
Natural Products That Changed Society


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
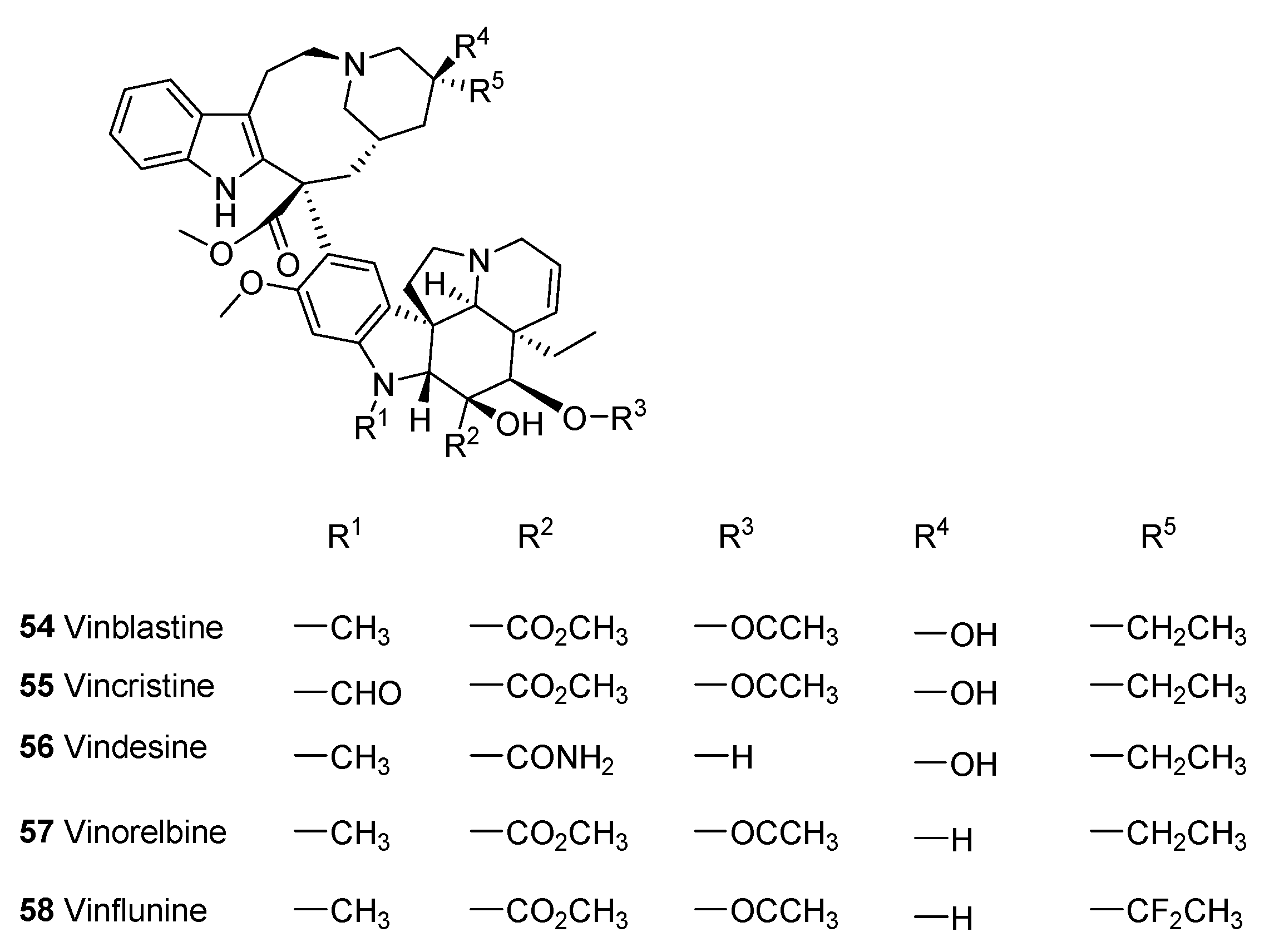{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
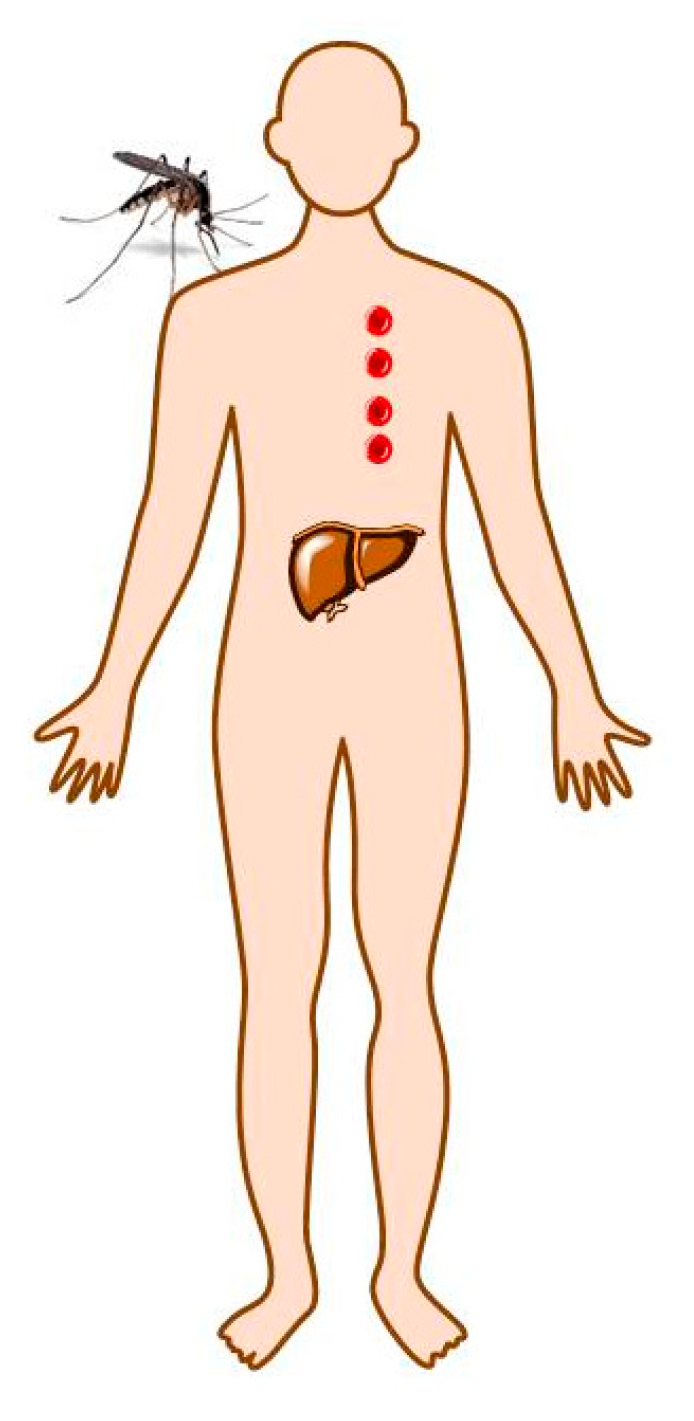
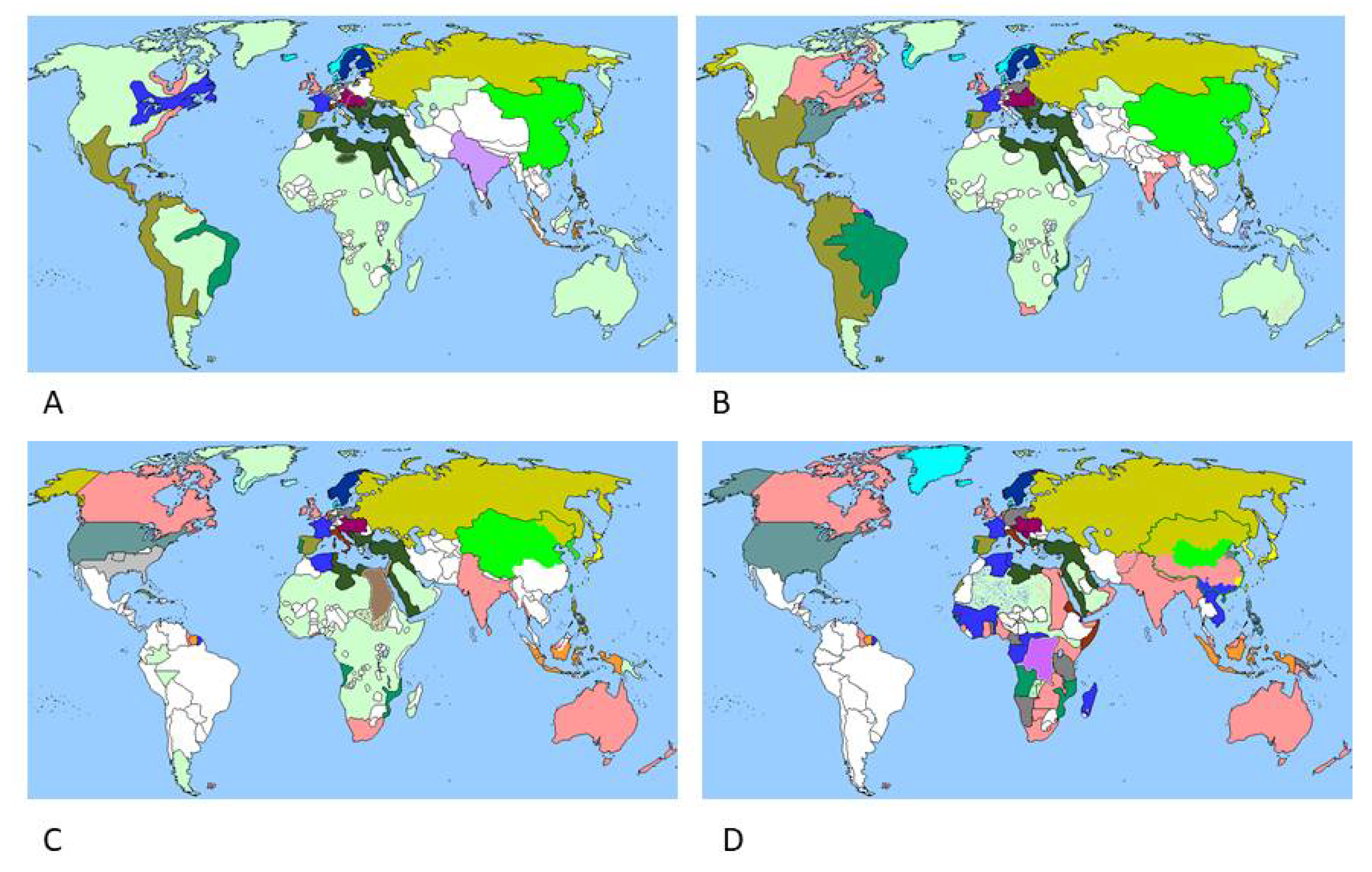
2. Malaria
2.1. Drugs to Treat and Prevent Malaria
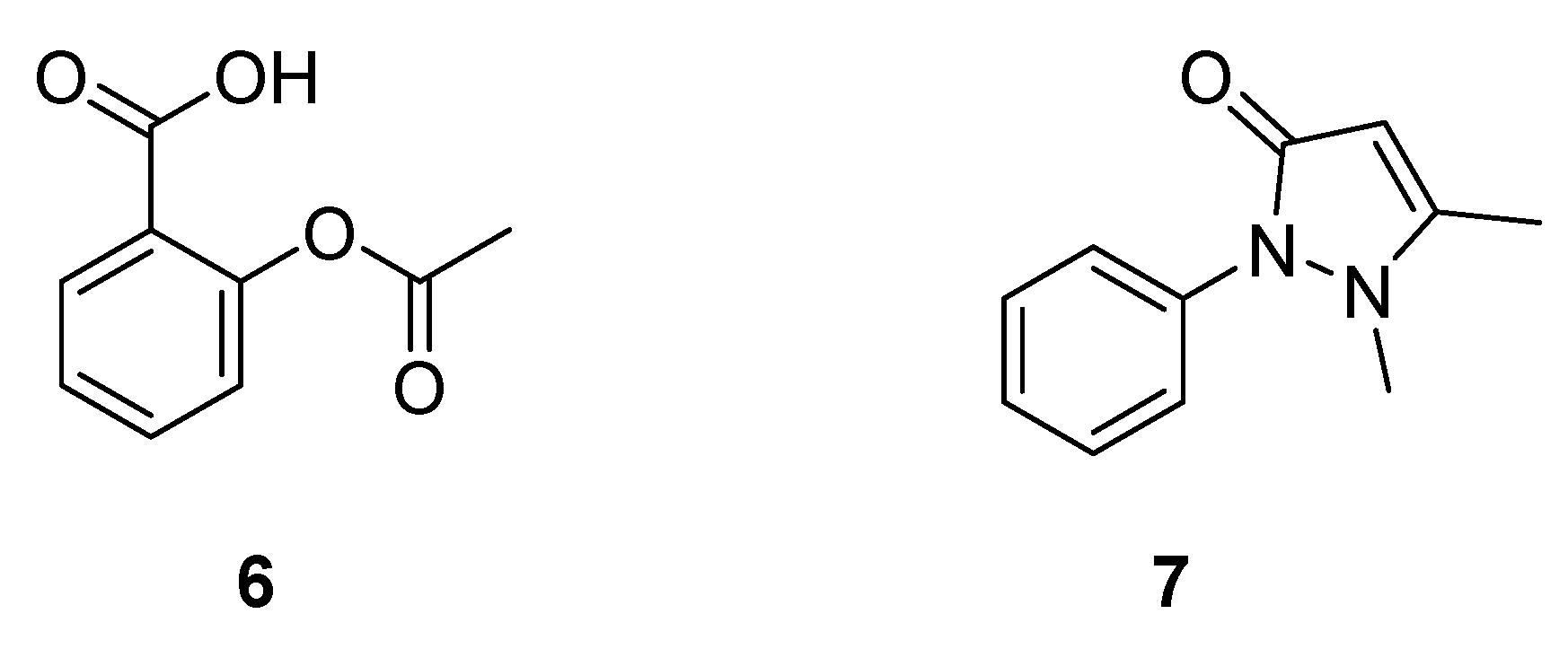
2.1.1. Cinchona Bark
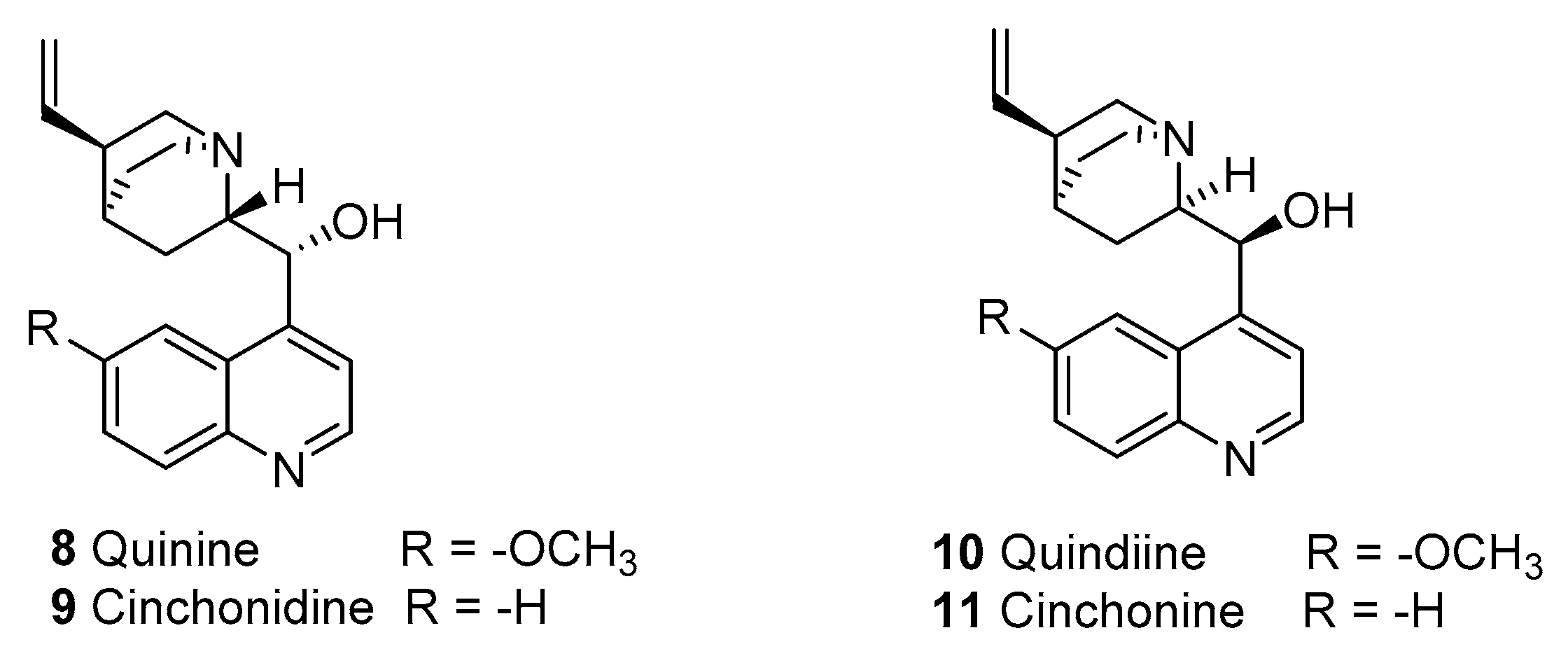
2.1.2. Quinine
2.1.3. Quinine Resistance
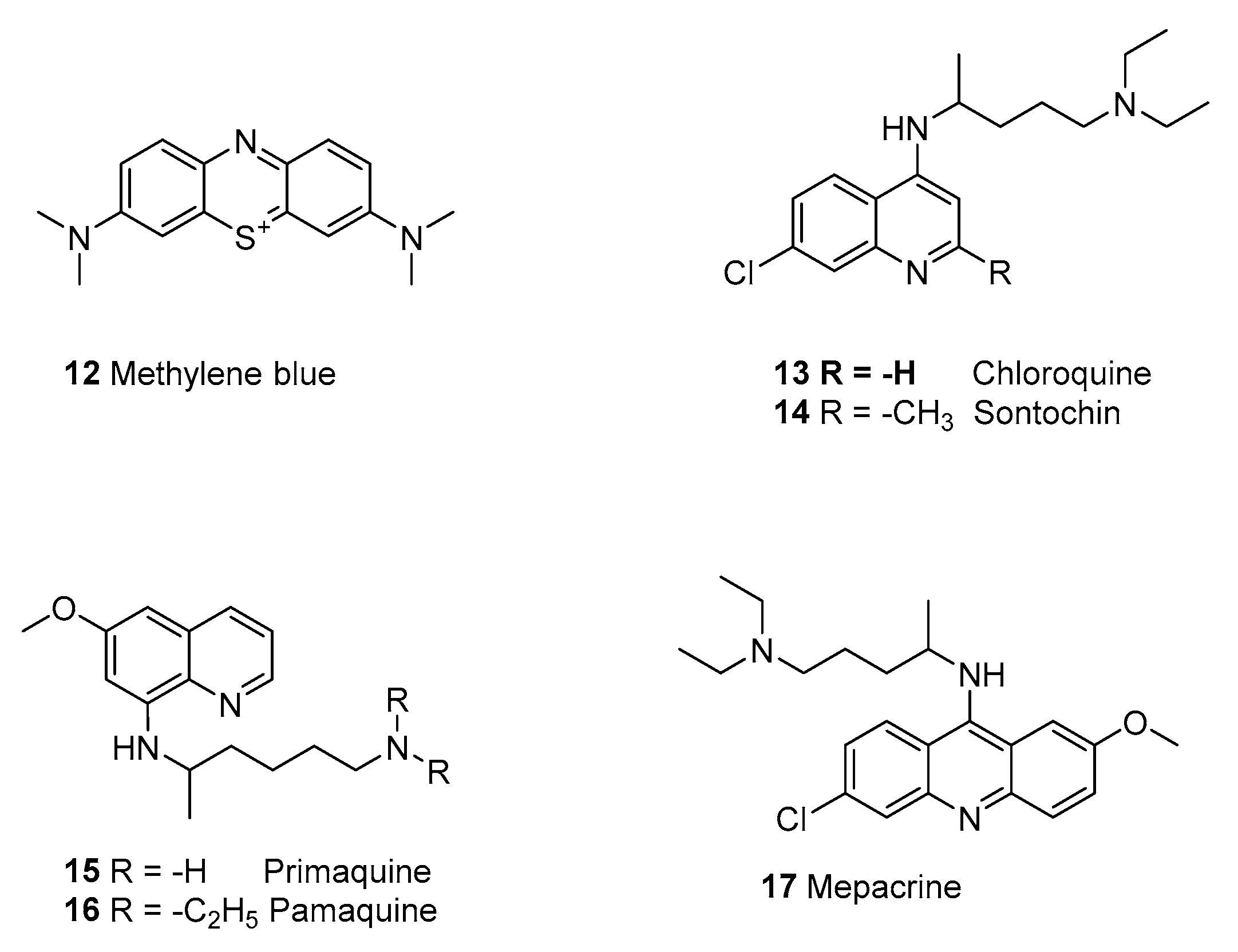
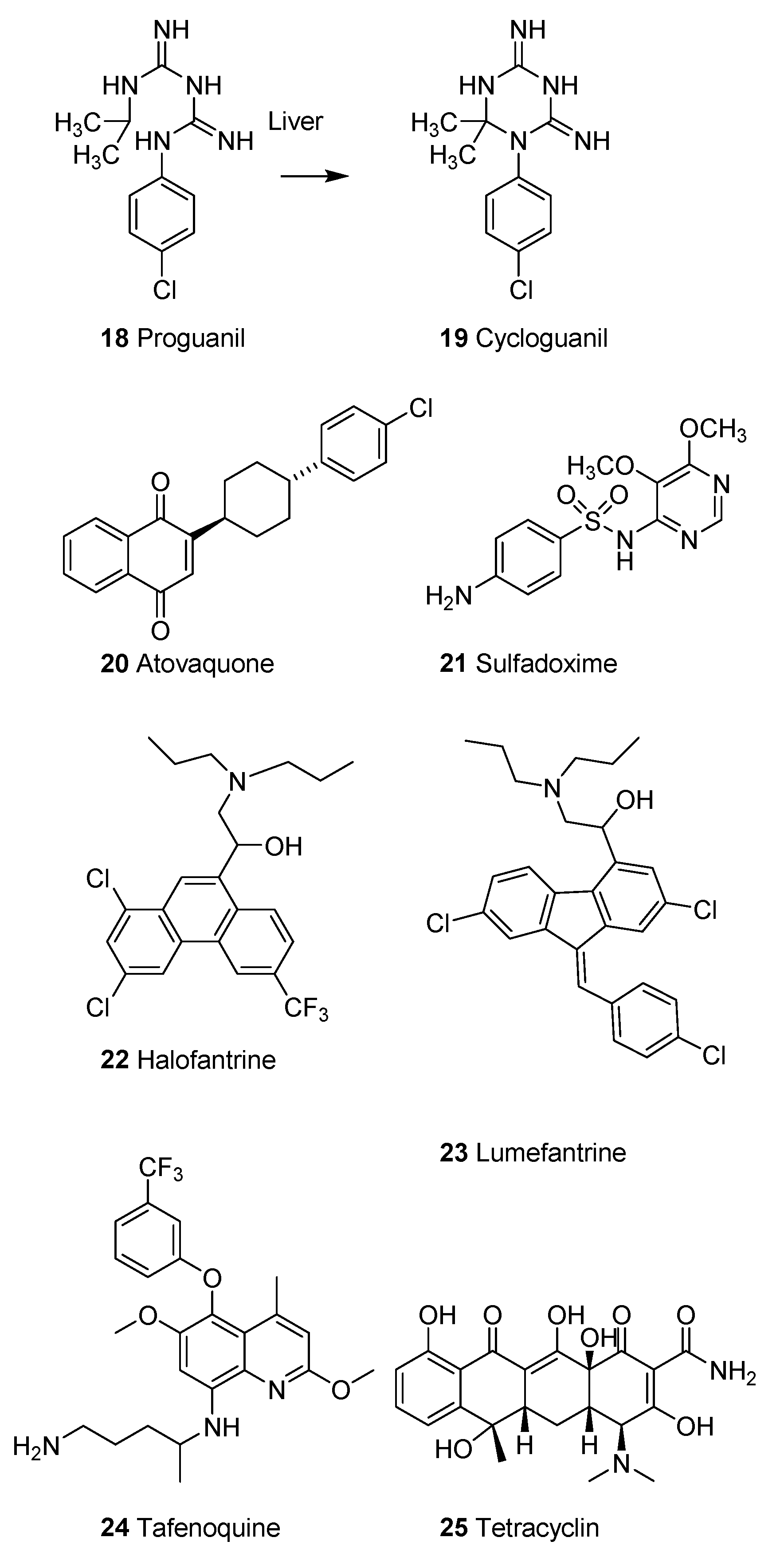
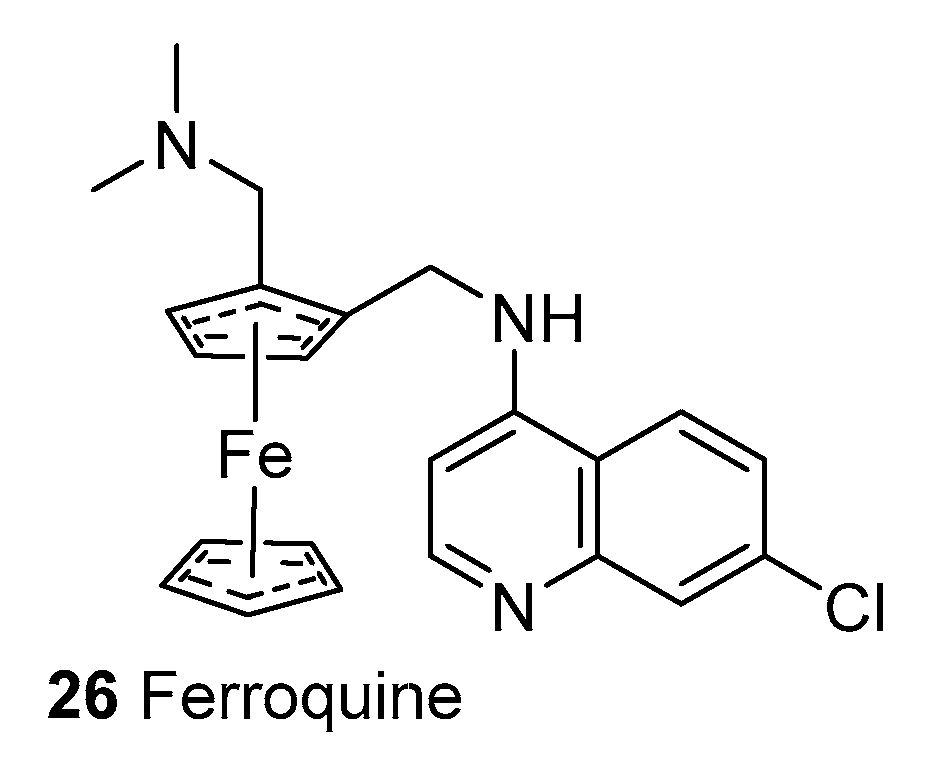
2.1.4. Synthetic Malaria Drugs
2.1.5. Malaria during the Pacific War
2.1.6. Malaria Control
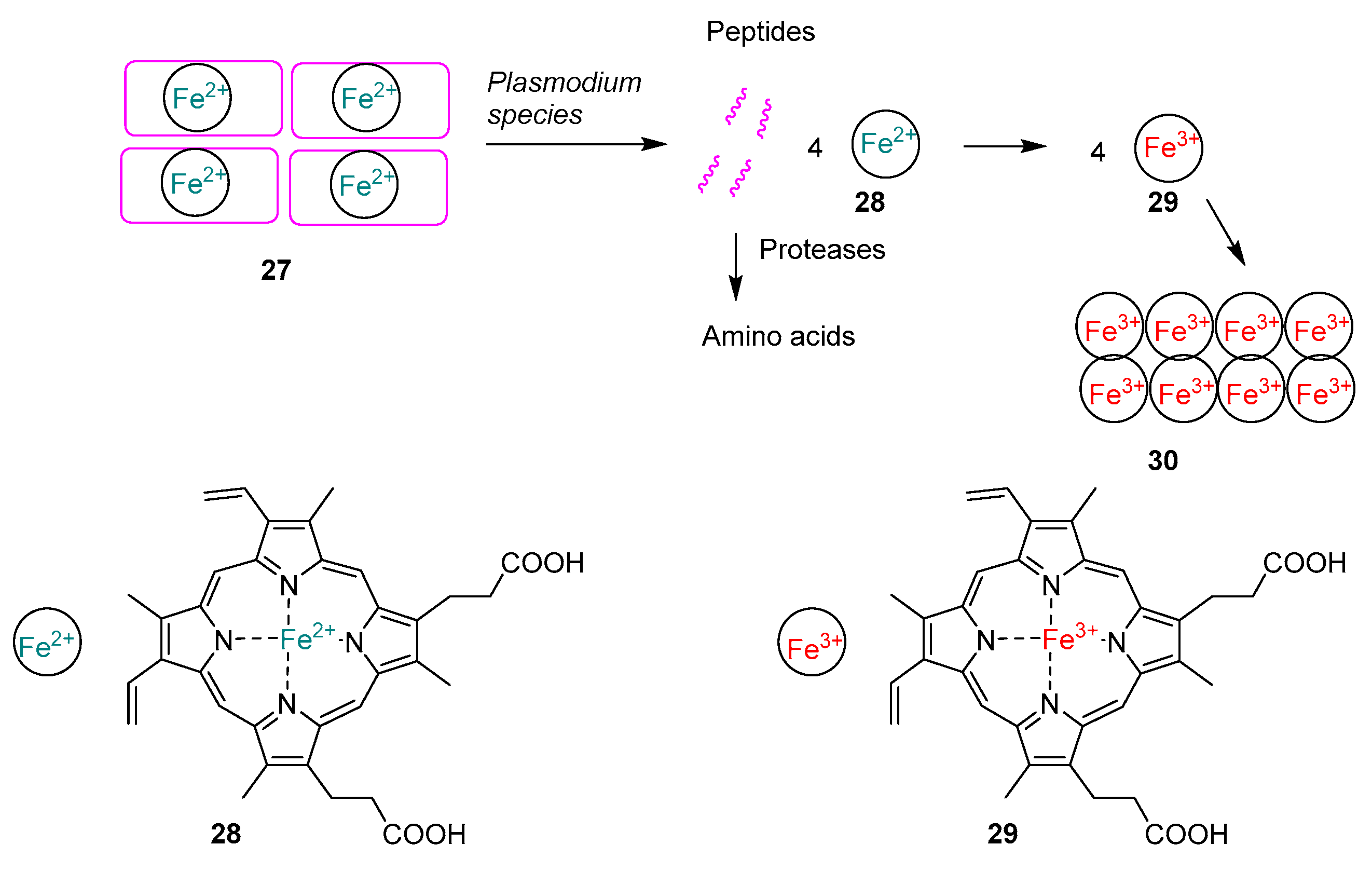
2.1.7. Mechanism of Action of Quinolines
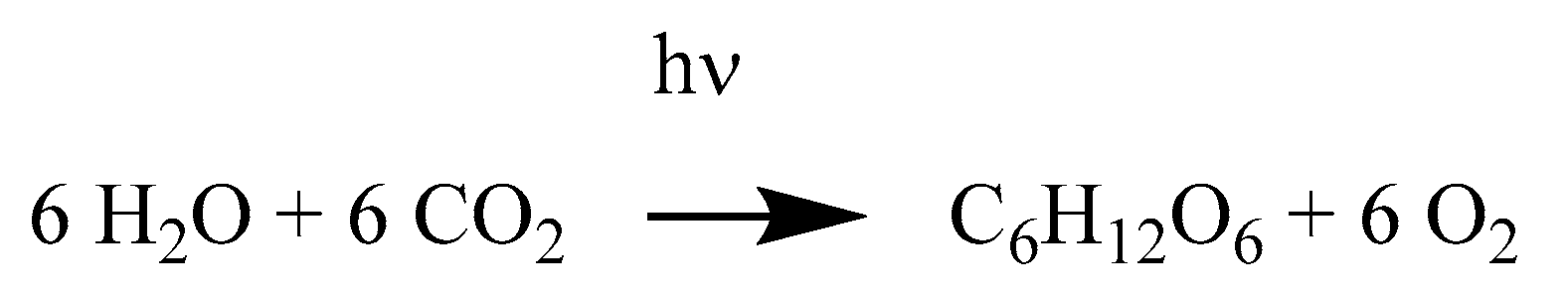
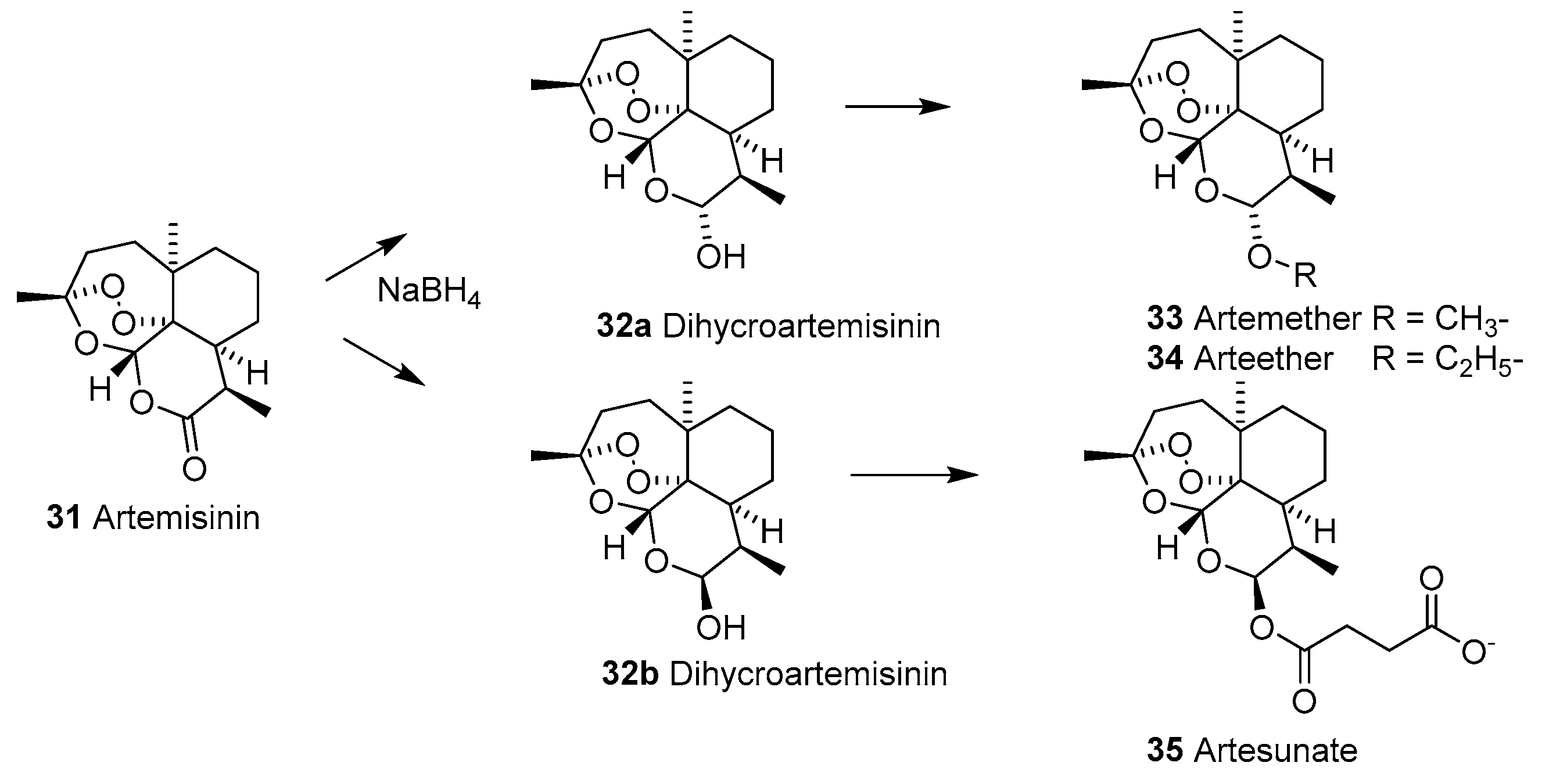
2.1.8. Artemisinins
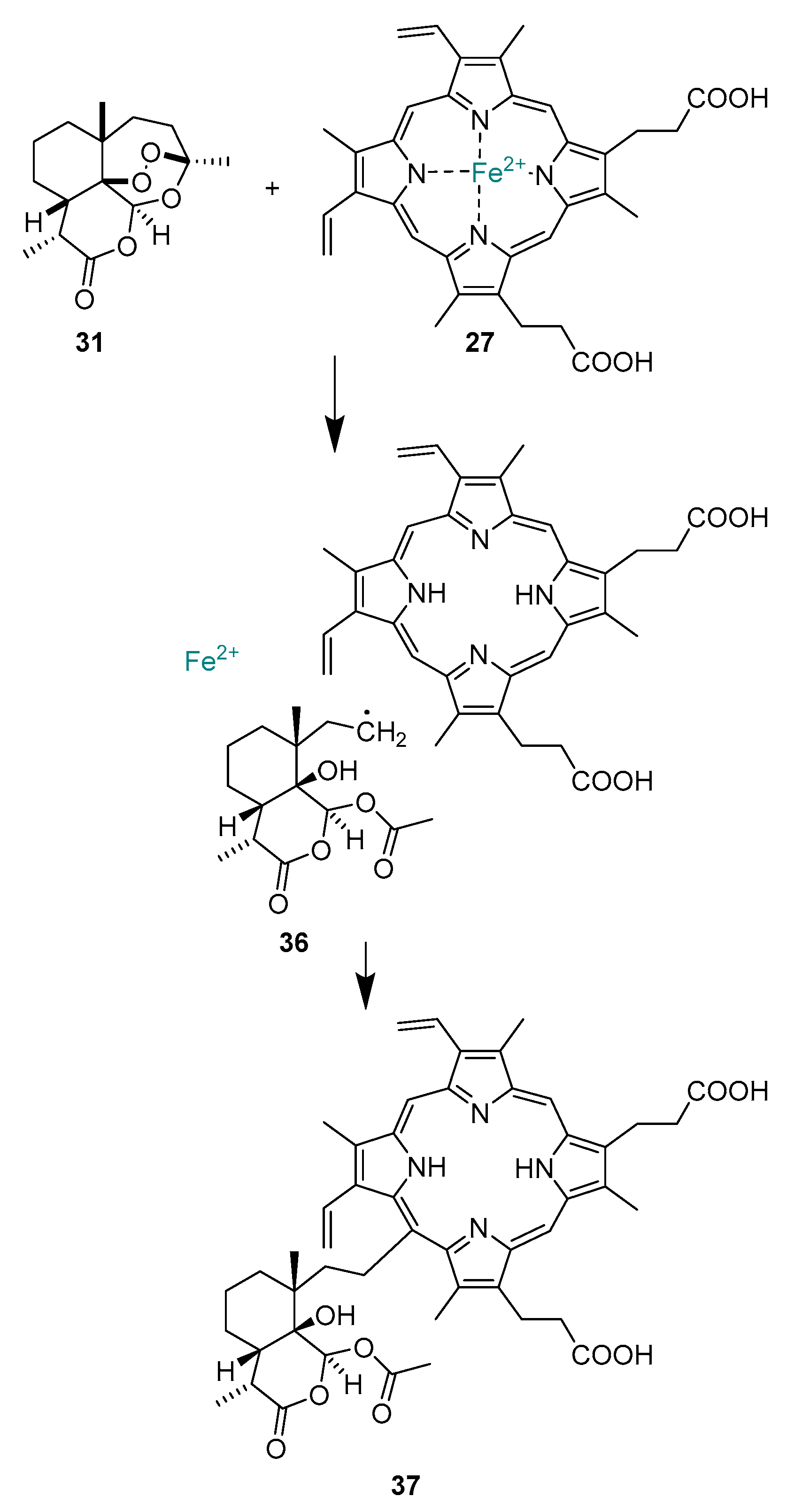
2.1.9. Mechanism of Action of Artemisinins
2.1.10. Clinical Use of Artemisinins
2.1.11. Resistance toward Artemsinins
2.1.12. Sustainable Supply of Artemisnin
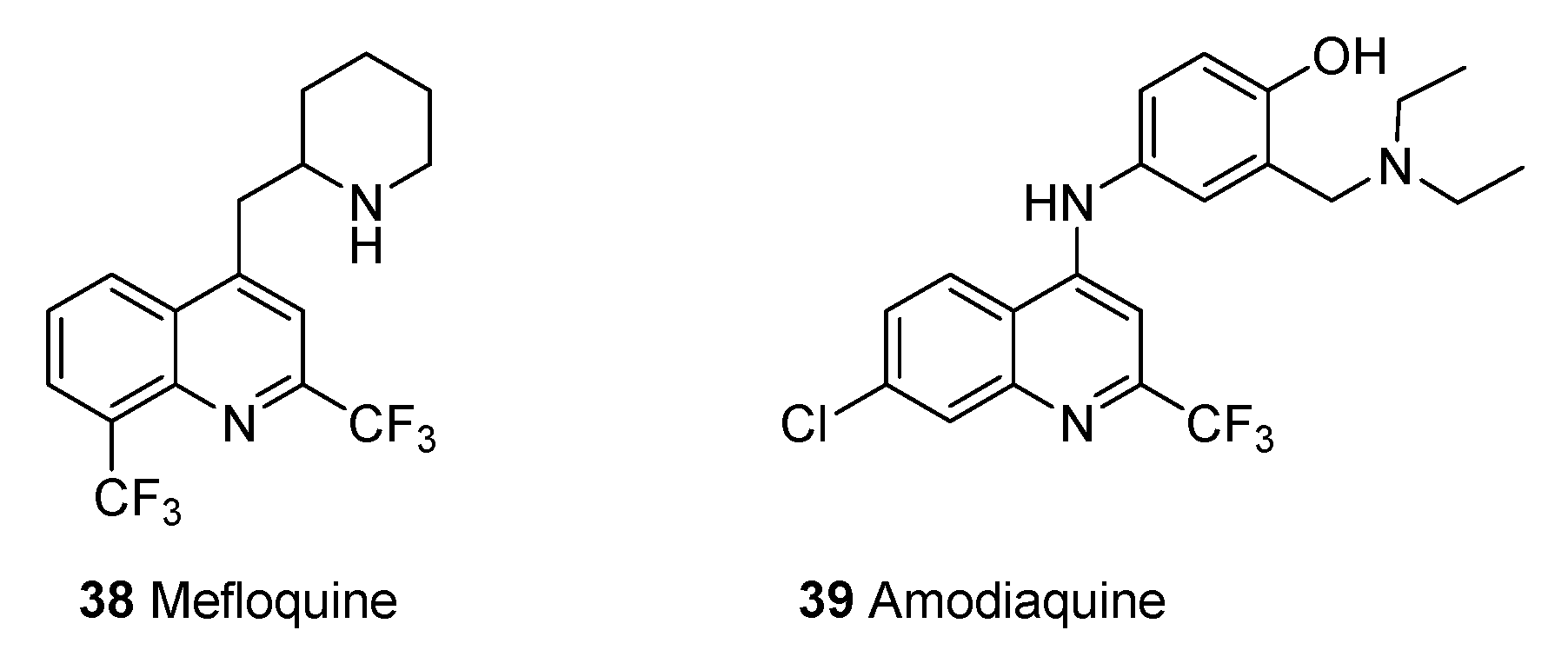
2.1.13. New Drugs for Treatment and Prophylaxis of Malaria
3. Onchocerciasis
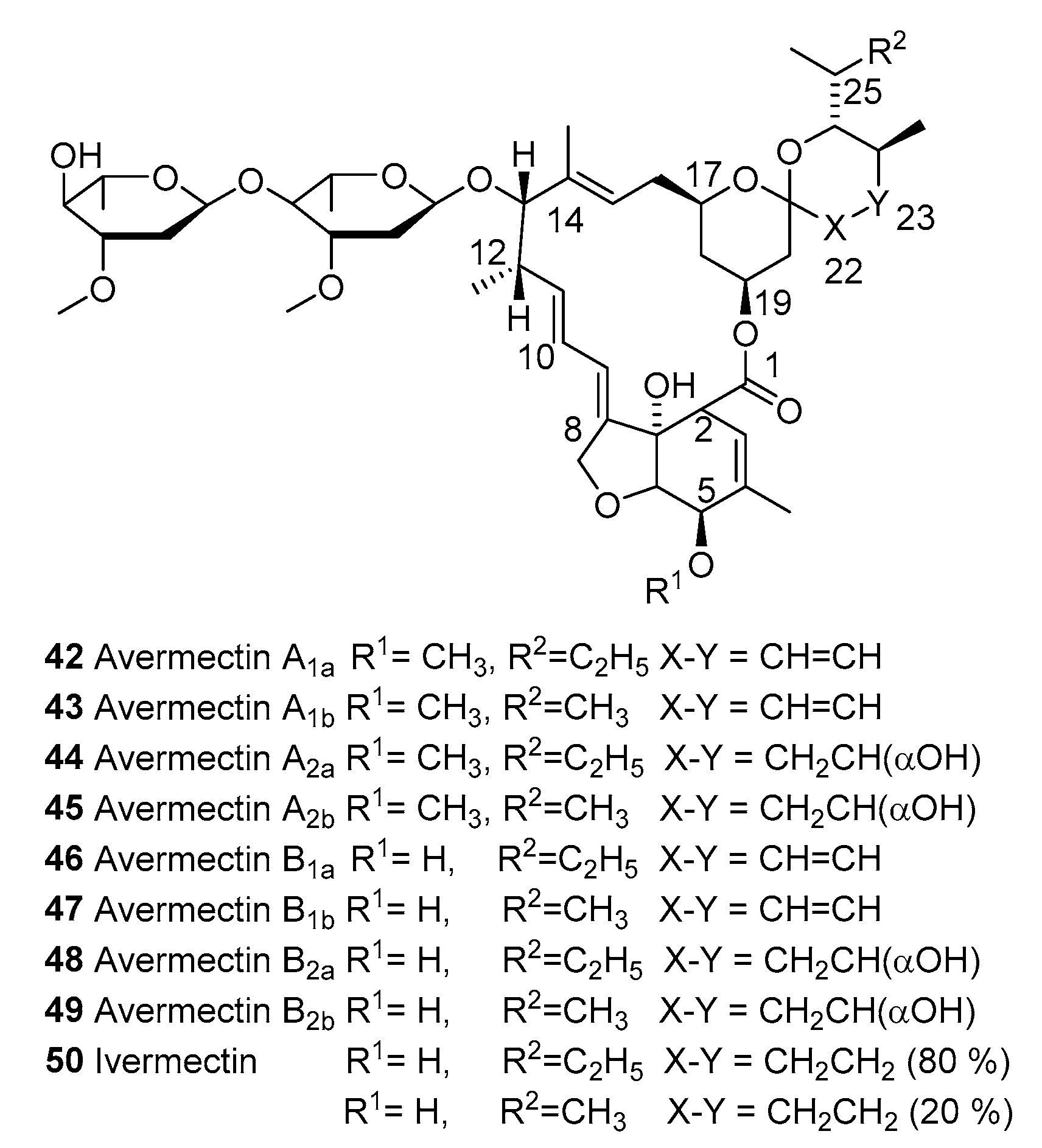
3.1. Ivermectin
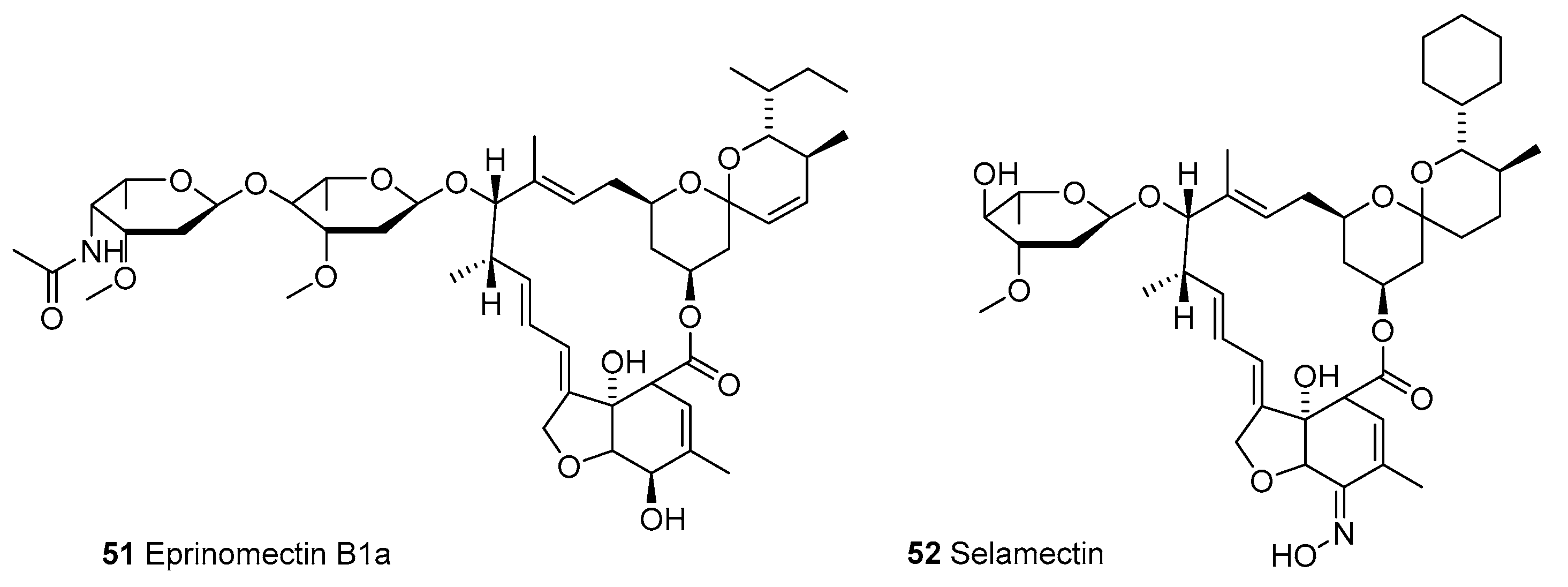
3.1.1. Macrocyclic Latones Veterinary Use
3.1.2. Ivermectin for Human Use
