
Pharmacological Inhibition of WIP1 Sensitizes Acute Myeloid Leukemia Cells to the MDM2 Inhibitor Nutlin-3a


 , , ,
, , ,  ,
, 
 , ,
, ,  ,
, 

Abstract
1. Introduction
2. Experimental Section
2.1. Human AML Cell Lines
2.2. Human Primary Cells
2.3. Drugs
2.4. Cell Viability Assay
2.5. Annexin V-Propidium IODIDE Staining of Apoptotic Cells
2.6. RNA Extraction and Gene Expression Profiling (GEP)
2.7. Analysis of Public GEP and RNA-Sequencing Cohorts
2.8. Western Blots Analysis
2.9. Statistical Analyses
3. Results
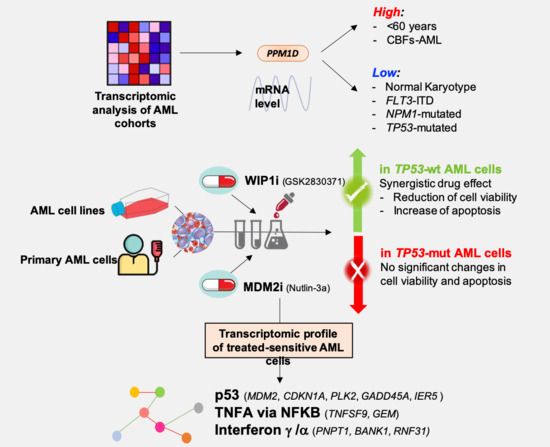
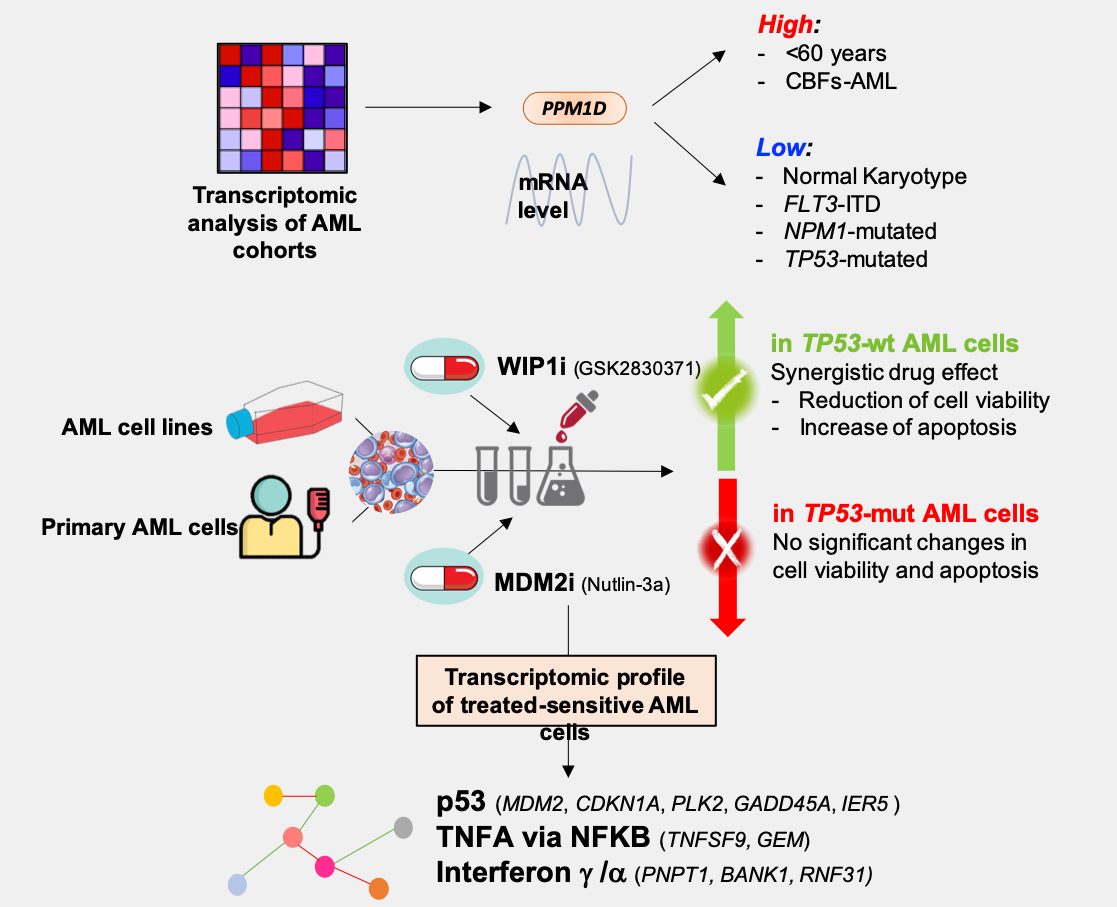
3.1. PPM1D mRNA Levels Differ among Age, Cytogenetic and Mutational Subgroups in AML
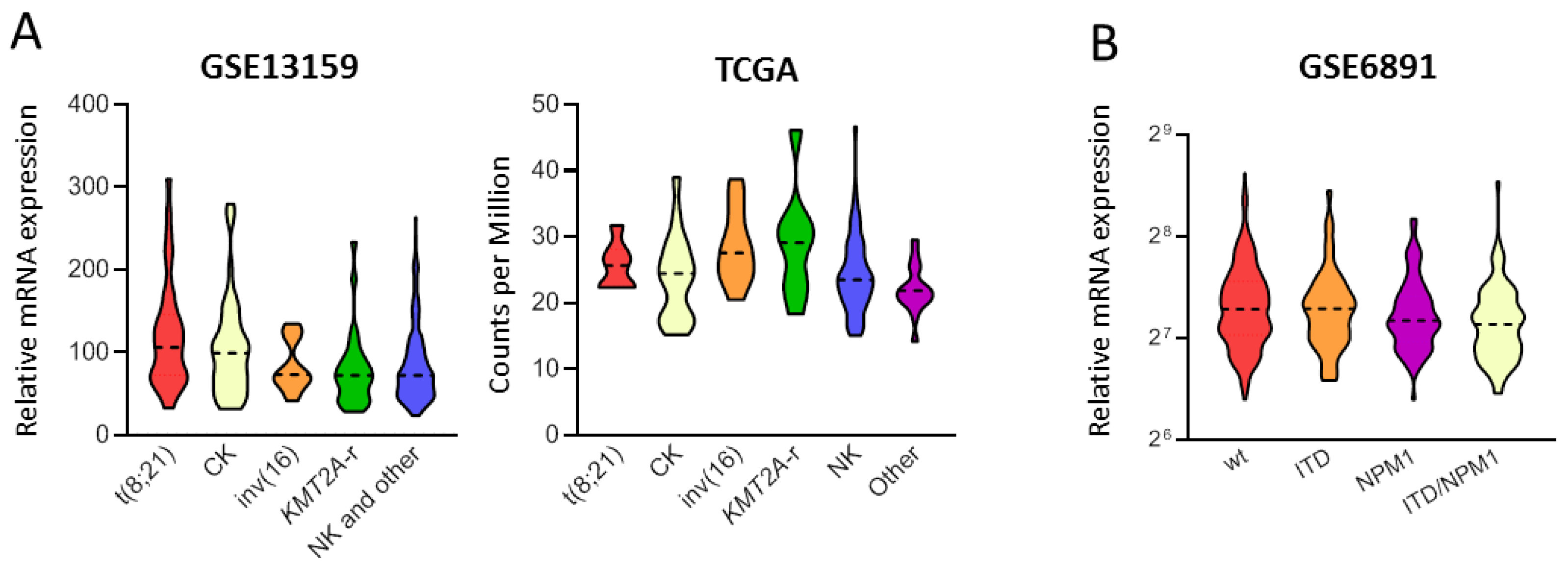
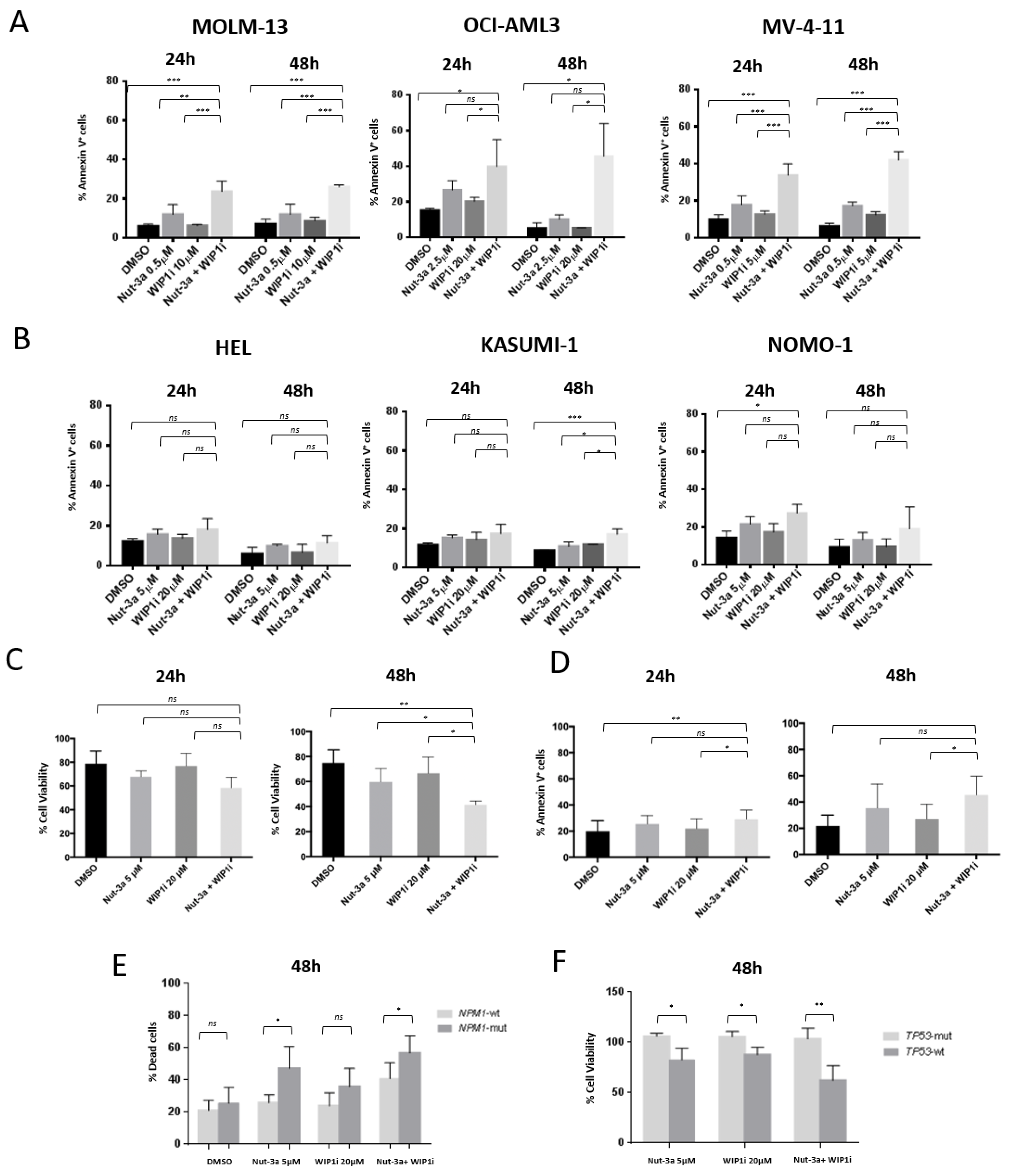
3.2. Combined Inhibition of Nut-3a and WIP1i Synergistically Reduces AML Cells Viability
3.3. WIP1i Sensitizes TP53-wt AML Cells to Nut-3a-Induced Apoptosis
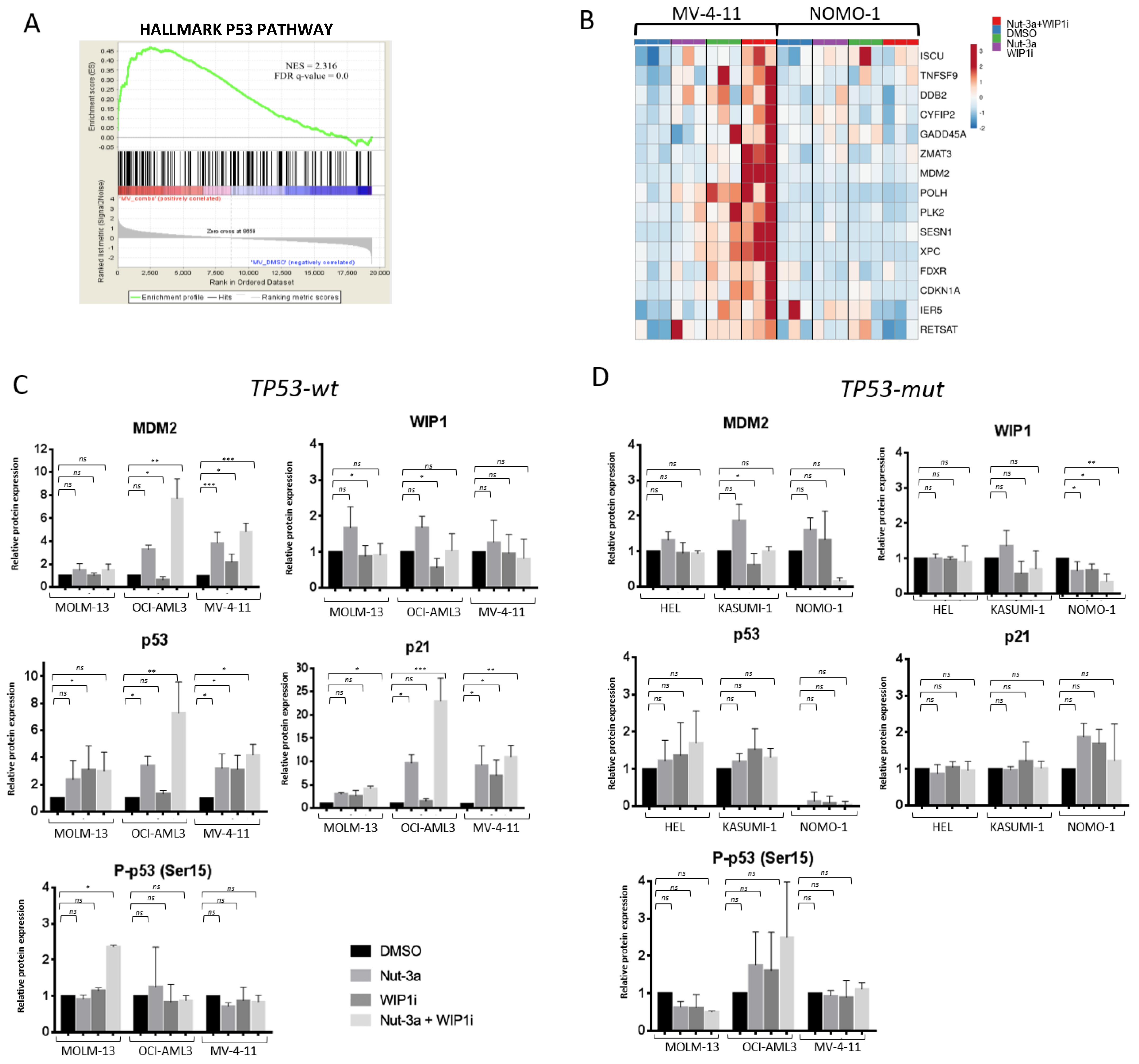
3.4. The Inhibition of WIP1 and MDM2 Altered the Expression of p53 Pathway-Related Genes in TP53-wt Cells
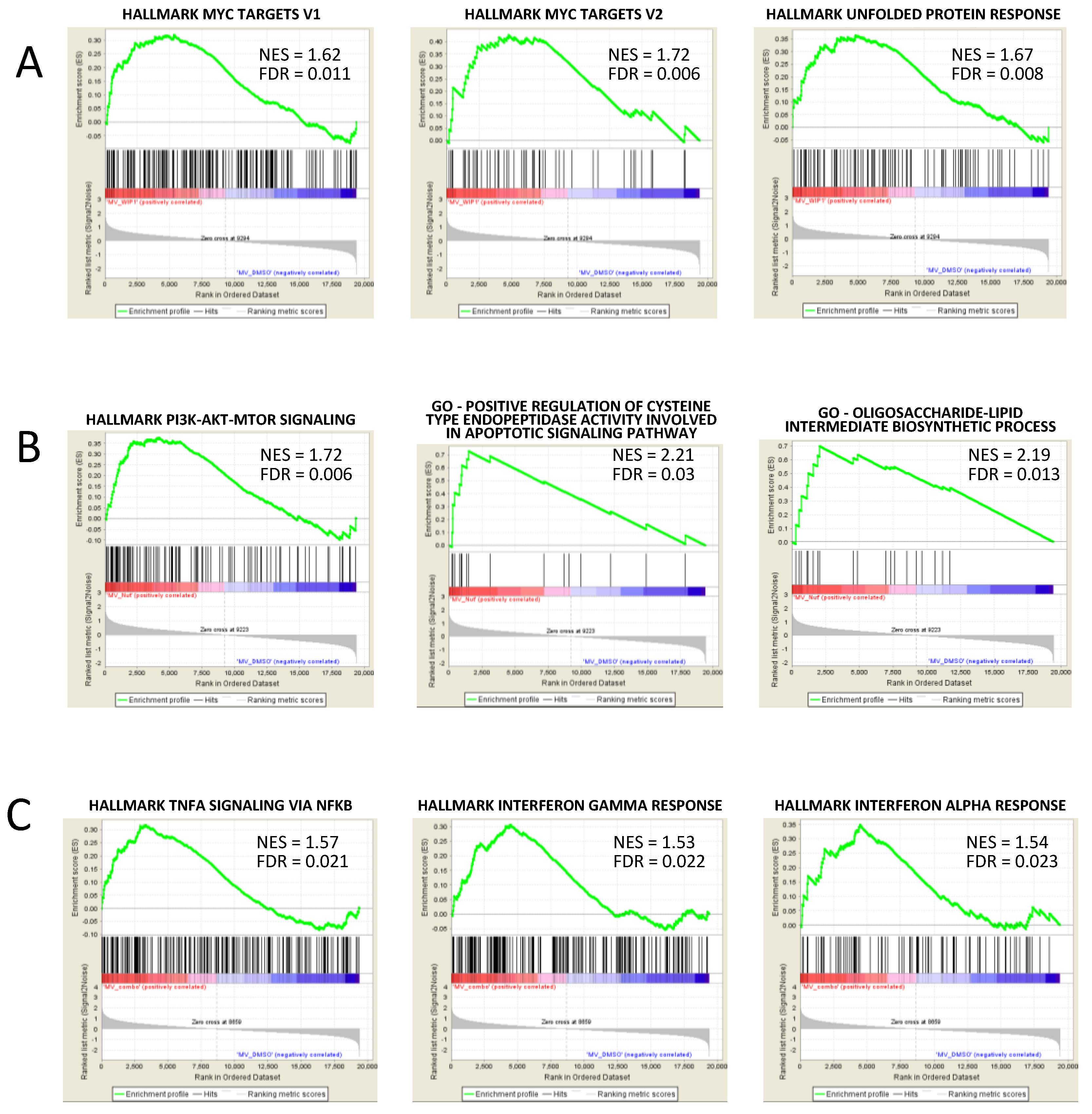
3.5. GSEA Analysis Showed the Enrichment of Single Agent- and Drug Combination-Specific Genes and Pathways in MV-4-11 and NOMO-1 Cells
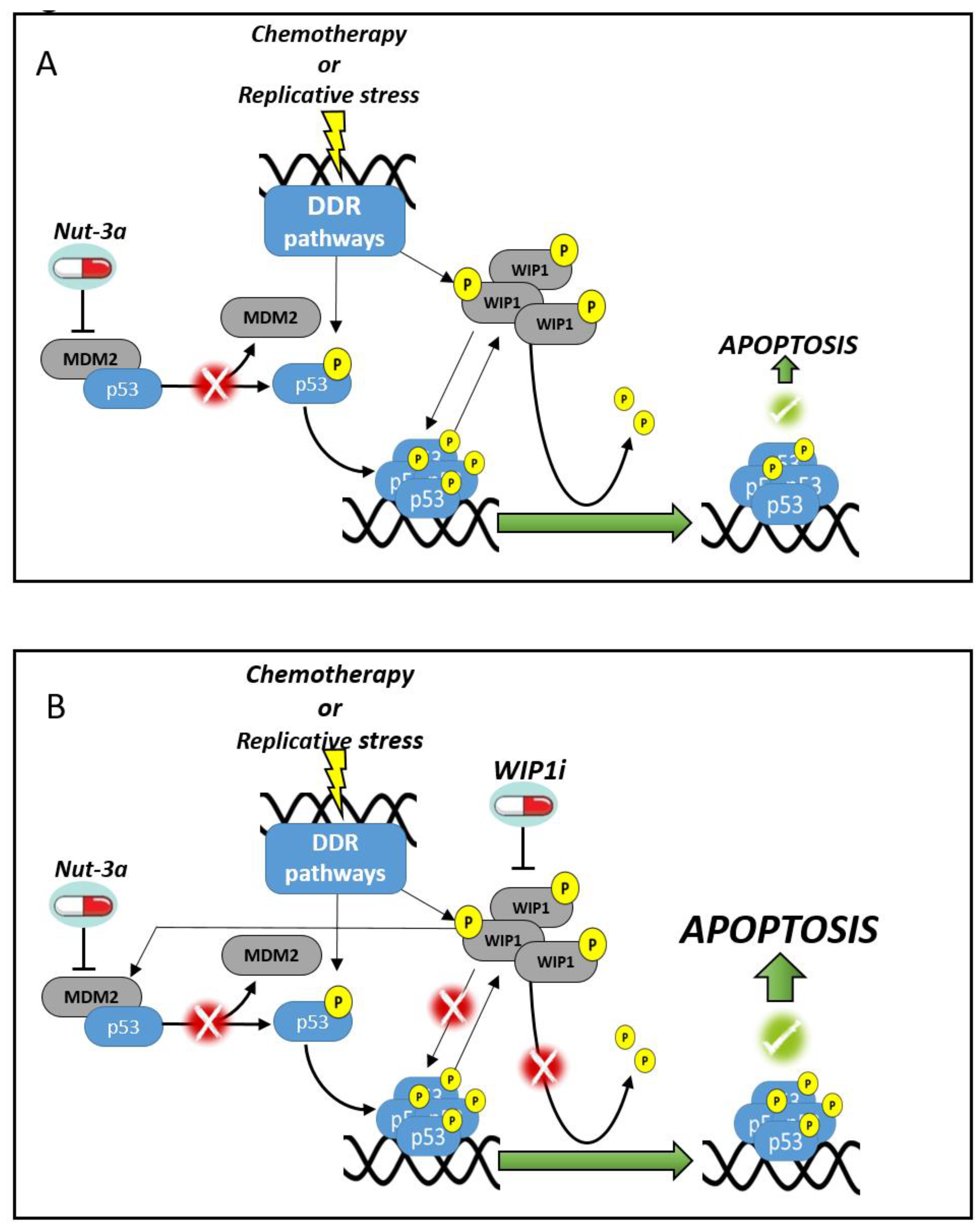
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bhattacharya, D.; Hiregange, D.; Rao, B.J. ATR kinase regulates its attenuation via PPM1D phosphatase recruitment to chromatin during recovery from DNA replication stress signalling. J. Biosci. 2018, 43, 25–47. [Google Scholar] [CrossRef] [PubMed]
- Macurek, L.; Benada, J.; Müllers, E.; Halim, V.A.; Krejčíková, K.; Burdová, K.; Pecháčková, S.; Hodný, Z.; Lindqvist, A.; Medema, R.H.; et al. Downregulation of Wip1 phosphatase modulates the cellular threshold of DNA damage signaling in mitosis. Cell Cycle 2013, 12, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Cha, H.; Lowe, J.M.; Li, H.; Lee, J.S.; Belova, G.I.; Bulavin, D.V.; Fornace, A.J. Wip1 Directly Dephosphorylates -H2AX and Attenuates the DNA Damage Response. Cancer Res. 2010, 70, 4112–4122. [Google Scholar] [CrossRef] [PubMed]
- Brazina, J.; Svadlenka, J.; Macurek, L.; Andera, L.; Hodny, Z.; Bartek, J.; Hanzlikova, H. DNA damage-induced regulatory interplay between DAXX, p53, ATM kinase and Wip1 phosphatase. Cell Cycle 2015, 14, 375–387. [Google Scholar] [CrossRef] [PubMed]
- Shreeram, S.; Demidov, O.N.; Hee, W.K.; Yamaguchi, H.; Onishi, N.; Kek, C.; Timofeev, O.N.; Dudgeon, C.; Fornace, A.J.; Anderson, C.W.; et al. Wip1 Phosphatase Modulates ATM-Dependent Signaling Pathways. Mol. Cell 2006, 23, 757–764. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, H.; Benada, J.; Müllers, E.; Akopyan, K.; Burdova, K.; Koolmeister, T.; Helleday, T.; Medema, R.H.; Macurek, L.; Lindqvist, A. ATM /Wip1 activities at chromatin control Plk1 re-activation to determine G2 checkpoint duration. EMBO J. 2017, 36, 2161–2176. [Google Scholar] [CrossRef]
