biomedicines 9, no. 5: 491. https://doi.org/10.3390/biomedicines9050491
APA StyleFernández-Irigoyen, J., Cartas-Cejudo, P., Iruarrizaga-Lejarreta, M., & Santamaría, E. (2021). Alteration in the Cerebrospinal Fluid Lipidome in Parkinson’s Disease: A Post-Mortem Pilot Study. Biomedicines, 9(5), 491. https://doi.org/10.3390/biomedicines9050491