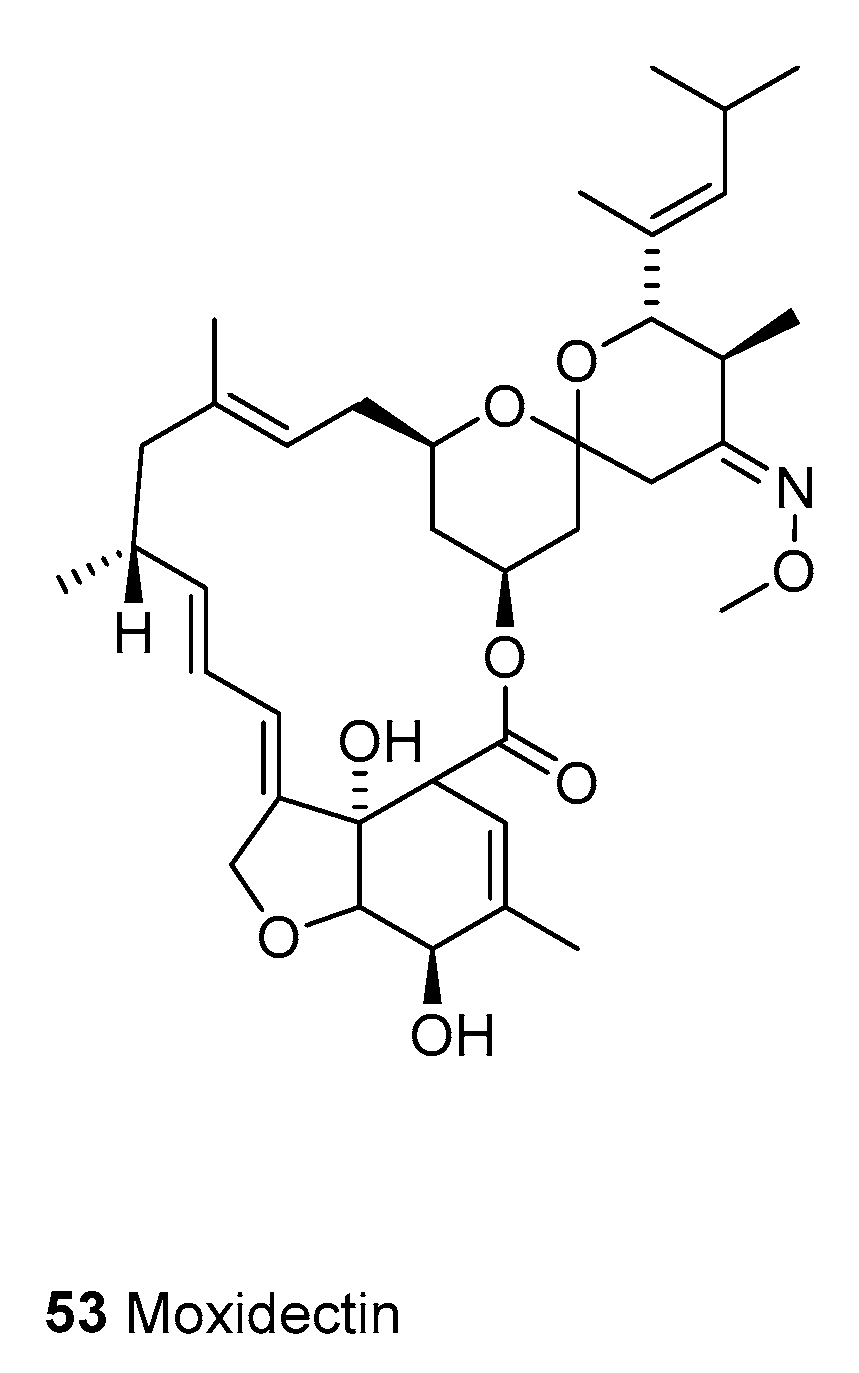
3.1.3. Moxidectin
4. Lymphatic Filariasis
5. Cancer Diseases
5.1. Vinca Alkaloids
5.1.1. Clinical Use
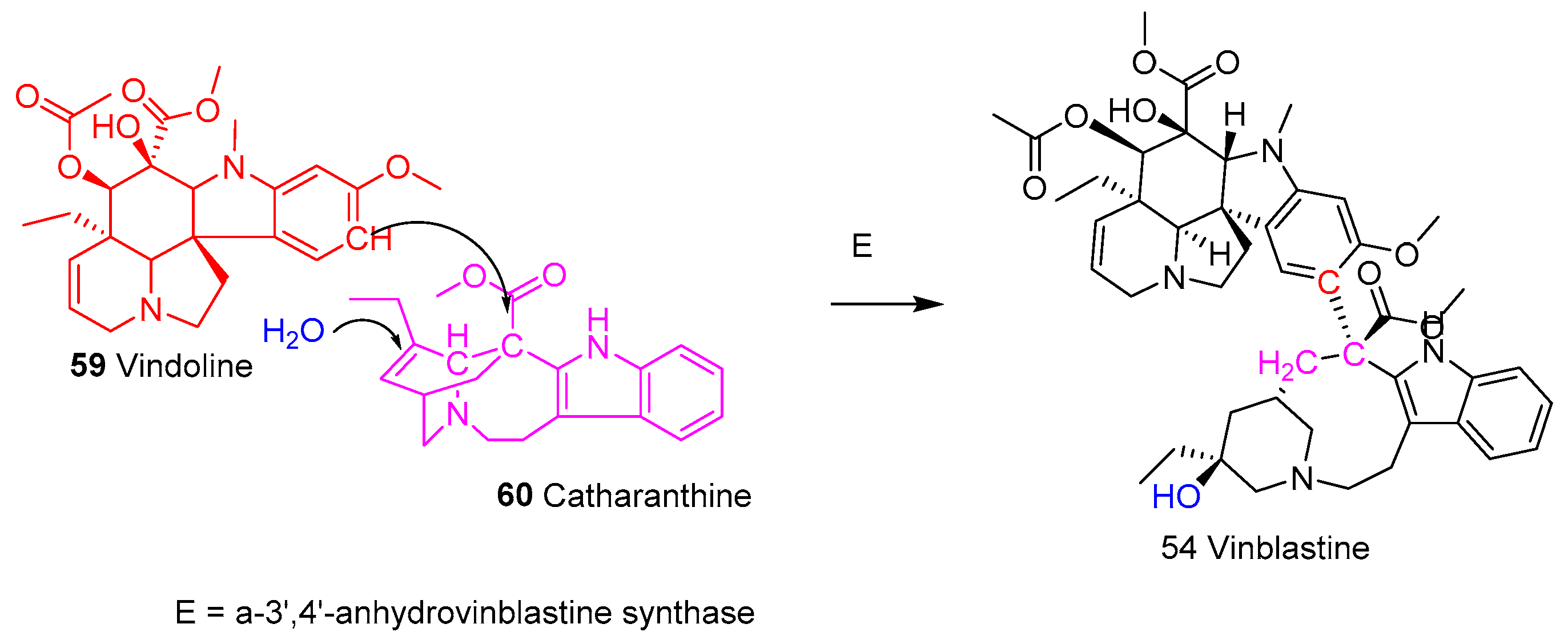
5.1.2. Mechanism of Action
5.1.3. Analogues of Vincalkaloids
5.1.4. Sustainable Production of Vinca Alkaloids
6. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Cragg, G.M.; Grothaus, P.G.; Newman, D.J. Impact of natural products on developing new anti-cancer agents. Chem. Rev. 2009, 109, 3012–3043. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural products as drugs and leads to drugs: The historical perspective. In Natural Product Chemistry for Drug Discovery; Royal Society of Chemistry: London, UK, 2010; pp. 3–27. [Google Scholar]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs over the Nearly Four Decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef] [PubMed]
- Franzyk, H.; Christensen, S.B. Targeting Toxins towards Tumors. Molecules 2021, 26, 1292. [Google Scholar] [CrossRef] [PubMed]
- Hyde, K.D.; Xu, J.; Rapior, S.; Jeewon, R.; Lumyong, S.; Niego, A.G.A.T.; Abeywickrama, P.D.; Aluthmuhandiram, J.V.S.; Brahamanage, R.S.; Brooks, S.; et al. The amazing potential of fungi: 50 ways we can exploit fungi industrially. Fungal Divers. 2019, 97, 1–136. [Google Scholar]
- Blunt, J.W.; Carroll, A.R.; Copp, B.R.; Davis, R.A.; Keyzers, R.A.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2018, 35, 8–53. [Google Scholar] [CrossRef] [PubMed]