- Choi, B.-K.; Fujiwara, K.; Dayaram, T.; Darlington, Y.; Dickerson, J.; Goodell, M.A.; Donehower, L.A. WIP1 dephosphorylation of p27 Kip1 Serine 140 destabilizes p27 Kip1 and reverses anti-proliferative effects of ATM phosphorylation. Cell Cycle 2020, 1–13. [Google Scholar] [CrossRef]
- Rodríguez, A.; Jesús Naveja, J.; Torres, L.; De Teresa, B.G.; Juárez-Figueroa, U.; Ayala-Zambrano, C.; Azpeitia, E.; Mendoza, L.; Frías, S. WIP1 contributes to the adaptation of fanconi anemia cells to DNA damage as determined by the regulatory network of the fanconi anemia and checkpoint recovery pathways. Front. Genet. 2019, 10, 411. [Google Scholar] [CrossRef]
- Lu, X. PPM1D dephosphorylates Chk1 and p53 and abrogates cell cycle checkpoints. Genes Dev. 2005, 19, 1162–1174. [Google Scholar] [CrossRef]
- Zhang, X.; Lin, L.; Guo, H.; Yang, J.; Jones, S.N.; Jochemsen, A.; Lu, X. Phosphorylation and Degradation of MdmX Is Inhibited by Wip1 Phosphatase in the DNA Damage Response. Cancer Res. 2009, 69, 7960–7968. [Google Scholar] [CrossRef]
- Lu, X.; Ma, O.; Nguyen, T.-A.; Jones, S.N.; Oren, M.; Donehower, L.A. The Wip1 Phosphatase Acts as a Gatekeeper in the p53-Mdm2 Autoregulatory Loop. Cancer Cell 2007, 12, 342–354. [Google Scholar] [CrossRef]
- Goloudina, A.R.; Kochetkova, E.Y.; Pospelova, T.V.; Demidov, O.N. Wip1 phosphatase: Between p53 and MAPK kinases pathways. Oncotarget 2016, 7, 31563. [Google Scholar] [CrossRef] [PubMed]
- Lindqvist, A.; de Bruijn, M.; Macurek, L.; Brás, A.; Mensinga, A.; Bruinsma, W.; Voets, O.; Kranenburg, O.; Medema, R.H. Wip1 confers G2 checkpoint recovery competence by counteracting p53-dependent transcriptional repression. EMBO J. 2009, 28, 3196–3206. [Google Scholar] [CrossRef] [PubMed]
- Castellino, R.C.; De Bortoli, M.; Lu, X.; Moon, S.-H.; Nguyen, T.-A.; Shepard, M.A.; Rao, P.H.; Donehower, L.A.; Kim, J.Y.H. Medulloblastomas overexpress the p53-inactivating oncogene WIP1/PPM1D. J. Neurooncol. 2008, 86, 245–256. [Google Scholar] [CrossRef]
- Bedognetti, D.; Hendrickx, W.; Marincola, F.M.; Miller, L.D. Prognostic and predictive immune gene signatures in breast cancer. Curr. Opin. Oncol. 2015, 27, 433–444. [Google Scholar] [CrossRef] [PubMed]
- Kleiblova, P.; Shaltiel, I.A.; Benada, J.; Ševčík, J.; Pecháčková, S.; Pohlreich, P.; Voest, E.E.; Dundr, P.; Bartek, J.; Kleibl, Z.; et al. Gain-of-function mutations of PPM1D/Wip1 impair the p53-dependent G1 checkpoint. J. Cell Biol. 2013, 201, 511–521. [Google Scholar] [CrossRef] [PubMed]
- Peng, T.-S.; He, Y.-H.; Nie, T.; Hu, X.-D.; Lu, H.-Y.; Yi, J.; Shuai, Y.-F.; Luo, M. PPM1D is a prognostic marker and therapeutic target in colorectal cancer. Exp. Ther. Med. 2014, 8, 430–434. [Google Scholar] [CrossRef]
- Li, K.; Liu, Y.; Xu, S.; Wang, J. PPM1D Functions as Oncogene and is Associated with Poor Prognosis in Esophageal Squamous Cell Carcinoma. Pathol. Oncol. Res. 2018. [Google Scholar] [CrossRef]
- Ma, D.; Zhang, C.-J.; Chen, Z.-L.; Yang, H. Prognostic value of PPM1D in 800 gastric cancer patients. Mol. Med. Rep. 2014, 10, 191–194. [Google Scholar] [CrossRef] [PubMed]
- Jiao, L.; Shen, D.; Liu, G.; Jia, J.; Geng, J.; Wang, H.; Sun, Y. PPM1D as a novel biomarker for prostate cancer after radical prostatectomy. Anticancer Res. 2014, 34, 2919–2925. [Google Scholar]
- Wang, Z.-P.; Chen, S.-Y.; Tian, Y. Wild-type p53-induced phosphatase 1 is a prognostic marker and therapeutic target in bladder transitional cell carcinoma. Oncol. Lett. 2017, 13, 875–880. [Google Scholar] [CrossRef] [PubMed]
- Li, G.-B.; Zhang, X.-L.; Yuan, L.; Jiao, Q.-Q.; Liu, D.-J.; Liu, J. Protein Phosphatase Magnesium-Dependent 1δ (PPM1D) mRNA Expression Is a Prognosis Marker for Hepatocellular Carcinoma. PLoS ONE 2013, 8, e60775. [Google Scholar] [CrossRef]
- Swisher, E.M.; Harrell, M.I.; Norquist, B.M.; Walsh, T.; Brady, M.; Lee, M.; Hershberg, R.; Kalli, K.R.; Lankes, H.; Konnick, E.Q.; et al. Somatic Mosaic Mutations in PPM1D and TP53 in the Blood of Women With Ovarian Carcinoma. JAMA Oncol. 2016, 2, 370–372. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.; Won, D.; Lee, S.-T.; Choi, J.R. Somatic mosaic truncating mutations of PPM1D in blood can result from expansion of a mutant clone under selective pressure of chemotherapy. PLoS ONE 2019, 14, e0217521. [Google Scholar] [CrossRef]
- Hsu, J.I.; Dayaram, T.; Tovy, A.; De Braekeleer, E.; Jeong, M.; Wang, F.; Zhang, J.; Heffernan, T.P.; Gera, S.; Kovacs, J.J.; et al. PPM1D Mutations Drive Clonal Hematopoiesis in Response to Cytotoxic Chemotherapy. Cell Stem Cell 2018, 23, 700–713.e6. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-Related Clonal Hematopoiesis Associated with Adverse Outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef] [PubMed]
- Genovese, G.; Kähler, A.K.; Handsaker, R.E.; Lindberg, J.; Rose, S.A.; Bakhoum, S.F.; Chambert, K.; Mick, E.; Neale, B.M.; Fromer, M.; et al. Clonal Hematopoiesis and Blood-Cancer Risk Inferred from Blood DNA Sequence. N. Engl. J. Med. 2014, 371, 2477–2487. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Lu, C.; Wang, J.; Mclellan, M.D.; Kimberly, J.; Wendl, M.C.; Mcmichael, J.F.; Schmidt, H.K.; Miller, C.A.; Ozenberger, B.A.; et al. Age-related cancer mutations associated with clonal hematopoietic expansion. Nat. Med. 2015, 20, 1472–1478. [Google Scholar] [CrossRef] [PubMed]