- White, N.J. Malaria. In Mansons Tropical Diseases, 23rd ed.; Farrar, J., Hotez, P., Junghanss, T., Kang, G., Lalloo, D., White, N.J., Eds.; Elsevier: Amsterdam, The Netherlands, 2014; Volume 9, pp. 523–601. [Google Scholar]
- Seca, A.M.L.; Pinto, D.C.G.A. Plant secondary metabolites as anticancer agents: Successes in clinical trials and therapeutic application. Int. J. Mol. Sci. 2018, 19, 263. [Google Scholar] [CrossRef]
- Trendafilova, A.; Moujir, L.M.; Sousa, P.M.C.; Seca, A.M.L. Research Advances on Health Effects of Edible Artemisia Species and Some Sesquiterpene Lactones Constituents. Foods 2020, 10, 65. [Google Scholar] [CrossRef]
- Omura, S. Ivermectin: 25 years and still going strong. Int. J. Antimicrob. Agents 2008, 31, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Martino, E.; Casamassima, G.; Castiglione, S.; Cellupica, E.; Pantalone, S.; Papagni, F.; Rui, M.; Siciliano, A.M.; Collina, S. Vinca alkaloids and analogues as anti-cancer agents: Looking back, peering ahead. Bioorg. Med. Chem. Lett. 2018, 28, 2816–2826. [Google Scholar] [CrossRef]
- Singer, C.; Underwood, E.A. A Short History of Medicine, 2nd ed.; Clarendon Press: Oxford, UK, 1962. [Google Scholar]
- Wöhler, F. Ueber künstliche Bildung des Harnstoffs. Ann. Phys. Chem. 1828, 12, 253–257. [Google Scholar] [CrossRef]