- Kahn, J.D.; Miller, P.G.; Silver, A.J.; Sellar, R.S.; Bhatt, S.; Gibson, C.; McConkey, M.; Adams, D.; Mar, B.; Mertins, P.; et al. PPM1D truncating mutations confer resistance to chemotherapy and sensitivity to PPM1D inhibition in hematopoietic cells. Blood 2018, 132, 1095–1105. [Google Scholar] [CrossRef]
- Esfandiari, A.; Hawthorne, T.A.; Nakjang, S.; Lunec, J. Chemical Inhibition of Wild-Type p53-Induced Phosphatase 1 (WIP1/PPM1D) by GSK2830371 Potentiates the Sensitivity to MDM2 Inhibitors in a p53-Dependent Manner. Mol. Cancer Ther. 2016, 15, 379–391. [Google Scholar] [CrossRef]
- Zhang, M.; Xu, E.; Zhang, J.; Chen, X. PPM1D phosphatase, a target of p53 and RBM38 RNA-binding protein, inhibits p53 mRNA translation via dephosphorylation of RBM38. Oncogene 2015, 34, 5900–5901. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Esfandiari, A.; Ho, Y.; Wang, N.; Mahdi, A.K.; Aptullahoglu, E.; Lovat, P.; Lunec, J. Targeting negative regulation of p53 by MDM2 and WIP1 as a therapeutic strategy in cutaneous melanoma. Nat. Publ. Gr. 2017, 118, 495–508. [Google Scholar] [CrossRef] [PubMed]
- Pechackova, S.; Burdova, K.; Benada, J.; Kleiblova, P.; Jenikova, G.; Macurek, L. Inhibition of WIP1 phosphatase sensitizes breast cancer cells to genotoxic stress and to MDM2 antagonist nutlin-3. Oncotarget 2016, 7, 14458–14475. [Google Scholar] [CrossRef] [PubMed]
- Rücker, F.G.; Schlenk, R.F.; Bullinger, L.; Kayser, S.; Teleanu, V.; Kett, H.; Habdank, M.; Kugler, C.M.; Holzmann, K.; Gaidzik, V.I.; et al. TP53 alterations in acute myeloid leukemia with complex karyotype correlate with specific copy number alterations, monosomal karyotype, and dismal outcome. Blood 2012, 119, 2114–2121. [Google Scholar] [CrossRef]
- Simonetti, G.; Padella, A.; do Valle, I.F.; Fontana, M.C.; Fonzi, E.; Bruno, S.; Baldazzi, C.; Guadagnuolo, V.; Manfrini, M.; Ferrari, A.; et al. Aneuploid acute myeloid leukemia exhibits a signature of genomic alterations in the cell cycle and protein degradation machinery. Cancer 2018. [Google Scholar] [CrossRef]
- Prokocimer, M.; Molchadsky, A.; Rotter, V. Dysfunctional diversity of p53 proteins in adult acute myeloid leukemia: Projections on diagnostic workup and therapy. Blood 2017, 130, 699–712. [Google Scholar] [CrossRef]
- Kojima, K.; Konopleva, M.; Samudio, I.J.; Shikami, M.; Cabreira-Hansen, M.; McQueen, T.; Ruvolo, V.; Tsao, T.; Zeng, Z.; Vassilev, L.T.; et al. MDM2 antagonists induce p53-dependent apoptosis in AML: Implications for leukemia therapy. Blood 2005, 106, 3150–3159. [Google Scholar] [CrossRef] [PubMed]
- Seipel, K.; Marques, M.A.T.; Sidler, C.; Mueller, B.U.; Pabst, T. The Cellular p53 Inhibitor MDM2 and the Growth Factor Receptor FLT3 as Biomarkers for Treatment Responses to the MDM2-Inhibitor Idasanutlin and the MEK1 Inhibitor Cobimetinib in Acute Myeloid Leukemia. Cancers (Basel) 2018, 10, 170. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, C.; Friess, T.; Birzele, F.; Kiialainen, A.; Dangl, M. Superior anti-tumor activity of the MDM2 antagonist idasanutlin and the Bcl-2 inhibitor venetoclax in p53 wild-type acute myeloid leukemia models. J. Hematol. Oncol. 2016, 9, 50. [Google Scholar] [CrossRef]
- Daver, N.G.; Pollyea, D.A.; Garcia, J.S.; Jonas, B.A.; Yee, K.W.L.; Fenaux, P.; Assouline, S.; Vey, N.; Olin, R.; Roboz, G.J.; et al. Safety, Efficacy, Pharmacokinetic (PK) and Biomarker Analyses of BCL2 Inhibitor Venetoclax (Ven) Plus MDM2 Inhibitor Idasanutlin (idasa) in Patients (pts) with Relapsed or Refractory (R/R) AML: A Phase Ib, Non-Randomized, Open-Label Study. Blood 2018, 132, 767. [Google Scholar] [CrossRef]
- Reis, B.; Jukofsky, L.; Chen, G.; Martinelli, G.; Zhong, H.; So, V.; Dickinson, M.J.; Drummond, M.; Assouline, S.; Hashemyan, M.; et al. Acute myeloid leukemia patients’ clinical response to idasanutlin (RG7388) is associated with pre-treatment MDM2 protein expression in leukemic blasts. Haematologica 2016, 101, e185–e188. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, R.; De Matteis, S.; Carloni, S.; Bruno, S.; Abbati, G.; Capelli, L.; Ghetti, M.; Bochicchio, M.; Liverani, C.; Mercatali, L.; et al. Kevetrin induces apoptosis in TP53 wild-type and mutant acute myeloid leukemia cells. Oncol. Rep. 2020, 44, 1561–1573. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.-I.; Lin, T.-C.; Chou, W.-C.; Tang, J.-L.; Lin, D.-T.; Tien, H.-F. A novel fluorescence-based multiplex PCR assay for rapid simultaneous detection of CEBPA mutations and NPM mutations in patients with acute myeloid leukemias. Leukemia 2006, 20, 1899–1903. [Google Scholar] [CrossRef] [PubMed]
- Thiede, C.; Steudel, C.; Mohr, B.; Schaich, M.; Schäkel, U.; Platzbecker, U.; Wermke, M.; Bornhäuser, M.; Ritter, M.; Neubauer, A.; et al. Analysis of FLT3-activating mutations in 979 patients with acute myelogenous leukemia: Association with FAB subtypes and identification of subgroups with poor prognosis. Blood 2002, 99, 4326–4335. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.C. Drug combination studies and their synergy quantification using the chou-talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.W.; Wouters, B.J.; Erpelinck, C.A.J.; Abbas, S.; Beverloo, H.B.; Lugthart, S.; Löwenberg, B.; Delwel, R.; Valk, P.J.M. Prediction of molecular subtypes in acute myeloid leukemia based on gene expression profiling. Haematologica 2009, 94, 131–134. [Google Scholar] [CrossRef] [PubMed]
- Kohlmann, A.; Kipps, T.J.; Rassenti, L.Z.; Downing, J.R.; Shurtleff, S.A.; Mills, K.I.; Gilkes, A.F.; Hofmann, W.-K.; Basso, G.; Dell’orto, M.C.; et al. An international standardization programme towards the application of gene expression profiling in routine leukaemia diagnostics: The Microarray Innovations in LEukemia study prephase. Br. J. Haematol. 2008, 142, 802–807. [Google Scholar] [CrossRef]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef]