- McKie, D. Wöhlers “synthetic” urea and the rejection of vitalism: A chemical legend. Nature 1944, 153, 608–610. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Sorenson, E.J. Classics in Total Synthesis: Targets, Strategies, Methods; Wiley: Weinheim, Germany, 1996. [Google Scholar]
- Nicolaou, K.C.; Snyder, S.A. Classics in Total Synthesis II: More Targets, Stratgeis and Methods; Wiley: Weinheim, Germany, 2003. [Google Scholar]
- Nicolaou, K.C.; Montagnon, T. Molcules that Changed the World; Wiley: Weinheim, Germany, 2008; p. 366. [Google Scholar]
- Nicolaou, K.C.; Chen, J.S. Classics in Total Synthesis III: Further Targets, Strategies, Methods; Wiley: Weinheim, Germany, 2011. [Google Scholar]
- Mahdi, J.G.; Mahdi, A.J.; Bowen, I.D. The historical analysis of aspirin discovery, its relation to the willow tree and antiproliferative and anticancer potential. Cell Prolif. 2006, 39, 147–155. [Google Scholar] [CrossRef]
- Marko, V. Fraom Aspitin to Viagara; Springer Nature: Cham, Schwitzerland, 2020. [Google Scholar]
- Appendino, G.; Pollastro, F. Plants: Revamping the oldest source of medicines with modern science. In Natural Product Chemistry for Drug Discovery; Royal Society of Chemistry: London, UK, 2010; pp. 140–173. [Google Scholar]
- Dewick, P.M. Medicinal Natural Products, 3rd ed.; John Wiley and Sons Ltd.: Chicester, UK, 2009. [Google Scholar]