- Fischer, M. Census and evaluation of p53 target genes. Oncogene 2017, 36, 3943–3956. [Google Scholar] [CrossRef] [PubMed]
- Zajkowicz, A.; Butkiewicz, D.; Drosik, A.; Giglok, M.; Suwiński, R.; Rusin, M. Truncating mutations of PPM1D are found in blood DNA samples of lung cancer patients. Br. J. Cancer 2015, 112, 1114–1120. [Google Scholar] [CrossRef]
- Clausse, V.; Goloudina, A.R.; Uyanik, B.; Kochetkova, E.Y.; Richaud, S.; Fedorova, O.A.; Hammann, A.; Bardou, M.; Barlev, N.A.; Garrido, C.; et al. Wee1 inhibition potentiates Wip1-dependent p53-negative tumor cell death during chemotherapy. Cell Death Dis. 2016, 7, e2195. [Google Scholar] [CrossRef] [PubMed]
- Yu, E.; Ahn, Y.S.; Jang, S.J.; Kim, M.-J.; Yoon, H.S.; Gong, G.; Choi, J. Overexpression of the wip1 gene abrogates the p38 MAPK/p53/Wip1 pathway and silences p16 expression in human breast cancers. Breast Cancer Res. Treat. 2007, 101, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Uyanik, B.; Grigorash, B.B.; Goloudina, A.R.; Demidov, O.N. DNA damage-induced phosphatase Wip1 in regulation of hematopoiesis, immune system and inflammation. Cell Death Discov. 2017, 3, 17018. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Hu, J.; He, D.; Chen, Q.; Liu, S.; Zhu, X.; Li, B. Potentiality of Protein phosphatase Mg 2+ /Mn 2+ dependent 1D as a biomarker for predicting prognosis in acute myeloid leukemia patients. J. Clin. Lab. Anal. 2020, 34, e23171. [Google Scholar] [CrossRef] [PubMed]
- Kozakai, Y.; Kamada, R.; Furuta, J.; Kiyota, Y.; Chuman, Y.; Sakaguchi, K. PPM1D controls nucleolar formation by up-regulating phosphorylation of nucleophosmin. Sci. Rep. 2016, 6, 33272. [Google Scholar] [CrossRef]
- Chan, N.; Meng Lim, T. Cytoplasmic nucleophosmin has elevated T199 phosphorylation upon which G2/M phase progression is dependent. Sci. Rep. 2015, 5, 11777. [Google Scholar] [CrossRef]
- Krześniak, M.; Zajkowicz, A.; Gdowicz-Kłosok, A.; Głowala-Kosińska, M.; Łasut-Szyszka, B.; Rusin, M. Synergistic activation of p53 by actinomycin D and nutlin-3a is associated with the upregulation of crucial regulators and effectors of innate immunity. Cell. Signal. 2020, 69, 109552. [Google Scholar] [CrossRef]
- Gamble, L.D.; Kees, U.R.; Tweddle, D.A.; Lunec, J. MYCN sensitizes neuroblastoma to the MDM2-p53 antagonists Nutlin-3 and MI-63. Oncogene 2012, 31, 752–763. [Google Scholar] [CrossRef]
- Phesse, T.J.; Myant, K.B.; Cole, A.M.; Ridgway, R.A.; Pearson, H.; Muncan, V.; van den Brink, G.R.; Vousden, K.H.; Sears, R.; Vassilev, L.T.; et al. Endogenous c-Myc is essential for p53-induced apoptosis in response to DNA damage in vivo. Cell Death Differ. 2014, 21, 956–966. [Google Scholar] [CrossRef]