- Hayashi, Y. Time Economy in Total Synthesis. J. Org. Chem. 2021, 86, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Seeman, J.I. The Woodward-Doering/Rabe-Kindler total synthesis of quinine: Setting the record straight. Angew. Chem. Int. Ed. 2007, 46, 1378–1413. [Google Scholar] [CrossRef] [PubMed]
- Barber, J. Photosystem II: The water splitting enzyme of photosynthesis and the origin of oxygen in our atmosphere. Q. Rev. Biophys. 2016, 49, e14. [Google Scholar] [CrossRef]
- Oro, J. Chemical evolution and the origin of life. Adv. Space Res. 1983, 3, 77–94. [Google Scholar] [CrossRef]
- Nye, E.R. Alphonse Laveran (1845-1922): Discoverer of the malarial parasite and Nobel laureate, 1907. J. Med. Biogr. 2002, 10, 81–87. [Google Scholar] [CrossRef]
- Yoeli, M. Sir Ronald Ross and the evolution of malaria research. Bull. N. Y. Acad. Med. 1973, 49, 722–735. [Google Scholar]
- Hay, S.I.; Guerra, C.A.; Tatem, A.J.; Noor, A.M.; Snow, R.W. The global distribution and population at risk of malaria: Past, present, and future. Lancet Infect. Dis. 2004, 4, 327–336. [Google Scholar] [CrossRef]
- WHO. World Malaria Report; WHO: Geneva, Switzerland, 2019. [Google Scholar]
- Curtin, P.D. The End of the “White Man’s Grave”? Nineteenth-Century Mortality in West Africa. J. Interdiscip. Hist. 1990, 21, 63–88. [Google Scholar] [CrossRef]