- Cheng, Q.; Chen, J. Mechanism of p53 stabilization by ATM after DNA damage. Cell Cycle 2010, 9, 472–478. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | GSE6891 [48] (n = 499) | GSE13159 [49] (n = 458) | TCGA [50] (n = 178) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| PPM1D Median | n (%) | p-Value | PPM1D Median | n (%) | p-Value | PPM1D Median (Min–Max) | n (%) | p-Value | |
| (Min–Max) | (Min–Max) | ||||||||
| Age ‡ | |||||||||
| <60-years | 151.6 (83.9–393.3) | 417 (84.8) | 0.03 | NA | NA | NA | 25.6 (15.1–46.7) | 92 (51.7) | 0.004 |
| ≥60-years | 146.7 (94.5–234.4) | 75 (15.2) | 22.6 (14.2–39.4) | 86 (48.3) | |||||
| NA = 7 | |||||||||
| Cytogenetic group ‡ | |||||||||
| t(8;21) | 169.8 (89.9–318.9) | 38 (9.0) | 0.185 | 106.2 (32.6–309.9) | 35 (7.6) | <0.001 | 25.7 (22.3–31.7) | 7 (4.2) | 0.031 |
| inv(16)/t(16;16) | 156.3 (95.8–319.7) | 42 (10.0) | 73.2 (41.4–134.6) | 27 (5.9) | 27.6 (20.5–38.7) | 11 (6.6) | |||
| NK | 147.5 (83.9–371.3) | 171 (40.5) | NA | 0 | 23.5 (15.1–46.7) | 96 (57.8) | |||
| CK | 146.4 (97.9–259.7) | 34 (8.1) | 99.3 (31.5–279.4) | 45 (9.8) | 24.4 (15.2–39.0) | 21 (12.7) | |||
| KMT2A-r | 139.6 (94.3–256.3) | 17 (4.0) | 72.2 (28.2–233.4) | 29 (6.3) | 29.1 (18.3–46.1) | 10 (6.0) | |||
| Other | 143.1 (84.1–393.3) | 120 (28.4) | NA | 0 | 21.9 (14.2–29.5) | 21 (12.7) | |||
| Normal/Other * | 72.2 (23.4–263.2) | 322 (21.8) | |||||||
| NA = 77 | NA = 12 | ||||||||
| FLT3-ITD ‡ | |||||||||
| FLT3-ITD+ | 146.1 (87.6–371.3) | 134 (26.9) | 0.017 | NA | NA | NA | 24.2 (14.2–46.7) | 32 (18.4) | 0.87 |
| FLT3-ITD− | 152.2 (83.9–393.3) | 365 (73.1) | 23.2 (15.2–39.0) | 143 (81.7) | |||||
| NA = 3 | |||||||||
| NPM1 status ‡ | |||||||||
| NPM1-mut | 143.1 (83.9–371.3) | 159 (39.9) | 0.002 | NA | NA | NA | 23.7 (16.1–46.7) | 53 (30.3) | 0.69 |
| NPM1-wt | 155.8 (84.1–393.3) | 340 (68.1) | 24.0 (14.2–46.1) | 122 (69.7) | |||||
| NA = 3 | |||||||||
| Pathway Name | NES | FDR | Reatment Comparison |
|---|---|---|---|
| Hallmark of p53 Pathway | 2.31 | ≤0.001 | Nut3a+WIP1i vs. DMSO |
| 2.13 | ≤0.001 | Nut3a vs. DMSO | |
| 1.49 | 0.029 | WIP1i vs. DMSO | |
| Hallmark of Protein Secretion | 1.98 | ≤0.001 | Nut3a+WIP1i vs. DMSO |
| 2.24 | ≤0.001 | Nut3a vs. DMSO | |
| 1.73 | 0.005 | WIP1i vs. DMSO | |
| Hallmark of Oxidative Phosphorylation | 1.87 | 0.001 | Nut3a+WIP1i vs. DMSO |
| 1.94 | ≤0.001 | Nut3a vs. DMSO | |
| 2.31 | 0.005 | WIP1i vs. DMSO | |
| Hallmark of Apoptosis | 1.70 | 0.009 | Nut3a+WIP1i vs. DMSO |
| 1.70 | 0.003 | Nut3a vs. DMSO | |
| 1.52 | 0.024 | WIP1i vs. DMSO | |
| Hallmark of mTORC1 Signaling | 1.75 | 0.002 | Nut3a vs. DMSO |
| 1.88 | 0.003 | WIP1i vs. DMSO | |
| Hallmark of Fatty Acid Metabolism | 1.75 | 0.002 | Nut3a vs. DMSO |
| 1.62 | 0.010 | WIP1i vs. DMSO | |