- Rocco, F. The Miraculous Fever-Tree. Malaria, Medicine and the Cure that Changed the World; HarperCollins: London, UK, 2003. [Google Scholar]
- Naranjo, P. Epidemic hecatomb in the New World. Allergy Proc. 1992, 13, 237–241. [Google Scholar] [CrossRef]
- Gachelin, G.; Garner, P.; Ferroni, E.; Trohler, U.; Chalmers, I. Evaluating Cinchona bark and quinine for treating and preventing malaria. J. R. Soc. Med. 2017, 110, 31–40. [Google Scholar] [CrossRef]
- Gachelin, G.; Garner, P.; Ferroni, E.; Trohler, U.; Chalmers, I. Evaluating Cinchona bark and quinine for treating and preventing malaria. J. R. Soc. Med. 2017, 110, 73–82. [Google Scholar] [CrossRef]
- Meshnick, S.R.; Dobson, M.J. The History of Antimalarial Drugs. In Antimalarial Chemotherapy; Rosenthal, P.J., Ed.; Humana Press: Towota, NJ, USA, 2001; pp. 15–25. [Google Scholar]
- McHale, D. The cinchona tree. Biologist 1986, 33, 45–53. [Google Scholar]
- Pabst, G. Köhler’s Medizinal-Pflanzen; Verlag von Fr. Eugen Köhler: Gera-Untermhaus, Germany, 1887; Volume 1. [Google Scholar]
- Nair, K.P.P. Cinchona (Cinchona sp.). In The Agronomy and Economy of Important Tree Crops of the Developing World; Nair, K.P.P., Ed.; Elsevier: Amsterdam, The Netherlands, 2010. [Google Scholar]
- Cross, S.H. Quinine, Production and Marketing; Bureau of Foreign and Domestic Commerce: Washington, DC, USA, 1924.
- Williams, D. Clements Robert Markham and the Introduction of the Cinchona Tree into British India, 1861. Georg. J. 1962, 128, 431–442. [Google Scholar] [CrossRef]
- Efferth, T.; Banerjee, M.; Paul, N.W.; Abdelfatah, S.; Arend, J.; Hamdoun, S.; Hamm, R.; Hong, C.; Kadioglu, O.; Nass, J.; et al. Biopiracy of natural products and good bioprospecting practice. Phytomedicine 2016, 23, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Cragg, G.M.; Katz, F.; Newman, D.J.; Rosenthal, J. The impact of the United Nations Convention on Biological Diversity on natural products research. Nat. Prod. Rep. 2012, 29, 1407–1423. [Google Scholar] [CrossRef]
- Rivero-Cruz, I.; Cristians, S.; Ovalle-Magallanes, B.; Mata, R. Mexican copalchis of the Rubiaceae family: More than a century of pharmacological and chemical investigations. Phytochem. Rev. 2019, 18, 1435–1455. [Google Scholar] [CrossRef]
- Maldonado, C.; Barnes, C.J.; Cornett, C.; Holmfred, E.; Hansen, S.H.; Persson, C.; Antonelli, A.; Rønsted, N. Phylogeny Predicts the Quantity of Antimalarial Alkaloids within the Iconic Yellow Cinchona Bark (Rubiaceae: Cinchona calisaya). Front. Plant Sci. 2017, 8, 391. [Google Scholar] [CrossRef]
- Canales, N.A.; Gress Hansen, T.N.; Cornett, C.; Walker, K.; Driver, F.; Antonelli, A.; Maldonado, C.; Nesbitt, M.; Barnes, C.J.; Roensted, N. Historical chemical annotations of Cinchona bark collections are comparable to results from current day high-pressure liquid chromatography technologies. J. Ethnopharmacol. 2020, 249, 112375. [Google Scholar] [CrossRef] [PubMed]
- Martin, W.E.; Gandara, J.A. Alkaloid content of Ecuadoran and other American cinchona barks. Bot. Gaz. 1945, 107, 184–199. [Google Scholar] [CrossRef]
- Karamanou, M.; Tsoucalas, G.; Pantos, K.; Androutsos, G. Isolating colchicine in 19th century: An old drug revisited. Curr. Pharm. Des. 2018, 24, 654–658. [Google Scholar] [CrossRef]
- Mitcham, J.C. Patrolling the White Man’s Grave: The impact of disease on Anglo-American naval operations against the slave trade, 1841–1862. North. Mar. 2010, 20, 37–56. [Google Scholar]
- Holland, J.H. Ledger Bark and Red Bark. Bull. Misc. Inf. 1932, 1932, 1–17. [Google Scholar] [CrossRef]
- Streller, S.; Roth, K. Eine Rinde erobert die Welt. Chem. Unserer Zeit 2012, 46, 228–247. [Google Scholar] [CrossRef]
- Prelog, V. Häfliger, Über China-alkaloide. Helv. Chim Acta 1950, 23, 257–2578. [Google Scholar]
- Baird, J.K. Effectiveness of antimalarial drugs. N. Engl. J. Med. 2005, 352, 1565–1577. [Google Scholar] [CrossRef]
- Kaufman, T.S.; Ruveda, E.A. The quest for quinine: Those who won the battles and those who won the war. Angew. Chem. Int. Ed. 2005, 44, 854–885. [Google Scholar] [CrossRef] [PubMed]
- Gensini, G.F.; Conti, A.A.; Lippi, D. The contributions of Paul Ehrlich to infectious disease. J. Infect. 2007, 54, 221–224. [Google Scholar] [CrossRef]
- Winau, F.; Westphal, O.; Winau, R. Paul Ehrlich—In search of the magic bullet. Microbes Infect. 2004, 6, 786–789. [Google Scholar] [CrossRef] [PubMed]
- Coatney, G.R. Pitfalls in a discovery: The chronicle of chloroquine. Am. J. Trop. Med. Hyg. 1963, 12, 121–128. [Google Scholar] [CrossRef]
- Sharma, V.P. Re-emergence of malaria in India. Indian J. Med. Res. 1996, 103, 26–45. [Google Scholar] [PubMed]
- Wellems, T.E.; Plowe, C.V. Chloroquine-resistant malaria. J. Infect. Dis. 2001, 184, 770–776. [Google Scholar] [CrossRef] [PubMed]
- Ridley, R.G. Medical need, scientific opportunity and the drive for antimalarial drugs. Nature 2002, 415, 686–693. [Google Scholar] [CrossRef] [PubMed]
- Frosch, A.E.P.; Venkatesan, M.; Laufer, M.K. Patterns of chloroquine use and resistance in sub-Saharan Africa: A systematic review of household survey and molecular data. Malar. J. 2011, 10, 116. [Google Scholar] [CrossRef] [PubMed]
- Trape, J.F. The public health impact of chloroquine resistance in Africa. Am. J. Trop. Med. Hyg. 2001, 64, 12–17. [Google Scholar] [CrossRef]
- Ecker, A.; Lehane, A.M.; Clain, J.; Fidock, D.A. PfCRT and its role in antimalarial drug resistance. Trends Parasitol. 2012, 28, 504–514. [Google Scholar] [CrossRef]
- Shanks, G.D.; Biomndo, K.; Hay, S.I.; Snow, R.W. Changing patterns of clinical malaria since 1965 among a tea estate population located in the Kenyan highlands. Trans. R. Soc. Trop. Med. Hyg. 2000, 94, 253–255. [Google Scholar] [CrossRef]
- Sidhu, A.b.S.; Verdier-Pinard, D.; Fidock, D.A. Chloroquine resistance in Plasmodium falciparum malaria parasites conferred by pfcrt mutations. Science 2002, 298, 210–213. [Google Scholar] [CrossRef] [PubMed]
- Dorsey, G.; Fidock, D.A.; Wellems, T.E.; Rosenthal, P.J. Mechanisms of Quinoline Resistance. In Antimalarial Chemotherapy; Rosenthal, P.J., Ed.; Humana Press: Totowa, NJ, USA, 2001; pp. 153–172. [Google Scholar]
- Dahl, E.L.; Shock, J.L.; Shenai, B.R.; Gut, J.; DeRisi, J.L.; Rosenthal, P.J. Tetracyclines specifically target the apicoplast of the malaria parasite Plasmodium falciparum. Antimicrob.Agents Chemother. 2006, 50, 3124–3131. [Google Scholar] [CrossRef]
- Berman, J.; Brown, T.; Dow, G.; Toovey, S. Tafenoquine and primaquine do not exhibit clinical neurologic signs associated with central nervous system lesions in the same manner as earlier 8-aminoquinolines. Malar. J. 2018, 17, 407. [Google Scholar] [CrossRef] [PubMed]
- Araujo, E.; Braun, M.-G.; Dragovich, P.S.; Converso, A.; Nantermet, P.G.; Roecker, A.J.; Dhar, T.G.M.; Haile, P.; Hurtley, A.; Merritt, J.R.; et al. To Market, To Market—2018: Small Molecules. In 2019 Medicinal Chemistry Reviews—2019; Bonson, J.J., Ed.; American Chemical Society: Washington, DC, USA, 2019; Volume 54, pp. 469–596. [Google Scholar]
- Christensen, S.B. Negelected Diseases. In Textbook of Drug Desing and Discovery, 4th ed.; Krogsgaard-Larsen, P., Strømgård, K., Madsen, U., Eds.; CRC Press: Boca Raton, FL, USA, 2010; pp. 314–358. [Google Scholar]
- Uddin, T.; McFadden, G.I.; Goodman, C.D. Validation of putative apicoplast-targeting drugs using a chemical supplementation assay in cultured human malaria parasites. Antimicrob. Agents Chemother. 2018, 62, 1–17. [Google Scholar] [CrossRef]
- Schlitzer, M. Malaria Chemotherapeutics Part I: History of Antimalarial Drug Development, Currently Used Therapeutics, and Drugs in Clinical Development. ChemMedChem 2007, 2, 944–986. [Google Scholar] [CrossRef] [PubMed]
- Mathews, E.S.; John, A.R.O. Tackling resistance: Emerging antimalarials and new parasite targets in the era of elimination. F1000Research 2018, 7, 1170. [Google Scholar] [CrossRef] [PubMed]
- Blank, B.R.; Gonciarz, R.L.; Talukder, P.; Gut, J.; Legac, J.; Rosenthal, P.J.; Renslo, A.R. Antimalarial Trioxolanes with Superior Drug-Like Properties and In Vivo Efficacy. ACS Infect. Dis. 2020, 6, 1827–1835. [Google Scholar] [CrossRef] [PubMed]
- Biot, C.; Nosten, F.; Fraisse, L.; Ter-Minassian, D.; Khalife, J.; Dive, D. The antimalarial ferroquine: From bench to clinic. Parasite 2011, 18, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Tilley, L.; Loria, P.; Foley, M. Chloroquine and Other Quinoline Antimalarials. In Antimalarial Chemotherapy; Rosenthal, P.J., Ed.; Humana Press: Totowa, NJ, USA, 2001; pp. 87–121. [Google Scholar]
- Haynes, R.K.; Cheu, K.-W.; N’Da, D.; Coghi, P.; Monti, D. Considerations on the Mechanism of Action of Artemisinin Antimalarials: Part 1—The ‘Carbon Radical’ and ‘Heme’ Hypotheses. Infect. Dis. Drug Targets 2013, 13, 217–277. [Google Scholar] [CrossRef] [PubMed]
- Brocks, D.R.; Mehvar, R. Stereoselectivity in the pharmacodynamics and pharmacokinetics of the chiral antimalarial drugs. Clin. Pharmacokinet. 2003, 42, 1359–1382. [Google Scholar] [CrossRef]
- Cooper, R.; Deakin, J. Botanical Miracles. Chemistry of Plants that Changed the World; CRC Press: Boca Raton, FL, USA, 2016; p. 271. [Google Scholar]
- Tu, Y. The discovery of artemisinin (qinghaosu) and gifts from Chinese medicine. Nat. Med. 2011, 17, 1217–1220. [Google Scholar] [CrossRef]
- Klayman, D.L. Qinhaosu (Artemisinin): An Antimalarial Drug from China. Science 1985, 228, 1049–1055. [Google Scholar] [CrossRef]
- Group, Q.A.C.R. Antimalaria studies on qinhaosu. Chin. Med. J. 1979, 92, 811–816. [Google Scholar]
- Vajs, V.; Jokic, A.; Milosavljevic, S. Artemisinin Story from the Balkans. Nat. Prod. Commun. 2017, 12. [Google Scholar] [CrossRef]
- Christensen, S.B.; Bygbjerg, I.C. Neglected Diseases. In Textbook of Drug Design and Discovery, 5th ed.; Strømgård, K., Krogsgaard-Larsen, P., Madsen, U., Eds.; CRC Press: Boca Raton, FL, USA, 2017; pp. 347–368. [Google Scholar]
- Lu, F.; He, X.-L.; Richard, C.; Cao, J. A brief history of artemisinin: Modes of action and mechanisms of resistance. Chin. J. Nat. Med. 2019, 17, 331–336. [Google Scholar] [CrossRef]
- Ikram, N.K.B.K.; Simonsen, H.T. A Review of Biotechnological Artemisinin Production in Plants. Front. Plant Sci. 2017, 8, 1966. [Google Scholar] [CrossRef] [PubMed]