| Hallmark of DNA Repair | 1.58 | 0.014 | Nut3a vs. DMSO |
| 1.63 | 0.011 | WIP1i vs. DMSO | |
| Hallmark of Peroxisome | 1.52 | 0.024 | Nut3a vs. DMSO |
| 1.80 | 0.005 | WIP1i vs. DMSO | |
| Hallmark of TNFA Signaling Via NFKB | 1.57 | 0.021 | Nut3a+WIP1i vs. DMSO |
| Hallmark of Interferon Gamma Response | 1.53 | 0.022 | Nut3a+WIP1i vs. DMSO |
| Hallmark of Interferon Alpha Response | 1.54 | 0.023 | Nut3a+WIP1i vs. DMSO |
| Hallmark of PI3K AKT mTOR Signaling | 1.73 | 0.002 | Nut3a vs. DMSO |
| Positive Regulation Of Cysteine Type Endopeptidase Activity Involved In Apoptotic Signaling Pathway | 2.21 | 0.030 | Nut3a vs. DMSO |
| Oligosaccharide-Lipid Intermediate Biosynthetic Process | 2.19 | 0.013 | Nut3a vs. DMSO |
| Hallmark of MYC Targets_V2 | 1.72 | 0.006 | WIP1i vs. DMSO |
| Hallmark of Unfolded Protein Response | 1.67 | 0.008 | WIP1i vs. DMSO |
| Hallmark of MYC Targets_V1 | 1.62 | 0.011 | WIP1i vs. DMSO |
| Pathway Name | NES | FDR | Treatment Comparison |
|---|---|---|---|
| Hallmark of Inflammatory Response | 1.99 | 0.001 | Nut3a+WIP1i vs. DMSO |
| 1.53 | 0.043 | Nut3a vs. DMSO | |
| Hallmark of TNFA Signaling Via NFKB | 2.02 | 0.002 | Nut3a+WIP1i vs. DMSO |
| 1.52 | 0.022 | Nut3a vs. DMSO |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fontana, M.C.; Nanni, J.; Ghelli Luserna di Rorà, A.; Petracci, E.; Padella, A.; Ghetti, M.; Ferrari, A.; Marconi, G.; Soverini, S.; Iacobucci, I.; et al. Pharmacological Inhibition of WIP1 Sensitizes Acute Myeloid Leukemia Cells to the MDM2 Inhibitor Nutlin-3a. Biomedicines 2021, 9, 388. https://doi.org/10.3390/biomedicines9040388
Fontana MC, Nanni J, Ghelli Luserna di Rorà A, Petracci E, Padella A, Ghetti M, Ferrari A, Marconi G, Soverini S, Iacobucci I, et al. Pharmacological Inhibition of WIP1 Sensitizes Acute Myeloid Leukemia Cells to the MDM2 Inhibitor Nutlin-3a. Biomedicines. 2021; 9(4):388. https://doi.org/10.3390/biomedicines9040388
Chicago/Turabian StyleFontana, Maria Chiara, Jacopo Nanni, Andrea Ghelli Luserna di Rorà, Elisabetta Petracci, Antonella Padella, Martina Ghetti, Anna Ferrari, Giovanni Marconi, Simona Soverini, Ilaria Iacobucci, and et al. 2021. "Pharmacological Inhibition of WIP1 Sensitizes Acute Myeloid Leukemia Cells to the MDM2 Inhibitor Nutlin-3a" Biomedicines 9, no. 4: 388. https://doi.org/10.3390/biomedicines9040388
APA StyleFontana, M. C., Nanni, J., Ghelli Luserna di Rorà, A., Petracci, E., Padella, A., Ghetti, M., Ferrari, A., Marconi, G., Soverini, S., Iacobucci, I., Papayannidis, C., Curti, A., Audisio, E., Giannini, M. B., Rondoni, M., Lanza, F., Cavo, M., Martinelli, G., & Simonetti, G. (2021). Pharmacological Inhibition of WIP1 Sensitizes Acute Myeloid Leukemia Cells to the MDM2 Inhibitor Nutlin-3a. Biomedicines, 9(4), 388. https://doi.org/10.3390/biomedicines9040388