- Giannangelo, C.; Siddiqui, G.; De Paoli, A.; Anderson, B.M.; Edgington-Mitchell, L.E.; Charman, S.A.; Creek, D.J. System-wide biochemical analysis reveals ozonide antimalarials initially act by disrupting Plasmodium falciparum haemoglobin digestion. PLoS Pathog. 2020, 16, e1008485. [Google Scholar] [CrossRef] [PubMed]
- Simonsen, P.E.; Fischer, P.U.; Hoerauf, A.; Weil, G.J. The Filariases. In Manson’s Tropical Diseases; Farrar, J., Hotez, P., Junghanss, T., Kang, G., Lalloo, D., White, N., Eds.; Elservier: Amsterdam, The Netherlands, 2014; pp. 737–765.e5. [Google Scholar]
- Hopkins, A.D. Onchocerciasis: Status and progress towards elimination. CAB Rev. 2020, 15, 021. [Google Scholar] [CrossRef]
- Omura, S. A Splendid Gift from the Earth: The Origins and Impact of the Avermectins (Nobel Lecture). Angew. Chem. Int. Ed. 2016, 55, 10190–10209. [Google Scholar] [CrossRef] [PubMed]
- Laing, R.; Gillan, V.; Devaney, E. Ivermectin—Old Drug, New Tricks? Trends Parasitol. 2017, 33, 463–472. [Google Scholar] [CrossRef]
- Omura, S.; Crump, A. The live an times of ivermectin—A success story. Nat. Rev. Microbiol. 2004, 2, 984–989. [Google Scholar] [CrossRef]
- Prichard, R.; Menez, C.; Lespine, A. Moxidectin and the avermectins: Consanguinity but not identity. Int. J. Parasitol. Drugs Drug Resist. 2012, 2, 134–153. [Google Scholar] [CrossRef]
- Shoop, W.L.; Egerton, J.R.; Eary, C.H.; Haines, H.W.; Michael, B.F.; Mrozik, H.; Eskola, P.; Fisher, M.H.; Slayton, L.; Ostlind, D.A.; et al. Eprinomectin: A novel avermectin for use as a topical endectocide for cattle. Int. J. Parasitol. 1996, 26, 1237–1242. [Google Scholar] [CrossRef]
- Bishop, B.F.; Bruce, C.I.; Evans, N.A.; Goudie, A.C.; Gration, K.A.F.; Gibson, S.P.; Pacey, M.S.; Perry, D.A.; Walshe, N.D.A.; Witty, M.J. Selamectin: A novel broad-spectrum endectocide for dogs and cats. Vet. Parasitol. 2000, 91, 163–176. [Google Scholar] [CrossRef]
- Lee, S.R.; Kim, H.U.; Kang, H.M.; Choi, I.S. Preparation of Selamectin intermediate and process for Selamectin. KR2019056863A, 2019. [Google Scholar]
- Vercruysse, J.; Rew, R.S. Macrocyclic Lactones in Antiparasitic Therapy; CABI Publishing: Wallingford, UK, 2002. [Google Scholar]
- Milton, P.; Hamley, J.I.D.; Walker, M.; Basanez, M.-G. Moxidectin: An oral treatment for human onchocerciasis. Expert Rev. Anti Infect. Ther. 2020, 18, 1067–1081. [Google Scholar] [CrossRef]
- Brattig, N.W.; Cheke, R.A.; Garms, R. Onchocerciasis (River Blindness)—More than a Century of Research and Control. Acta Trop. 2020, 218, 105677. [Google Scholar] [CrossRef]
- Lakwo, T.L.; Garms, R.; Rubaale, T.; Katabarwa, M.; Walsh, F.; Habomugisha, P.; Oguttu, D.; Unnasch, T.; Namanya, H.; Tukesiga, E.; et al. The disappearance of onchocerciasis from the Itwara focus, western Uganda after elimination of the vector Simulium neavei and 19 years of annual ivermectin treatments. Acta Trop. 2013, 126, 218–221. [Google Scholar] [CrossRef] [PubMed]
- Sainas, S.; Dosio, F.; Boschi, D.; Lolli, M.L. Targeting Human Onchocerciasis: Recent Advances Beyond Ivermectin. Ann. Rep. Med. Chem. 2018, 51, 1–38. [Google Scholar]
- Verdu, J.R.; Cortez, V.; Martinez-Pinna, J.; Ortiz, A.J.; Lumaret, J.-P.; Lobo, J.M.; Sanchez-Pinero, F.; Numa, C. First assessment of the comparative toxicity of ivermectin and moxidectin in adult dung beetles: Sub-lethal symptoms and pre-lethal consequences. Sci. Rep. 2018, 8, 14885. [Google Scholar] [CrossRef] [PubMed]
- Awasthi, A.; Razzak, M.; Al-Kassas, R.; Harvey, J.; Garg, S. Analytical profile of moxidectin. Profiles Drug Subst. Excipients Relat. Methodol. 2013, 38, 315–366. [Google Scholar]
- WHO. Lymphatic Filariasis. 2020. Available online: https://www.who.int/news-room/fact-sheets/detail/lymphatic-filariasis (accessed on 1 April 2021).
- Björkling, F.; Moreira, J.; Stenvang, J. Anticancer Agents. In Textbook of Drug Design and Discovery, 5th ed.; Strømgaard, K., Krogsgaard-Larsen, P., Madsen, U., Eds.; CRC Press: Boca Raton, FL, USA, 2017; pp. 369–386. [Google Scholar]
- Xie, S.; Zhou, J. Harnessing Plant Biodiversity for the Discovery of Novel Anticancer Drugs Targeting Microtubules. Front. Plant. Sci. 2017, 8, 720. [Google Scholar] [CrossRef]
- Noble, R.L.; Beer, C.T.; Cutts, J.H. Role of chance observations in chemotherapy: Vinca rosea. Ann. N. Y. Acad. Sci. 1958, 76, 882–894. [Google Scholar] [CrossRef]
- Noble, R.L. The Discovery of the Vinca Alkaloids—Chemotherapeutic-Agents Against Cancer. Biochem. Cell Biol. Biochim. Biol. Cell. 1990, 68, 1344–1351. [Google Scholar] [CrossRef]
- Neuss, N.; Gorman, M.; Svoboda, G.H.; Maciak, G.; Beer, C.T. Vinca alkaloids. III. Characterization of leurosine and vincaleukoblastine, new alkaloids from Vinca rosea. J. Am. Chem. Soc. 1959, 81, 4754–4755. [Google Scholar] [CrossRef]
- Neuss, N.; Gorman, M.; Hargrove, W.; Cone, N.J.; Biemann, K.; Buechi, G.; Manning, R.E. Vinca alkaloids. XXI. Structures of the oncolytic alkaloids vinblastine and vincristine. J. Am. Chem. Soc. 1964, 86, 1440–1442. [Google Scholar] [CrossRef]
- Moncrief, W.J.; Lipscomb, W.N. Structure of leurocristine methiodide dihydrate by anomalous scattering methods; relation to leurocristine (vincristine) and vincaleukoblastine (vinblastine). Acta Crystallogr. 1966, 21, 322–331. [Google Scholar] [CrossRef]
- Barrales-Cureño, H.J.; Reyes, C.R.; García, I.V.; Valdez, L.G.L.; De Jesús, A.G.; Ruíz, J.A.C.; Herrera, L.M.S.; Caballero, M.C.C.; Magallón, J.A.S.; Perez, J.E.; et al. Alkaloids of Pharmacological Importance in Catharanthus roseus. In Alkaloids. Their Importance in Natur and Human Life; Kurek, J., Ed.; IntechOpen: London, UK, 2019. [Google Scholar]
- Gidding, C.E.; Kellie, S.J.; Kamps, W.A.; de Graaf, S.S. Vincristine revisited. Crit. Rev. Oncol. Hematol. 1999, 29, 267–287. [Google Scholar] [CrossRef]
- Jordan, M.A.; Wilson, L. Microtubules as a target for anticancer drugs. Nat. Rev. Cancer 2004, 4, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Almagro, L.; Fernandez-Perez, F.; Pedreno, M.A. Indole alkaloids from Catharanthus roseus: Bioproduction and their effect of human health. Molecules 2015, 20, 2973–3000. [Google Scholar] [CrossRef] [PubMed]
- Brady, S.F.; Pawluczyk, J.M.; Lumma, P.K.; Feng, D.M.; Wai, J.M.; Jones, R.; Feo-Jones, D.; Wong, B.K.; Miller-Stein, C.; Lin, J.H.; et al. Design and synthesis of a pro-drug of vinblastine targeted at treatment of prostate cancer with enhanced efficacy and reduced systemic toxicity. J. Med. Chem. 2002, 45, 4706–4715. [Google Scholar] [CrossRef] [PubMed]
- Verma, P.; Khan, S.A.; Parasharami, V.; Mathur, A.K. Biotechnological Interventions to Modulate Terpenoid Indole Alkaloid Pathway in Catharanthus roseus Using In Vitro Tools and Approaches. In Cathararanthus roseus. Current Research and Future Prospects; Naeem, M., Aftab, T., Masroor, M., Khan, A., Eds.; Springer International Publishing: Berlin/Heidelberg, Germany, 2017; pp. 247–275. [Google Scholar]
- Li, C.; Galani, S.; Hassan, F.-U.; Rashid, Z.; Naveed, M.; Fang, D.; Ashraf, A.; Qi, W.; Arif, A.; Saeed, M.; et al. Biotechnological approaches to the production of plant-derived promising anticancer agents: An update and overview. Biomed. Pharmacother. 2020, 132, 110918. [Google Scholar]
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Arya, S.S.; Rookes, J.E.; Cahill, D.M.; Lenka, S.K. Next-generation metabolic engineering approaches towards development of plant cell suspension cultures as specialized metabolite producing biofactories. Biotechnol. Adv. 2020, 45, 107635. [Google Scholar] [CrossRef]
- Atanasov, A.G.; Waltenberger, B.; Pferschy-Wenzig, E.-M.; Linder, T.; Wawrosch, C.; Uhrin, P.; Temml, V.; Wang, L.; Schwaiger, S.; Heiss, E.H.; et al. Discovery and resupply of pharmacologically active plant-derived natural products: A review. Biotechnol. Adv. 2015, 33, 1582–1614. [Google Scholar] [CrossRef] [PubMed]
- Mayer, A.M.S.; Glaser, K.B.; Cuevas, C.; Jacobs, R.S.; Kem, W.; Little, R.D.; McIntosh, J.M.; Newman, D.J.; Potts, B.C.; Shuster, D.E. The odyssey of marine pharmaceuticals: A current pipeline perspective. Trends Pharmacol. Sci. 2010, 31, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Li, S.-J.; Zhang, X.; Wang, X.-H.; Zhao, C.-Q. Novel natural compounds from endophytic fungi with anticancer activity. Eur. J. Med. Chem. 2018, 156, 316–343. [Google Scholar] [CrossRef]

























Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Christensen, S.B. Natural Products That Changed Society. Biomedicines 2021, 9, 472. https://doi.org/10.3390/biomedicines9050472
Christensen SB. Natural Products That Changed Society. Biomedicines. 2021; 9(5):472. https://doi.org/10.3390/biomedicines9050472
Chicago/Turabian StyleChristensen, Søren Brøgger. 2021. "Natural Products That Changed Society" Biomedicines 9, no. 5: 472. https://doi.org/10.3390/biomedicines9050472
APA StyleChristensen, S. B. (2021). Natural Products That Changed Society. Biomedicines, 9(5), 472. https://doi.org/10.3390/biomedicines9050472